
Longitudinal Dynamics of Human B-Cell Response at the Single-Cell Level in Response to Tdap Vaccination

 , , ,
, , ,  , and
, and 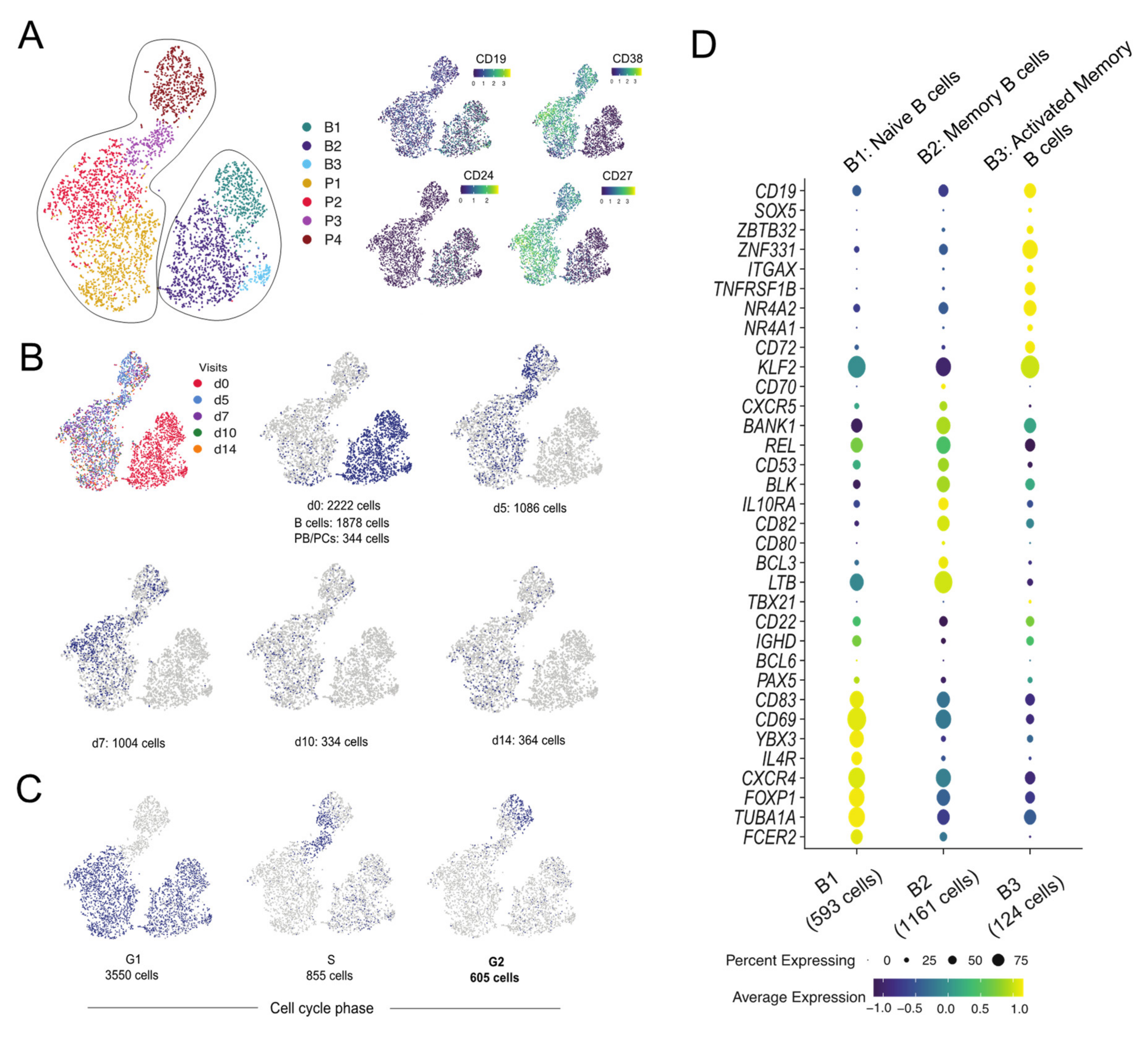{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Inclusion Criteria and Informed Consent from the Subject
2.2. High Throughput B-Cell and Plasmablast/Plasma Cell Sorting
2.3. 10x CITE-Seq Transcriptomics and scBCR-Rep Sequencing
2.4. Unsupervised Clustering of Single-Cell Transcriptomics Data
2.5. Differential Gene Expression Analysis
2.6. Identification of V(D)JC Genes in the Reconstructed BCRs
2.7. Identification of B-Cell Clonotypes
2.8. Analysis of scBCR-Rep and Clonotypes
2.9. Query Tool to Identify Anti-Toxoid BCRs
3. Results
3.1. Single-Cell Transcriptome Landscape of the Cells from a Vaccinated Individual
3.1.1. Majority of the PB/PCs Derived from d5 and d7 Post-Vaccination
3.1.2. Proliferating Cells Are Clustered in One Cluster
3.1.3. B1-B3 Clusters Correspond to Naive, Memory and Activated Memory B Cells, Respectively
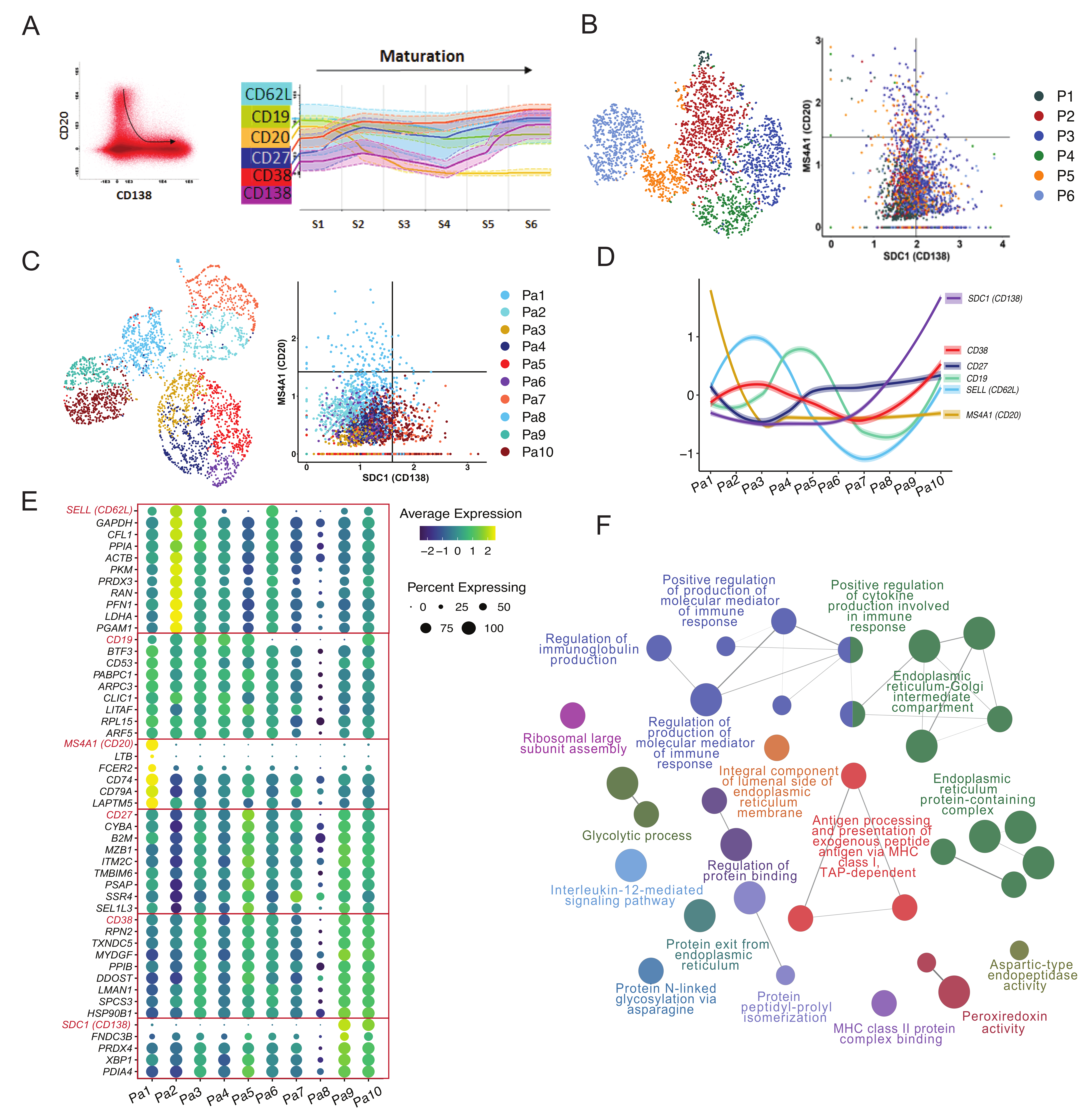
3.2. Flow Cytometry Guided Plasma Cell Maturation in the Single-Cell Transcriptome
3.2.1. Staging of PB/PCs Based on Flow Cytometry Markers
3.2.2. Pathways and Additional Membrane Markers Identified in Relation to the Maturation of PB/PCs
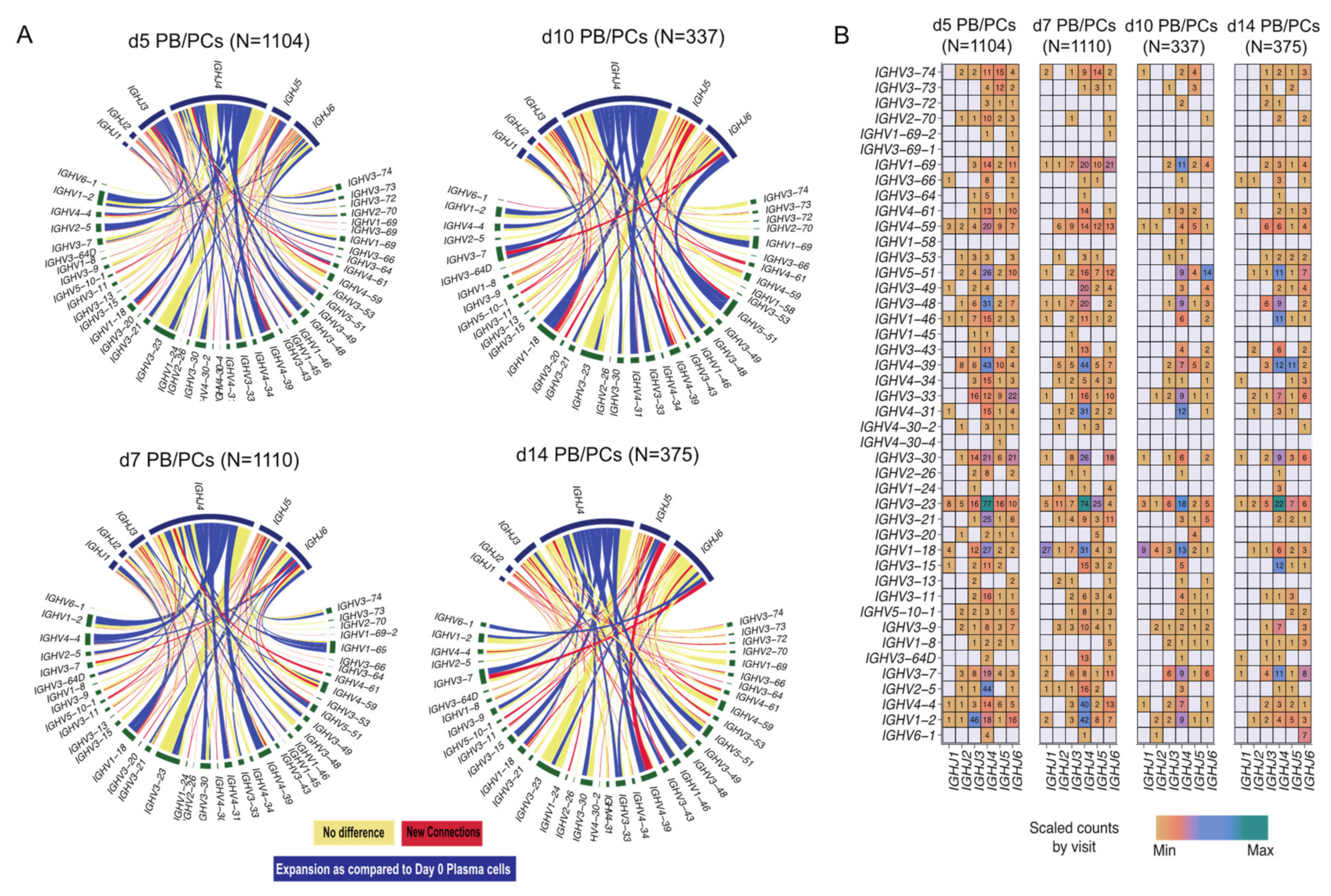
3.3. Characteristics of V(D)J Usage over Time upon Vaccination
3.3.1. Identification of Ig Heavy and Light Chains in Majority of the Sequenced Cells
3.3.2. Cells with Different IGHV and IGHJ Are Dominant at Different Time Points
3.3.3. IGHV3 and IGHJ4 Genes Predominant in d0 PB/PCs
3.3.4. Few Clonotypes Expand with Delay at d10
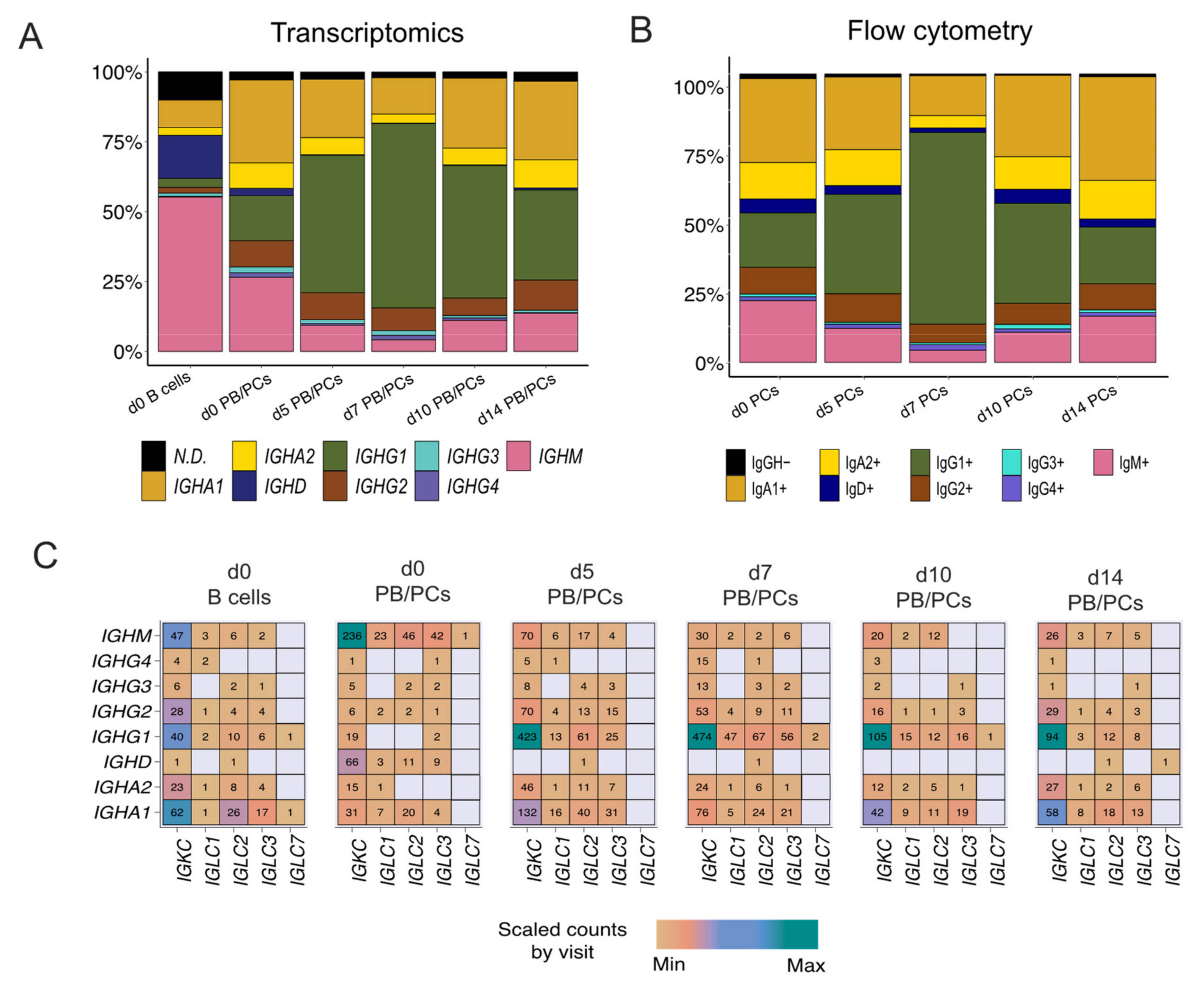
3.4. Constant Gene Usage over Time upon Vaccination
IGHG1 Constant Gene Predominant at the Peak of the PB/PCs
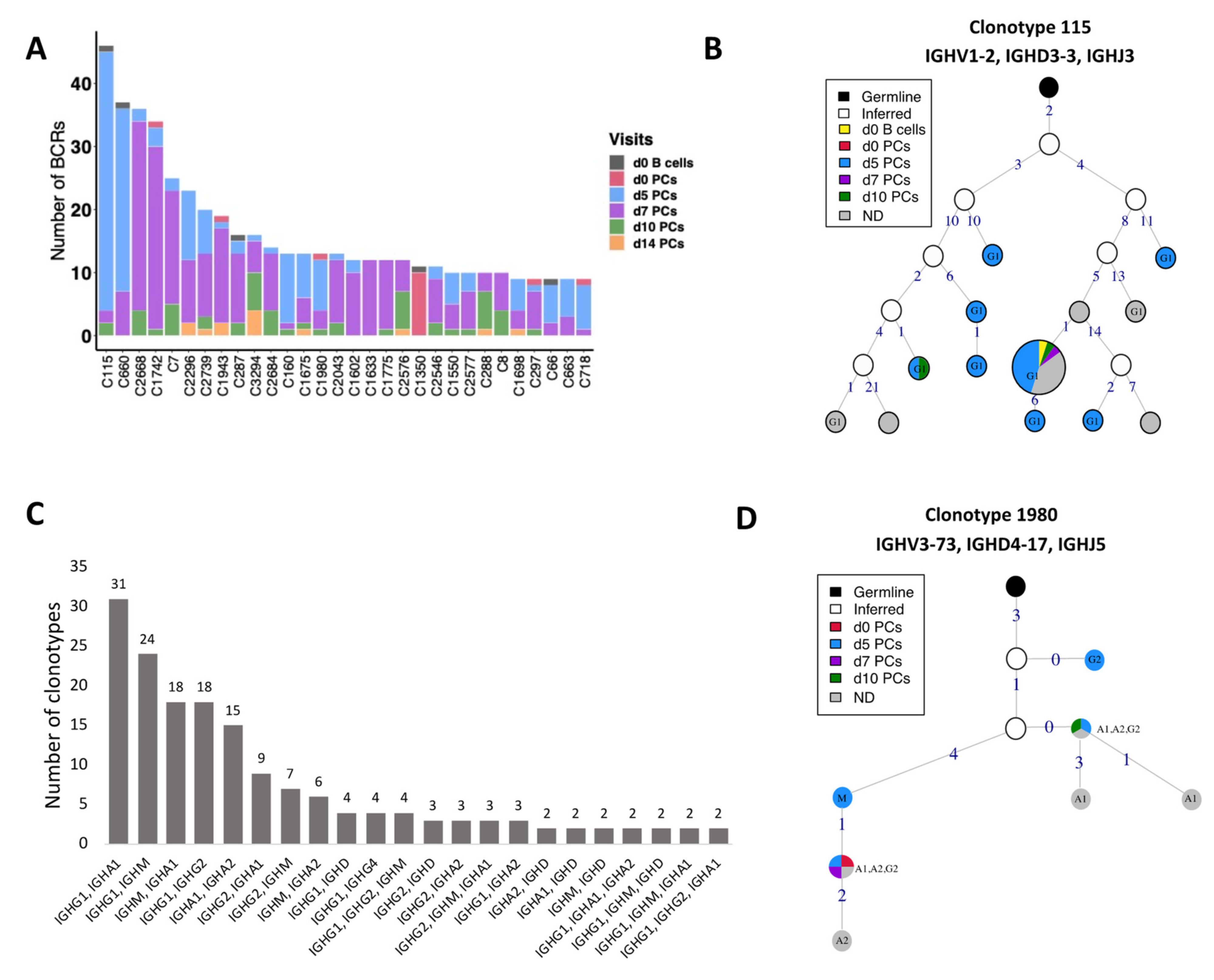
3.5. BCR Clonotypes over Time upon Vaccination
3.5.1. Identification of Clonotypes Based on V Family Grouping
3.5.2. CSR Events within the Clonotypes
3.5.3. Clonal Lineages of the Clonotypes with CSR Events
3.5.4. Comparable Length of Most of the Junctions
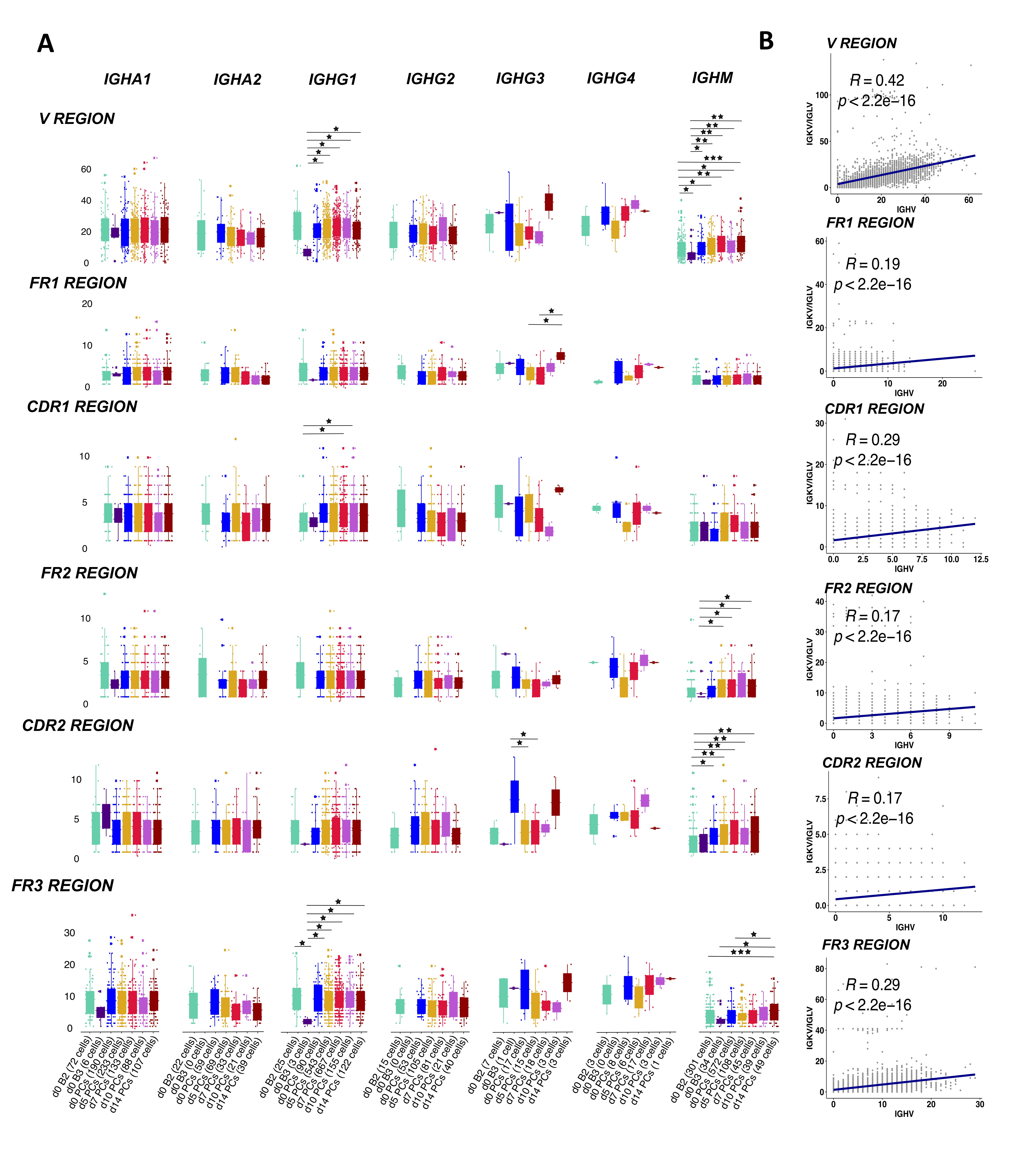
3.6. Mutation Load in BCRs upon Vaccination
3.6.1. Positively Correlated Mutations in Heavy and Light Chain Genes
3.6.2. Positive Selection of Mutations in the CDR Region over Time
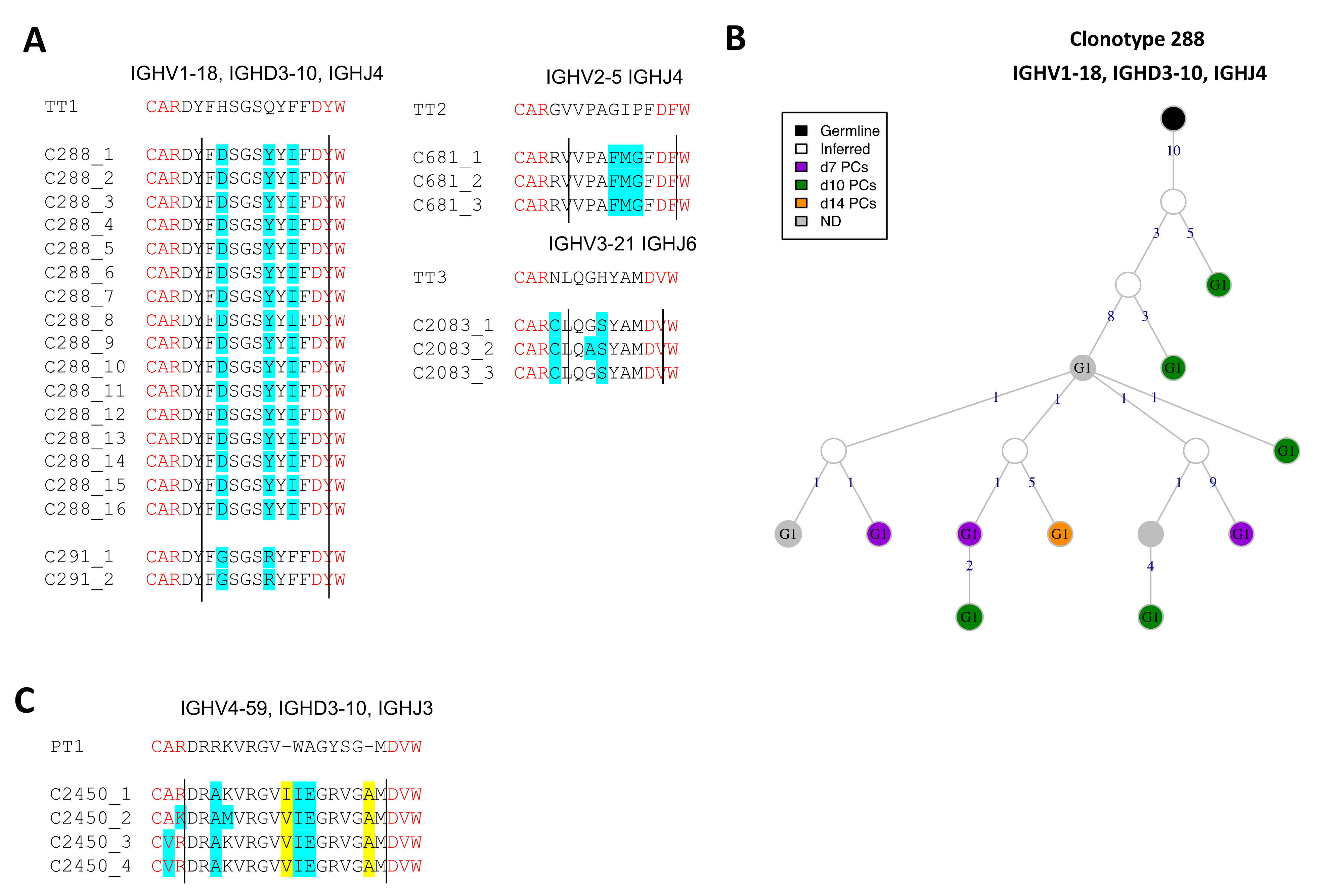
3.7. BCRs Specific to Vaccine Toxoids
3.7.1. Identification of Conserved Anti-TT BCRs
3.7.2. Identification of BCRs (Un-)Likely Related to Anti-PT BCRs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Eisen, H.N. Affinity enhancement of antibodies: How low-affinity antibodies produced early in immune responses are followed by high-affinity antibodies later and in memory B-cell responses. Cancer Immunol. Res. 2014, 2, 381–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammarlund, E.; Thomas, A.; Amanna, I.J.; Holden, L.A.; Slayden, O.D.; Park, B.; Gao, L.; Slifka, M.K. Plasma cell survival in the absence of B cell memory. Nat. Commun. 2017, 8, 1781. [Google Scholar] [CrossRef] [Green Version]
- Imani, F.; Kehoe, K.E. Infection of human B lymphocytes with MMR vaccine induces IgE class switching. Clin. Immunol. 2001, 100, 355–361. [Google Scholar] [CrossRef] [Green Version]
- Pan-Hammarström, Q.; Zhao, Y.; Hammarström, L. Class Switch Recombination: A Comparison Between Mouse and Human. Adv. Immunol. 2007, 93, 1–61. [Google Scholar]
- Diks, A.M.; Khatri, I.; Oosten, L.E.; De Mooij, B.; Groenland, R.J.; Teodosio, C.; Perez-Andres, M.; Orfao, A.; Berbers, G.A.M.; Zwaginga, J.J.; et al. Highly sensitive flow cytometry allows monitoring of changes in circulating immune cells in blood after Tdap booster vaccination. Front. Immunol. 2021, 12, 2091. [Google Scholar] [CrossRef]
- GlaxoSmithKline. BOOSTRIX (Tetanus Toxoid, Reduced Diphtheria Toxoid and Acellular Pertussis Vaccine, Adsorbed) Injectable Suspension, for Intramuscular Use; GlaxoSmithKline: Research Triangle Park, NC, USA, 2020. [Google Scholar]
- Ellebedy, A.H.; Jackson, K.J.L.; Kissick, H.T.; Nakaya, H.I.; Davis, C.W.; Roskin, K.M.; McElroy, A.K.; Oshansky, C.M.; Elbein, R.; Thomas, S.; et al. Defining antigen-specific plasmablast and memory B cell subsets in human blood after viral infection or vaccination. Nat. Immunol. 2016, 17, 1226–1234. [Google Scholar] [CrossRef]
- Wrammert, J.; Smith, K.; Miller, J.; Langley, W.A.; Kokko, K.; Larsen, C.; Zheng, N.Y.; Mays, I.; Garman, L.; Helms, C.; et al. Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature 2008, 453, 667–671. [Google Scholar] [CrossRef] [Green Version]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M.; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902.e21. [Google Scholar] [CrossRef] [PubMed]
- Mylka, V.; Aerts, J.; Matetovici, I.; Poovathingal, S.; Vandamme, N.; Seurinck, R.; Hulselmans, G.; Van Den Hoecke, S.; Wils, H.; Reumers, J.; et al. Comparative analysis of antibody-and lipid-based multiplexing methods for single-cell RNA-seq. bioRxiv 2020. [Google Scholar] [CrossRef]
- Kernfeld, E.M.; Genga, R.M.J.; Neherin, K.; Magaletta, M.E.; Xu, P.; Maehr, R. A Single-Cell Transcriptomic Atlas of Thymus Organogenesis Resolves Cell Types and Developmental Maturation. Immunity 2018, 48, 1258–1270.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [Green Version]
- Bischof, J.; Ibrahim, S.M. bcRep: R Package for Comprehensive Analysis of B Cell Receptor Repertoire Data. PLoS ONE 2016, 11, e0161569. [Google Scholar] [CrossRef]
- Zuckerman, N.S.; Hazanov, H.; Barak, M.; Edelman, H.; Hess, S.; Shcolnik, H.; Dunn-Walters, D.; Mehr, R. Somatic hypermutation and antigen-driven selection of B cells are altered in autoimmune diseases. J. Autoimmun. 2010, 35, 325–335. [Google Scholar] [CrossRef]
- Stern, J.N.H.; Yaari, G.; Vander Heiden, J.A.; Church, G.; Donahue, W.F.; Hintzen, R.Q.; Huttner, A.J.; Laman, J.D.; Nagra, R.M.; Nylander, A.; et al. B cells populating the multiple sclerosis brain mature in the draining cervical lymph nodes. Sci. Transl. Med. 2014, 6, 248ra107. [Google Scholar] [CrossRef] [Green Version]
- Berkowska, M.A.; Schickel, J.-N.; Grosserichter-Wagener, C.; de Ridder, D.; Ng, Y.S.; van Dongen, J.J.M.; Meffre, E.; van Zelm, M.C. Circulating Human CD27 − IgA + Memory B Cells Recognize Bacteria with Polyreactive Igs. J. Immunol. 2015, 195, 1417–1426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toellner, K.M. FOXP1 inhibits plasma cell differentiation. Blood 2015, 126, 2076–2077. [Google Scholar] [CrossRef] [Green Version]
- Hövelmeyer, N.; Wörns, M.A.; Reissig, S.; Adams-Quack, P.; Leclaire, J.; Hahn, M.; Wörtge, S.; Nikolaev, A.; Galle, P.R.; Waisman, A. Overexpression of Bcl-3 inhibits the development of marginal zone B cells. Eur. J. Immunol. 2014, 44, 545–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horns, F.; Vollmers, C.; Dekker, C.L.; Quake, S.R. Signatures of selection in the human antibody repertoire: Selective sweeps, competing subclones, and neutral drift. Proc. Natl. Acad. Sci. USA 2019, 116, 1261–1266. [Google Scholar] [CrossRef] [Green Version]
- Shi, Z.; Zhang, Q.; Yan, H.; Yang, Y.; Wang, P.; Zhang, Y.; Deng, Z.; Yu, M.; Zhou, W.; Wang, Q.; et al. More than one antibody of individual B cells revealed by single-cell immune profiling. Cell Discov. 2019, 5, 64. [Google Scholar] [CrossRef]
- Yaari, G.; Uduman, M.; Kleinstein, S.H. Quantifying selection in high-throughput Immunoglobulin sequencing data sets. Nucleic Acids Res. 2012, 40, e134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, L.; Lowther, D.E.; Meizlish, M.L.; Anderson, R.C.E.; Bruce, J.N.; Devine, L.; Huttner, A.J.; Kleinstein, S.H.; Lee, J.Y.; Stern, J.N.H.; et al. The immune cell infiltrate populating meningiomas is composed of mature, antigen-experienced T and B cells. Neuro. Oncol. 2013, 15, 1479–1490. [Google Scholar] [CrossRef] [Green Version]
- Sevigny, L.M.; Booth, B.J.; Rowley, K.J.; Leav, B.A.; Cheslock, P.S.; Garrity, K.A.; Sloan, S.E.; Thomas, W.; Babcock, G.J.; Wang, Y. Identification of a human monoclonal antibody to replace equine diphtheria antitoxin for treatment of diphtheria intoxication. Infect. Immun. 2013, 81, 3992–4000. [Google Scholar] [CrossRef] [Green Version]
- Lavinder, J.J.; Wine, Y.; Giesecke, C.; Ippolito, G.C.; Horton, A.P.; Lungu, O.I.; Hoi, K.H.; DeKosky, B.J.; Murrin, E.M.; Wirth, M.M.; et al. Identification and characterization of the constituent human serum antibodies elicited by vaccination. Proc. Natl. Acad. Sci. USA 2014, 111, 2259–2264. [Google Scholar] [CrossRef] [Green Version]
- Acquaye-Seedah, E.; Reczek, E.E.; Russell, H.H.; DiVenere, A.M.; Sandman, S.O.; Collins, J.H.; Stein, C.A.; Whitehead, T.A.; Maynard, J.A. Characterization of individual human antibodies that bind pertussis toxin stimulated by acellular immunization. Infect. Immun. 2018, 86, e00004-18. [Google Scholar] [CrossRef] [Green Version]
- Wec, A.Z.; Haslwanter, D.; Abdiche, Y.N.; Shehata, L.; Pedreño-Lopez, N.; Moyer, C.L.; Bornholdt, Z.A.; Lilov, A.; Nett, J.H.; Jangra, R.K.; et al. Longitudinal dynamics of the human B cell response to the yellow fever 17D vaccine. Proc. Natl. Acad. Sci. USA 2020, 117, 6675–6685. [Google Scholar] [CrossRef] [Green Version]
- Nourmohammad, A.; Otwinowski, J.; Łuksza, M.; Mora, T.; Walczak, A.M.; Leitner, T. Fierce Selection and Interference in B-Cell Repertoire Response to Chronic HIV-1. Mol. Biol. Evol. 2019, 36, 2184–2194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, Y.; O’neill, P.; Naradikian, M.S.; Scholz, J.L.; Cancro, M.P. A B-cell subset uniquely responsive to innate stimuli accumulates in aged mice. Blood J. Am. Soc. Hematol. 2011, 118, 1294–1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jash, A.; Wang, Y.; Weisel, F.J.; Scharer, C.D.; Boss, J.M.; Shlomchik, M.J.; Bhattacharya, D. ZBTB32 Restricts the Duration of Memory B Cell Recall Responses. J. Immunol. 2016, 197, 1159–1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masciarelli, S.; Sitia, R. Building and operating an antibody factory: Redox control during B to plasma cell terminal differentiation. Biochim. Biophys. Acta-Mol. Cell Res. 2008, 1783, 578–588. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Yuan, J.; Zhang, Z.; Chang, X. Cytoplasmic poly(A)-binding protein 1 (PABPC1) interacts with the RNA-binding protein hnRNPLL and thereby regulates immunoglobulin secretion in plasma cells. J. Biol. Chem. 2017, 292, 12285–12295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, S.; Shachar, I. Cytokines as regulators of proliferation and survival of healthy and malignant peripheral B cells. Cytokine 2012, 60, 13–22. [Google Scholar] [CrossRef]
- Gil-Yarom, N.; Radomir, L.; Sever, L.; Kramer, M.P.; Lewinsky, H.; Bornstein, C.; Blecher-Gonen, R.; Barnett-Itzhaki, Z.; Mirkin, V.; Friedlander, G.; et al. CD74 is a novel transcription regulator. Proc. Natl. Acad. Sci. USA 2017, 114, 562–567. [Google Scholar] [CrossRef] [Green Version]
- Andreani, V.; Ramamoorthy, S.; Pandey, A.; Lupar, E.; Nutt, S.L.; Lämmermann, T.; Grosschedl, R. Cochaperone Mzb1 is a key effector of Blimp1 in plasma cell differentiation and β1-integrin function. Proc. Natl. Acad. Sci. USA 2018, 115, E9630–E9639. [Google Scholar] [CrossRef] [Green Version]
- Lingwood, D.; McTamney, P.M.; Yassine, H.M.; Whittle, J.R.R.; Guo, X.; Boyington, J.C.; Wei, C.J.; Nabel, G.J. Structural and genetic basis for development of broadly neutralizing influenza antibodies. Nature 2012, 489, 566–570. [Google Scholar] [CrossRef]
- Lin, W.; Zhang, P.; Chen, H.; Chen, Y.; Yang, H.; Zheng, W.; Zhang, X.; Zhang, F.; Zhang, W.; Lipsky, P.E. Circulating plasmablasts/plasma cells: A potential biomarker for IgG4-related disease. Arthritis Res. Ther. 2017, 19, 25. [Google Scholar] [CrossRef] [Green Version]
- Shaffer, A.L.; Shapiro-Shelef, M.; Iwakoshi, N.N.; Lee, A.H.; Qian, S.B.; Zhao, H.; Yu, X.; Yang, L.; Tan, B.K.; Rosenwald, A.; et al. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity 2004, 21, 81–93. [Google Scholar] [CrossRef] [Green Version]
- Todd, D.J.; McHeyzer-Williams, L.J.; Kowal, C.; Lee, A.H.; Volpe, B.T.; Diamond, B.; McHeyzer-Williams, M.G.; Glimcher, L.H. XBP1 governs late events in plasma cell differentiation and is not required for antigen-specific memory B cell development. J. Exp. Med. 2009, 206, 2151–2159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergmann, H.; Yabas, M.; Short, A.; Miosge, L.; Barthel, N.; Teh, C.E.; Roots, C.M.; Bull, K.R.; Jeelall, Y.; Horikawa, K.; et al. B cell survival, surface BCR and BAFFR expression, CD74 metabolism, and CD8− dendritic cells require the intramembrane endopeptidase SPPL2A. J. Exp. Med. 2013, 210, 31. [Google Scholar] [CrossRef]
- Jasper, G.A.; Arun, I.; Venzon, D.; Kreitman, R.J.; Wayne, A.S.; Yuan, C.M.; Marti, G.E.; Stetler-Stevenson, M. Variables affecting the quantitation of CD22 in neoplastic B cells. Cytom. Part B Clin. Cytom. 2011, 80B, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Pappas, L.; Foglierini, M.; Piccoli, L.; Kallewaard, N.L.; Turrini, F.; Silacci, C.; Fernandez-Rodriguez, B.; Agatic, G.; Giacchetto-Sasselli, I.; Pellicciotta, G.; et al. Rapid development of broadly influenza neutralizing antibodies through redundant mutations. Nature 2014, 516, 418–422. [Google Scholar] [CrossRef] [PubMed]
- Galson, J.D.; Trück, J.; Clutterbuck, E.A.; Fowler, A.; Cerundolo, V.; Pollard, A.J.; Lunter, G.; Kelly, D.F. B-cell repertoire dynamics after sequential hepatitis B vaccination and evidence for cross-reactive B-cell activation. Genome Med. 2016, 8, 68. [Google Scholar] [CrossRef] [Green Version]
- Montague, Z.; Lv, H.; Otwinowski, J.; Wu, N.C.; Nourmohammad, A.; Ka, C.; Mok, P.; Dewitt, W.S.; Isacchini, G.; Yip, G.K.; et al. Dynamics of B cell repertoires and emergence of cross-reactive responses in patients with different severities of COVID-19. Cell Rep. 2021, 35, 109173. [Google Scholar] [CrossRef] [PubMed]
- Pinto, D.; Park, Y.J.; Beltramello, M.; Walls, A.C.; Tortorici, M.A.; Bianchi, S.; Jaconi, S.; Culap, K.; Zatta, F.; De Marco, A.; et al. Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody. Nature 2020, 583, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Rogers, T.F.; Zhao, F.; Huang, D.; Beutler, N.; Burns, A.; He, W.T.; Limbo, O.; Smith, C.; Song, G.; Woehl, J.; et al. Isolation of potent SARS-CoV-2 neutralizing antibodies and protection from disease in a small animal model. Science 2020, 369, 956–963. [Google Scholar] [CrossRef]
- Brouwer, P.J.M.; Caniels, T.G.; van der Straten, K.; Snitselaar, J.L.; Aldon, Y.; Bangaru, S.; Torres, J.L.; Okba, N.M.A.; Claireaux, M.; Kerster, G.; et al. Potent neutralizing antibodies from COVID-19 patients define multiple targets of vulnerability. Science 2020, 369, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Rue, M.; Zhou, J.; Wang, H.; Goldwasser, M.A.; Neuberg, D.; Dalton, V.; Zuckerman, D.; Lyons, C.; Silverman, L.B.; et al. Utilization of Ig heavy chain variable, diversity, and joining gene segments in children with B-lineage acute lymphoblastic leukemia: Implications for the mechanisms of VDJ recombination and for pathogenesis. Blood 2004, 103, 4602–4609. [Google Scholar] [CrossRef]
- Yamada, M.; Wasserman, R.; Reichard, B.A.; Shane, S.; Caton, A.J.; Rovera, G. Preferential utilization of specific immunoglobulin heavy chain diversity and joining segments in adult human peripheral blood B lymphocytes. J. Exp. Med. 1991, 173, 395–407. [Google Scholar] [CrossRef] [Green Version]
- Hong, B.; Wu, Y.; Li, W.; Wang, X.; Wen, Y.; Jiang, S.; Dimitrov, D.S.; Ying, T. In-depth analysis of human neonatal and adult IgM antibody repertoires. Front. Immunol. 2018, 9, 128. [Google Scholar] [CrossRef]
- Khatri, I.; Berkowska, M.A.; van den Akker, E.B.; Teodosio, C.; Reinders, M.J.T.; van Dongen, J.J.M. Population matched (pm) germline allelic variants of immunoglobulin (IG) loci: Relevance in infectious diseases and vaccination studies in human populations. Genes Immun. 2021, 22, 172–186. [Google Scholar] [CrossRef]
- Hesse, J.E.; Lieber, M.R.; Mizuuchi, K.; Gellert, M. V(D)J recombination: A functional definition of the joining signals. Genes Dev. 1989, 3, 1053–1061. [Google Scholar] [CrossRef] [Green Version]
- Belessi, C.; Stamatopoulos, K.; Hadzidimitriou, A.; Hatzi, K.; Smilevska, T.; Stavroyianni, N.; Marantidou, F.; Paterakis, G.; Fassas, A.; Anagnostopoulos, A.; et al. Analysis of expressed and non-expressed IGK locus rearrangements in chronic lymphocytic leukemia. Mol. Med. 2005, 11, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Bräuninger, A.; Goossens, T.; Rajewsky, K.; Küppers, R. Regulation of Immunoglobulin Light Chain Gene Rearrangements during Early B Cell Development in the Human. Eur. J. Immunol. 2001, 31, 3631–3637. [Google Scholar] [CrossRef]
- Korsmeyer, S.J.; Hieter, P.A.; Ravetch, J.V.; Poplack, D.G.; Waldmann, T.A.; Leder, P. Developmental hierarchy of immunoglobulin gene rearrangements in human leukemic pre-B-cells. Proc. Natl. Acad. Sci. USA 1981, 78, 7096–7100. [Google Scholar] [CrossRef] [Green Version]
- Odendahl, M.; Mei, H.; Hoyer, B.F.; Jacobi, A.M.; Hansen, A.; Muehlinghaus, G.; Berek, C.; Hiepe, F.; Manz, R.; Radbruch, A.; et al. Generation of migratory antigen-specific plasma blasts and mobilization of resident plasma cells in a secondary immune response. Blood 2005, 105, 1614–1621. [Google Scholar] [CrossRef]
- Galson, J.D.; Trück, J.; Fowler, A.; Clutterbuck, E.A.; Münz, M.; Cerundolo, V.; Reinhard, C.; van der Most, R.; Pollard, A.J.; Lunter, G.; et al. Analysis of B Cell Repertoire Dynamics Following Hepatitis B Vaccination in Humans, and Enrichment of Vaccine-specific Antibody Sequences. EBioMedicine 2015, 2, 2070–2079. [Google Scholar] [CrossRef] [Green Version]
- Horns, F.; Vollmers, C.; Croote, D.; Mackey, S.F.; Swan, G.E.; Dekker, C.L.; Davis, M.M.; Quake, S.R. Lineage tracing of human B cells reveals the in vivo landscape of human antibody class switching. Elife 2016, 5, e16578. [Google Scholar] [CrossRef] [PubMed]
- Neuberger, M.S.; Milstein, C. Somatic hypermutation. Curr. Opin. Immunol. 1995, 7, 248–254. [Google Scholar] [CrossRef]
- Storb, U.; Peters, A.; Klotz, E.; Rogerson, B.; Hackett, J. The mechanism of somatic hypermutation studied with transgenic and transfected target genes. Semin. Immunol. 1996, 8, 131–140. [Google Scholar] [CrossRef]
- Collins, A.M.; Watson, C.T. Immunoglobulin light chain gene rearrangements, receptor editing and the development of a self-tolerant antibody repertoire. Front. Immunol. 2018, 9, 2249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moticka, E.J.; Streilein, J.W. Hypothesis: Nonspecific polyclonal activation of memory B cells by antigen as a mechanism for the preservation of long term immunologic anamnesis. Cell. Immunol. 1978, 41, 406–413. [Google Scholar] [CrossRef]
- Blanco, E.; Pérez-Andrés, M.; Arriba-Méndez, S.; Contreras-Sanfeliciano, T.; Criado, I.; Pelak, O.; Serra-Caetano, A.; Romero, A.; Puig, N.; Remesal, A.; et al. Age-associated distribution of normal B-cell and plasma cell subsets in peripheral blood. J. Allergy Clin. Immunol. 2018, 141, 2208–2219.e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khatri, I.; Diks, A.M.; van den Akker, E.B.; Oosten, L.E.M.; Zwaginga, J.J.; Reinders, M.J.T.; van Dongen, J.J.M.; Berkowska, M.A. Longitudinal Dynamics of Human B-Cell Response at the Single-Cell Level in Response to Tdap Vaccination. Vaccines 2021, 9, 1352. https://0-doi-org.brum.beds.ac.uk/10.3390/vaccines9111352
Khatri I, Diks AM, van den Akker EB, Oosten LEM, Zwaginga JJ, Reinders MJT, van Dongen JJM, Berkowska MA. Longitudinal Dynamics of Human B-Cell Response at the Single-Cell Level in Response to Tdap Vaccination. Vaccines. 2021; 9(11):1352. https://0-doi-org.brum.beds.ac.uk/10.3390/vaccines9111352
Chicago/Turabian StyleKhatri, Indu, Annieck M. Diks, Erik B. van den Akker, Liesbeth E. M. Oosten, Jaap Jan Zwaginga, Marcel J. T. Reinders, Jacques J. M. van Dongen, and Magdalena A. Berkowska. 2021. "Longitudinal Dynamics of Human B-Cell Response at the Single-Cell Level in Response to Tdap Vaccination" Vaccines 9, no. 11: 1352. https://0-doi-org.brum.beds.ac.uk/10.3390/vaccines9111352