How Insertion of a Single Tryptophan in the N-Terminus of a Cecropin A-Melittin Hybrid Peptide Changes Its Antimicrobial and Biophysical Profile

 , ,
, ,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. General Reagents
2.2. Spectroscopic Measurements
2.3. Peptide Synthesis
2.4. Antibacterial Activity
2.5. Preparation of Large Unilamellar Vesicles
2.6. Photophysical Properties of Peptides in Aqueous Solution
2.7. Partition Constants
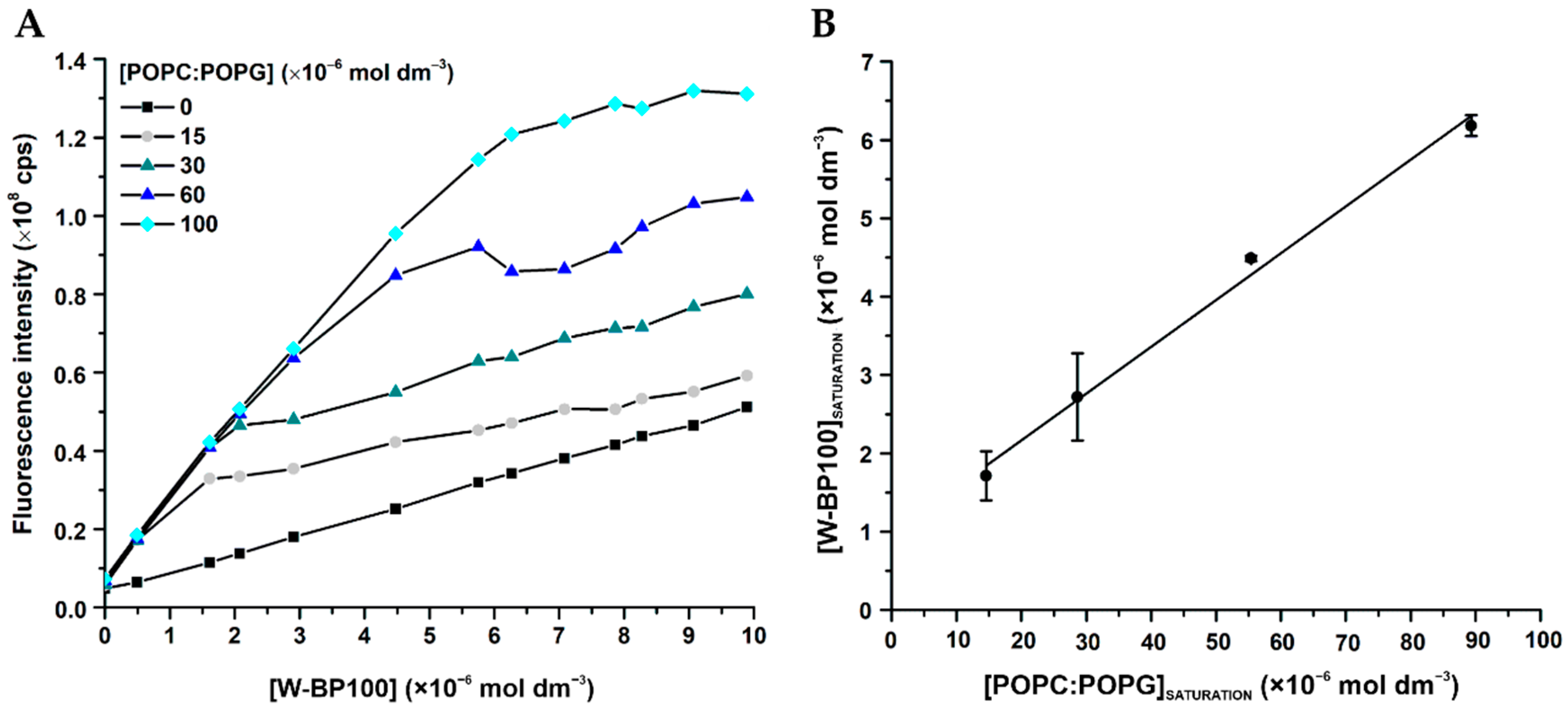
2.8. Membrane Saturation Studies
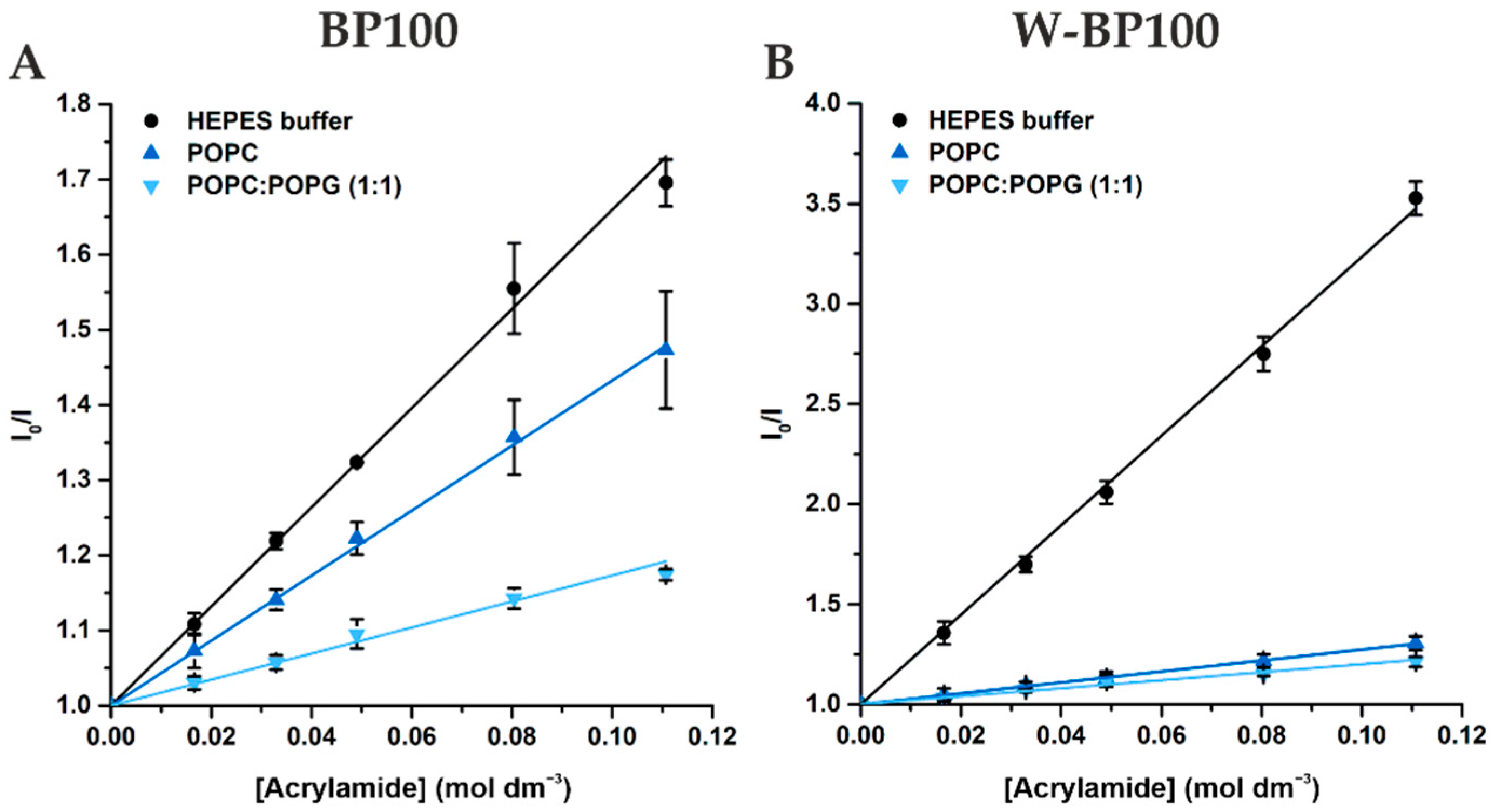
2.9. Fluorescence Acrylamide Quenching
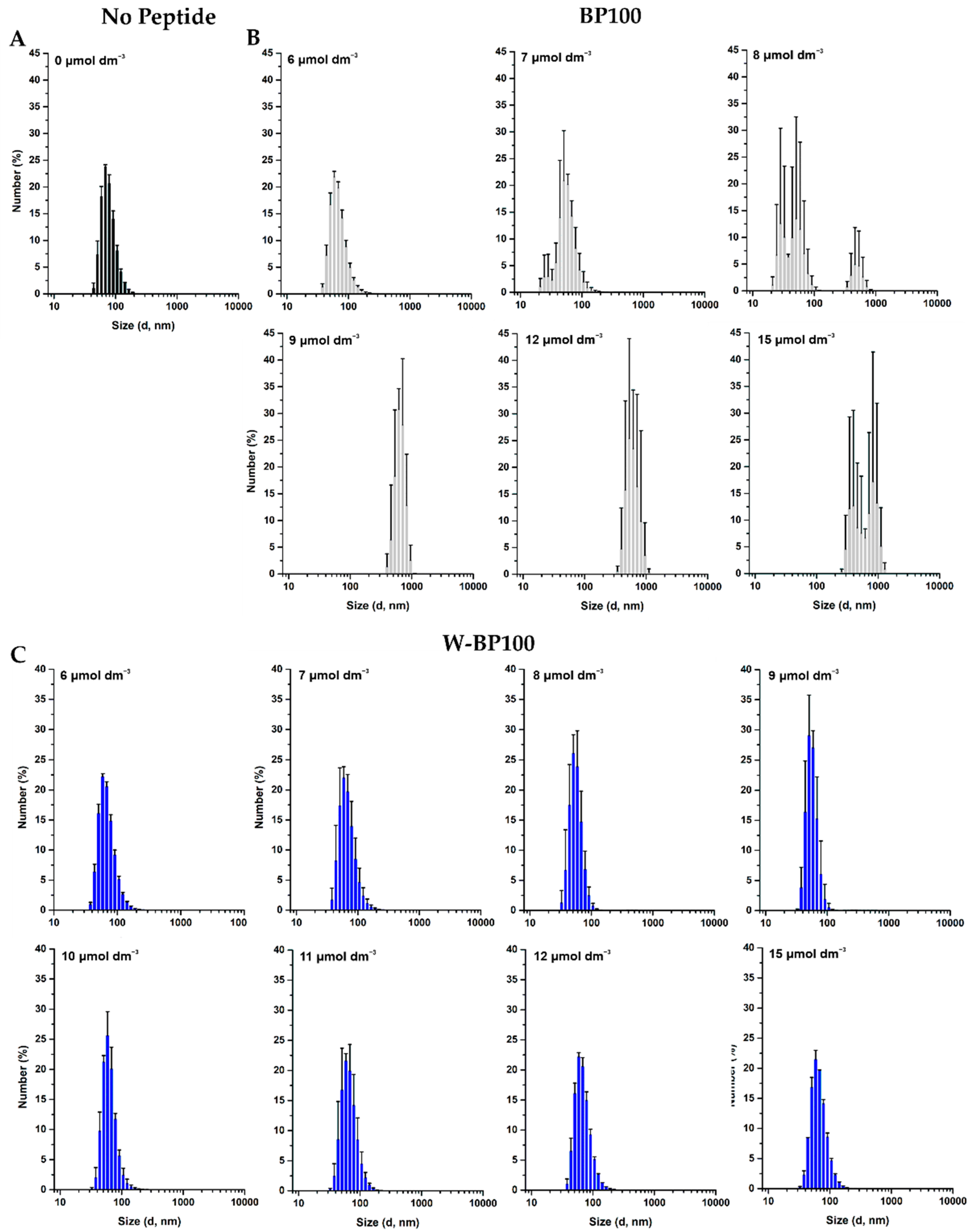
2.10. Aggregation of LUV Using DLS
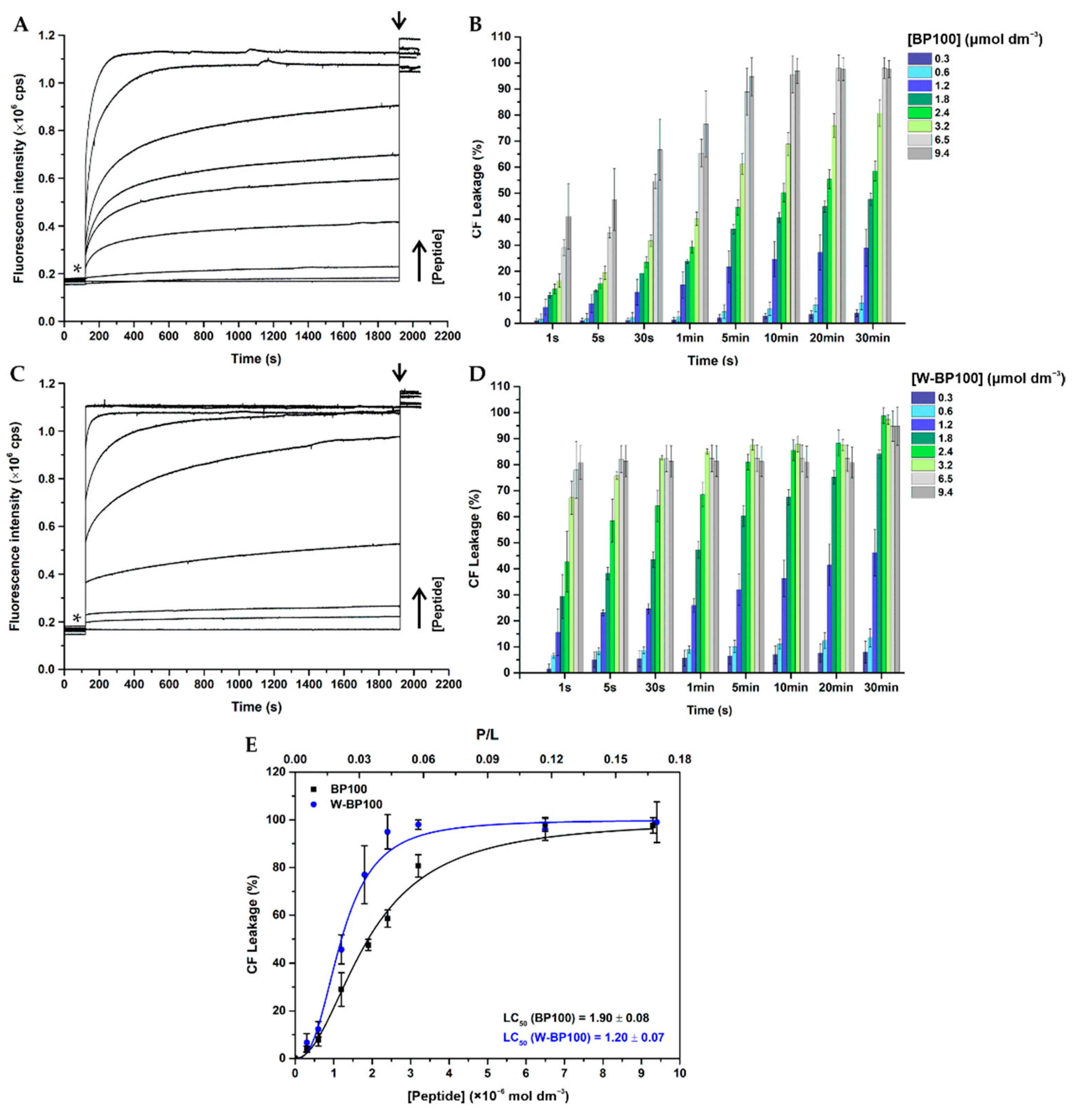
2.11. Membrane Permeabilization
3. Results and Discussion
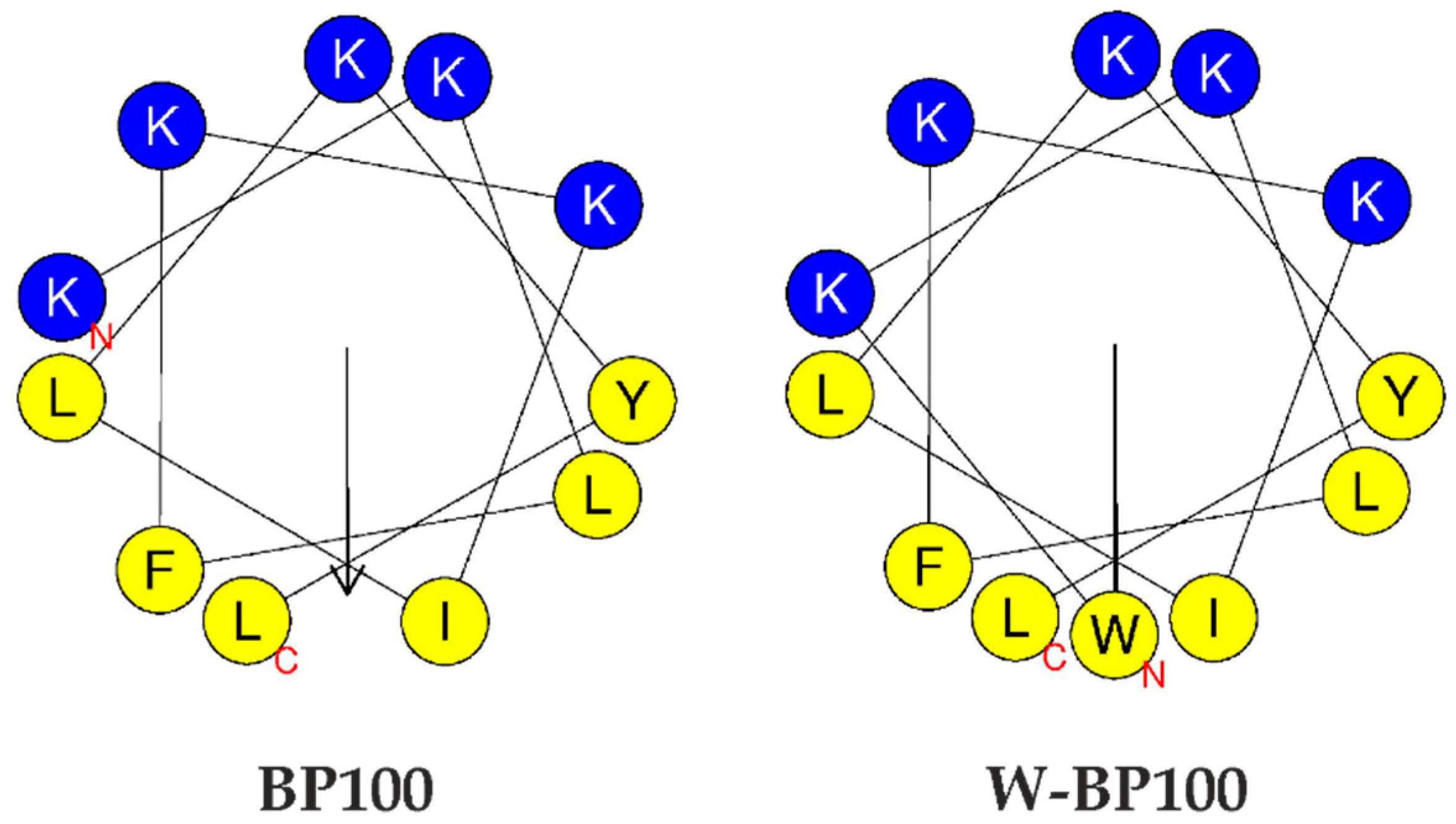
3.1. Peptide Synthesis and Photophysical Characterization
3.2. Antibacterial Activity of Peptides
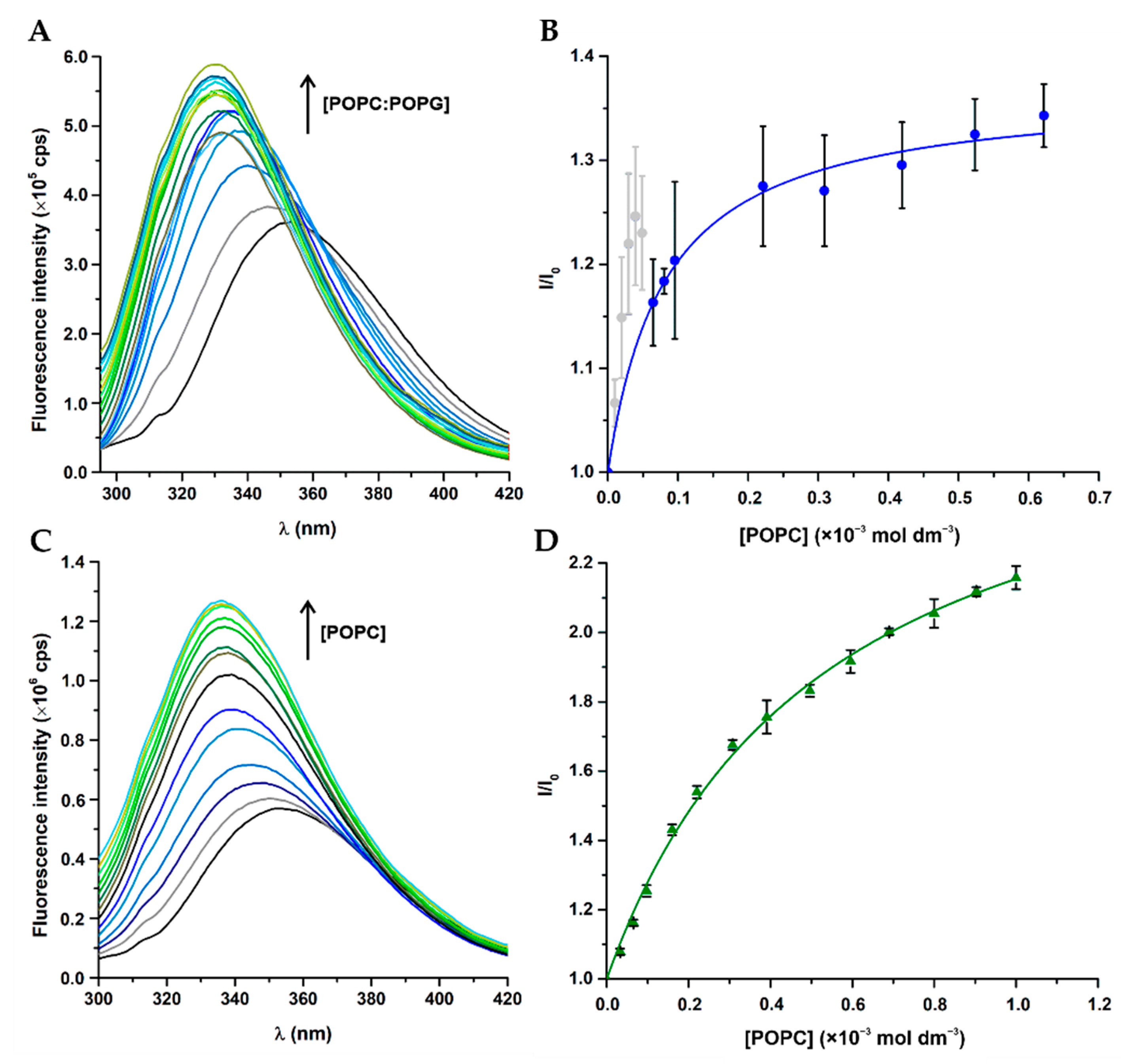
3.3. Peptide–Membrane Interactions with LUV
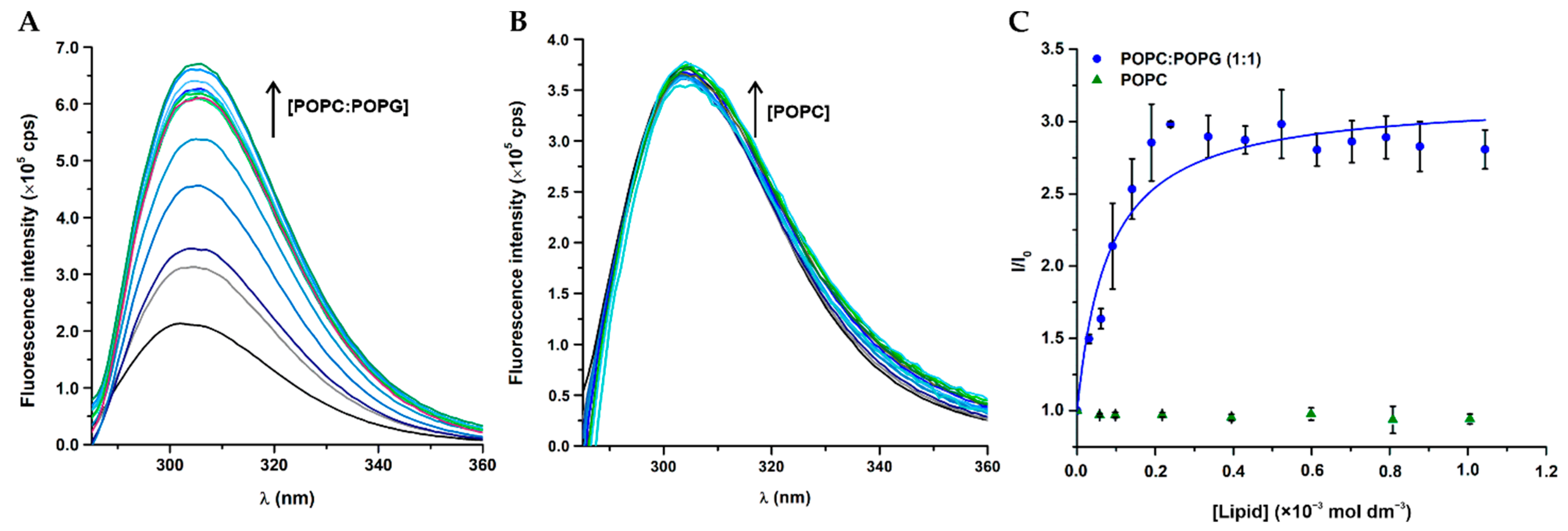
3.4. Membrane Location of Peptides in LUV
3.5. Aggregation and Membrane Permeabilization Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- WHO. Prioritization of Pathogens to Guide Discovery, Research and Development of New Antibiotics for Drug-Resistant Bacterial Infections, Including Tuberculosis (WHO/EMP/IAU/2017.12); Licence: CC BY-NC-SA 3.0 IGO; World Health Organization: Geneva, Switzerland, 2017; Available online: https://www.who.int/medicines/areas/rational_use/prioritization-of-pathogens/en/ (accessed on 4 April 2020).
- WHO. Antibacterial Agents in Clinical Development: An Analysis of the Antibacterial Clinical Development Pipeline; World Health Organization: Geneva, Switzerland, 2019; Licence: CC BY-NC-SA 3.0 IGO; Available online: https://apps.who.int/iris/handle/10665/330420 (accessed on 4 April 2020).
- Haney, E.F.; Straus, S.K.; Hancock, R.E.W. Reassessing the Host Defense Peptide Landscape. Front. Chem. 2019, 7, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Yeaman, M.R.; Yount, N.Y. Mechanisms of antimicrobial peptide action and resistance. Pharmacol. Rev. 2003, 55, 27–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mookherjee, N.; Anderson, M.A.; Haagsman, H.P.; Davidson, D.J. Antimicrobial host defence peptides: Functions and clinical potential. Nat. Rev. Drug. Discov. 2020. [Google Scholar] [CrossRef]
- Andreu, D.; Merrifield, R.B.; Steiner, H.; Boman, H.G. Solid-phase synthesis of cecropin A and related peptides. Proc. Natl. Acad. Sci. USA 1983, 80, 6475–6479. [Google Scholar] [CrossRef] [Green Version]
- Steiner, H.; Hultmark, D.; Engstrom, A.; Bennich, H.; Boman, H.G. Sequence and specificity of two antibacterial proteins involved in insect immunity. Nature 1981, 292, 246–248. [Google Scholar] [CrossRef]
- Habermann, E. Bee and wasp venoms. Science 1972, 177, 314–322. [Google Scholar] [CrossRef]
- Andreu, D.; Ubach, J.; Boman, A.; Wahlin, B.; Wade, D.; Merrifield, R.B.; Boman, H.G. Shortened cecropin A-melittin hybrids. Significant size reduction retains potent antibiotic activity. FEBS Lett. 1992, 296, 190–194. [Google Scholar] [CrossRef] [Green Version]
- Giacometti, A.; Cirioni, O.; Kamysz, W.; D’Amato, G.; Silvestri, C.; Simona Del Prete, M.; Lukasiak, J.; Scalise, G. In vitro activity and killing effect of the synthetic hybrid cecropin A-melittin peptide CA(1-7)M(2-9)NH(2) on methicillin-resistant nosocomial isolates of Staphylococcus aureus and interactions with clinically used antibiotics. Diagn. Microbiol. Infect. Dis. 2004, 49, 197–200. [Google Scholar] [CrossRef]
- Badosa, E.; Ferre, R.; Planas, M.; Feliu, L.; Besalu, E.; Cabrefiga, J.; Bardaji, E.; Montesinos, E. A library of linear undecapeptides with bactericidal activity against phytopathogenic bacteria. Peptides 2007, 28, 2276–2285. [Google Scholar] [CrossRef]
- Ferre, R.; Melo, M.N.; Correia, A.D.; Feliu, L.; Bardaji, E.; Planas, M.; Castanho, M. Synergistic effects of the membrane actions of cecropin-melittin antimicrobial hybrid peptide BP100. Biophys. J. 2009, 96, 1815–1827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alves, C.S.; Melo, M.N.; Franquelim, H.G.; Ferre, R.; Planas, M.; Feliu, L.; Bardaji, E.; Kowalczyk, W.; Andreu, D.; Santos, N.C.; et al. Escherichia coli cell surface perturbation and disruption induced by antimicrobial peptides BP100 and pepR. J. Biol. Chem. 2010, 285, 27536–27544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torcato, I.M.; Huang, Y.H.; Franquelim, H.G.; Gaspar, D.; Craik, D.J.; Castanho, M.A.; Troeira Henriques, S. Design and characterization of novel antimicrobial peptides, R-BP100 and RW-BP100, with activity against Gram-negative and Gram-positive bacteria. Biochim. Biophys. Acta 2013, 1828, 944–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carretero, G.P.B.; Saraiva, G.K.V.; Cauz, A.C.G.; Rodrigues, M.A.; Kiyota, S.; Riske, K.A.; Dos Santos, A.A.; Pinatto-Botelho, M.F.; Bemquerer, M.P.; Gueiros-Filho, F.J.; et al. Synthesis, biophysical and functional studies of two BP100 analogues modified by a hydrophobic chain and a cyclic peptide. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1502–1516. [Google Scholar] [CrossRef]
- Bastos, M.; Bai, G.; Gomes, P.; Andreu, D.; Goormaghtigh, E.; Prieto, M. Energetics and partition of two cecropin-melittin hybrid peptides to model membranes of different composition. Biophys. J. 2008, 94, 2128–2141. [Google Scholar] [CrossRef] [Green Version]
- Juvvadi, P.; Vunnam, S.; Merrifield, E.L.; Boman, H.G.; Merrifield, R.B. Hydrophobic effects on antibacterial and channel-forming properties of cecropin A-melittin hybrids. J. Pept. Sci. 1996, 2, 223–232. [Google Scholar] [CrossRef]
- Wei, S.Y.; Wu, J.M.; Kuo, Y.Y.; Chen, H.L.; Yip, B.S.; Tzeng, S.R.; Cheng, J.W. Solution structure of a novel tryptophan-rich peptide with bidirectional antimicrobial activity. J. Bacteriol. 2006, 188, 328–334. [Google Scholar] [CrossRef] [Green Version]
- Mishra, A.K.; Choi, J.; Moon, E.; Baek, K.H. Tryptophan-Rich and Proline-Rich Antimicrobial Peptides. Molecules 2018, 23, 815. [Google Scholar] [CrossRef] [Green Version]
- Arias, M.; Nguyen, L.T.; Kuczynski, A.M.; Lejon, T.; Vogel, H.J. Position-Dependent Influence of the Three Trp Residues on the Membrane Activity of the Antimicrobial Peptide, Tritrpticin. Antibiotics 2014, 3, 595–616. [Google Scholar] [CrossRef] [Green Version]
- Yau, W.M.; Wimley, W.C.; Gawrisch, K.; White, S.H. The preference of tryptophan for membrane interfaces. Biochemistry 1998, 37, 14713–14718. [Google Scholar] [CrossRef] [Green Version]
- Killian, J.A.; von Heijne, G. How proteins adapt to a membrane–water interface. Trends Biochem. Sci. 2000, 25, 429–434. [Google Scholar] [CrossRef]
- Bi, X.; Wang, C.; Dong, W.; Zhu, W.; Shang, D. Antimicrobial properties and interaction of two Trp-substituted cationic antimicrobial peptides with a lipid bilayer. J. Antibiot. 2014, 67, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Cox, L.; Celis, R.; Hermosin, M.C.; Cornejo, J.; Zsolnay, A.; Zeller, K. Effect of organic amendments on herbicide sorption as related to the nature of the dissolved organic matter. Environ. Sci. Technol. 2000, 34, 4600–4605. [Google Scholar] [CrossRef] [Green Version]
- Benoiton, N.L. Chemistry of Peptide Synthesis; CRC Press: Boca Raton, FL, USA, 2016. [Google Scholar]
- CLSI. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard—Ninth Edition. CLSI document M07-A9; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2012. [Google Scholar]
- Bessa, L.J.; Eaton, P.; Dematei, A.; Placido, A.; Vale, N.; Gomes, P.; Delerue-Matos, C.; Sa Leite, J.R.; Gameiro, P. Synergistic and antibiofilm properties of ocellatin peptides against multidrug-resistant Pseudomonas aeruginosa. Future Microbiol. 2018, 13, 151–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer, L.D.; Hope, M.J.; Cullis, P.R. Vesicles of variable sizes produced by a rapid extrusion procedure. Biochim. Biophys. Acta Biomembr. 1986, 858, 161–168. [Google Scholar] [CrossRef]
- Bartlett, G.R. Phosphorus assay in column chromatography. J. Biol. Chem. 1959, 234, 466–468. [Google Scholar] [CrossRef]
- Santos, N.C.; Prieto, M.; Castanho, M.A.R.B. Quantifying molecular partition into model systems of biomembranes: An emphasis on optical spectroscopic methods. Biochim. Biophys. Acta Biomembr. 2003, 1612, 123–135. [Google Scholar] [CrossRef] [Green Version]
- Melo, M.N.; Castanho, M.A. Omiganan interaction with bacterial membranes and cell wall models. Assigning a biological role to saturation. Biochim. Biophys. Acta 2007, 1768, 1277–1290. [Google Scholar] [CrossRef] [Green Version]
- Larsson, T.; Wedborg, M.; Turner, D. Correction of inner-filter effect in fluorescence excitation-emission matrix spectrometry using Raman scatter. Anal. Chim. Acta 2007, 583, 357–363. [Google Scholar] [CrossRef]
- Pace, C.N.; Vajdos, F.; Fee, L.; Grimsley, G.; Gray, T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995, 4, 2411–2423. [Google Scholar] [CrossRef] [Green Version]
- Ladokhin, A.S. Fluorescence Spectroscopy in Peptide and Protein Analysis. EAC 2006. [Google Scholar] [CrossRef]
- Alves, C.S.; Kairys, V.; Castanho, M.A.; Fernandes, M.X. Interaction of antimicrobial peptides, BP100 and pepR, with model membrane systems as explored by Brownian dynamics simulations on a coarse-grained model. Biopolymers 2012, 98, 294–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gautier, R.; Douguet, D.; Antonny, B.; Drin, G. HELIQUEST: A web server to screen sequences with specific alpha-helical properties. Bioinformatics 2008, 24, 2101–2102. [Google Scholar] [CrossRef] [PubMed]
- HELIQUEST. Available online: https://heliquest.ipmc.cnrs.fr (accessed on 25 January 2020).
- Chan, D.I.; Prenner, E.J.; Vogel, H.J. Tryptophan- and arginine-rich antimicrobial peptides: Structures and mechanisms of action. Biochim. Biophys. Acta 2006, 1758, 1184–1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dougherty, D.A. Cation-pi Interactions in Chemistry and Biology: A New View of Benzene, Phe, Tyr, and Trp. Science 1996, 271, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 3rd ed.; Springer: Boston, MA, USA, 2006. [Google Scholar]
- Melo, M.N.; Dugourd, D.; Castanho, M.A. Omiganan pentahydrochloride in the front line of clinical applications of antimicrobial peptides. Recent Pat. Antiinfect. Drug Discov. 2006, 1, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Connors, K.A. Binding Constants: The Measurement of Molecular Complex Stability, 1st ed.; Wiley-Interscience: New York, NY, USA, 1987. [Google Scholar]
- Subbalakshmi, C.; Krishnakumari, V.; Sitaram, N.; Nagaraj, R. Interaction of indolicidin, a 13-residue peptide rich in tryptophan and proline and its analogues with model membranes. J. Biosci. 1998, 23, 9–13. [Google Scholar] [CrossRef]
- Gazit, E.; Boman, A.; Boman, H.G.; Shai, Y. Interaction of the mammalian antibacterial peptide cecropin P1 with phospholipid vesicles. Biochemistry 1995, 34, 11479–11488. [Google Scholar] [CrossRef]
- Beschiaschvili, G.; Seelig, J. Melittin binding to mixed phosphatidylglycerol/phosphatidylcholine membranes. Biochemistry 1990, 29, 52–58. [Google Scholar] [CrossRef]
- Melo, M.N.; Ferre, R.; Castanho, M.A. Antimicrobial peptides: Linking partition, activity and high membrane-bound concentrations. Nat. Rev. Microbiol. 2009, 7, 245–250. [Google Scholar] [CrossRef]
- Misiewicz, J.; Afonin, S.; Grage, S.L.; van den Berg, J.; Strandberg, E.; Wadhwani, P.; Ulrich, A.S. Action of the multifunctional peptide BP100 on native biomembranes examined by solid-state NMR. J. Biomol. NMR 2015, 61, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Wadhwani, P.; Strandberg, E.; van den Berg, J.; Mink, C.; Burck, J.; Ciriello, R.A.; Ulrich, A.S. Dynamical structure of the short multifunctional peptide BP100 in membranes. Biochim. Biophys. Acta 2014, 1838, 940–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, P.; Franco, L.R.; Chaimovich, H.; Coutinho, K.; Cuccovia, I.M.; Lima, F.S. Binding and Flip as Initial Steps for BP-100 Antimicrobial Actions. Sci. Rep. 2019, 9, 8622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bi, X.; Wang, C.; Ma, L.; Sun, Y.; Shang, D. Investigation of the role of tryptophan residues in cationic antimicrobial peptides to determine the mechanism of antimicrobial action. J. Appl. Microbiol. 2013, 115, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Jacob, B.; Jang, M.; Kwak, C.; Lee, Y.; Son, K.; Lee, S.; Jung, I.D.; Jeong, M.S.; Kwon, S.H.; et al. Development of a novel short 12-meric papiliocin-derived peptide that is effective against Gram-negative sepsis. Sci. Rep. 2019, 9, 3817. [Google Scholar] [CrossRef] [Green Version]
- Deslouches, B.; Phadke, S.M.; Lazarevic, V.; Cascio, M.; Islam, K.; Montelaro, R.C.; Mietzner, T.A. De novo generation of cationic antimicrobial peptides: Influence of length and tryptophan substitution on antimicrobial activity. Antimicrob. Agents Chemother. 2005, 49, 316–322. [Google Scholar] [CrossRef] [Green Version]
- Khara, J.S.; Obuobi, S.; Wang, Y.; Hamilton, M.S.; Robertson, B.D.; Newton, S.M.; Yang, Y.Y.; Langford, P.R.; Ee, P.L.R. Disruption of drug-resistant biofilms using de novo designed short alpha-helical antimicrobial peptides with idealized facial amphiphilicity. Acta Biomater. 2017, 57, 103–114. [Google Scholar] [CrossRef]
- Bagheri, M.; Nikolenko, H.; Arasteh, S.; Rezaei, N.; Behzadi, M.; Dathe, M.; Hancock, R.E.W. Bacterial Aggregation Triggered by Fibril Forming Tryptophan-Rich Sequences: Effects of Peptide Side Chain and Membrane Phospholipids. ACS Appl. Mater. Interfaces 2020, 12, 26852–26867. [Google Scholar] [CrossRef]
- Gupta, K.; Singh, S.; van Hoek, M.L. Short, Synthetic Cationic Peptides Have Antibacterial Activity against Mycobacterium smegmatis by Forming Pores in Membrane and Synergizing with Antibiotics. Antibiotics 2015, 4, 358–378. [Google Scholar] [CrossRef] [Green Version]
- Manzini, M.C.; Perez, K.R.; Riske, K.A.; Bozelli, J.C., Jr.; Santos, T.L.; da Silva, M.A.; Saraiva, G.K.; Politi, M.J.; Valente, A.P.; Almeida, F.C.; et al. Peptide:lipid ratio and membrane surface charge determine the mechanism of action of the antimicrobial peptide BP100. Conformational and functional studies. Biochim. Biophys. Acta 2014, 1838, 1985–1999. [Google Scholar] [CrossRef] [Green Version]
- Melo, M.N.; Castanho, M.A. The Mechanism of Action of Antimicrobial Peptides: Lipid Vesicles vs. Bacteria. Front Immunol. 2012, 3, 236. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacteria | MIC µmol dm−3 (µg mL−1) | MBC µmol dm−3 (µg mL−1) | ||
|---|---|---|---|---|
| BP100 | W-BP100 | BP100 | W-BP100 | |
| Escherichia coli ATCC 25922 | 1.7 (2.4) | 0.75 (1.2) | 1.7 (2.4) | 0.75 (1.2) |
| Pseudomonas aeruginosa ATCC 27853 | 1.7 (2.4) | 1.5–3.0 (2.4–4.8) | 1.7 (2.4) | 1.5–3.0 (2.4–4.8) |
| Staphylococcus aureus ATCC 29213 | 27 (38) | 1.5 (2.4) | 27 (38) | 1.5 (2.4) |
| Enterococcus faecalis ATCC 29212 | 108–216 (154–307) | 3.0 (4.8) | 108–216 (154–307) | 3.0 (4.8) |
| Peptide | LUV | (mol dm−3) | Fitting Equation | |
|---|---|---|---|---|
| BP100 | POPC | n.o. | n.o. | n.o. 1 |
| POPC:POPG (1:1) | 16.2 ± 3.7 | 3.16 ± 0.12 | 1 | |
| W-BP100 | POPC | 2.4 ± 0.2 | 2.77 ± 0.06 | 1 |
| POPC:POPG (1:1) | 16.0 ± 1.8 | 1.37 ± 0.01 | 1 | |
| 80.6 ± 20.0 | - | 2 2 |
| Peptide | Medium | (mol−1 dm3) |
|---|---|---|
| BP100 | HEPES buffer | 6.60 ± 0.15 |
| POPC | 4.32 ± 0.04 | |
| POPC:POPG (1:1) | 1.73 ± 0.07 | |
| W-BP100 | HEPES buffer | 22.35 ± 0.28 |
| POPC | 2.73 ± 0.02 | |
| POPC:POPG (1:1) | 2.00 ± 0.09 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferreira, A.R.; Teixeira, C.; Sousa, C.F.; Bessa, L.J.; Gomes, P.; Gameiro, P. How Insertion of a Single Tryptophan in the N-Terminus of a Cecropin A-Melittin Hybrid Peptide Changes Its Antimicrobial and Biophysical Profile. Membranes 2021, 11, 48. https://0-doi-org.brum.beds.ac.uk/10.3390/membranes11010048
Ferreira AR, Teixeira C, Sousa CF, Bessa LJ, Gomes P, Gameiro P. How Insertion of a Single Tryptophan in the N-Terminus of a Cecropin A-Melittin Hybrid Peptide Changes Its Antimicrobial and Biophysical Profile. Membranes. 2021; 11(1):48. https://0-doi-org.brum.beds.ac.uk/10.3390/membranes11010048
Chicago/Turabian StyleFerreira, Ana Rita, Cátia Teixeira, Carla F. Sousa, Lucinda J. Bessa, Paula Gomes, and Paula Gameiro. 2021. "How Insertion of a Single Tryptophan in the N-Terminus of a Cecropin A-Melittin Hybrid Peptide Changes Its Antimicrobial and Biophysical Profile" Membranes 11, no. 1: 48. https://0-doi-org.brum.beds.ac.uk/10.3390/membranes11010048