SARS-CoV-2 Cellular Infection and Therapeutic Opportunities: Lessons Learned from Ebola Virus

 , ,
, , 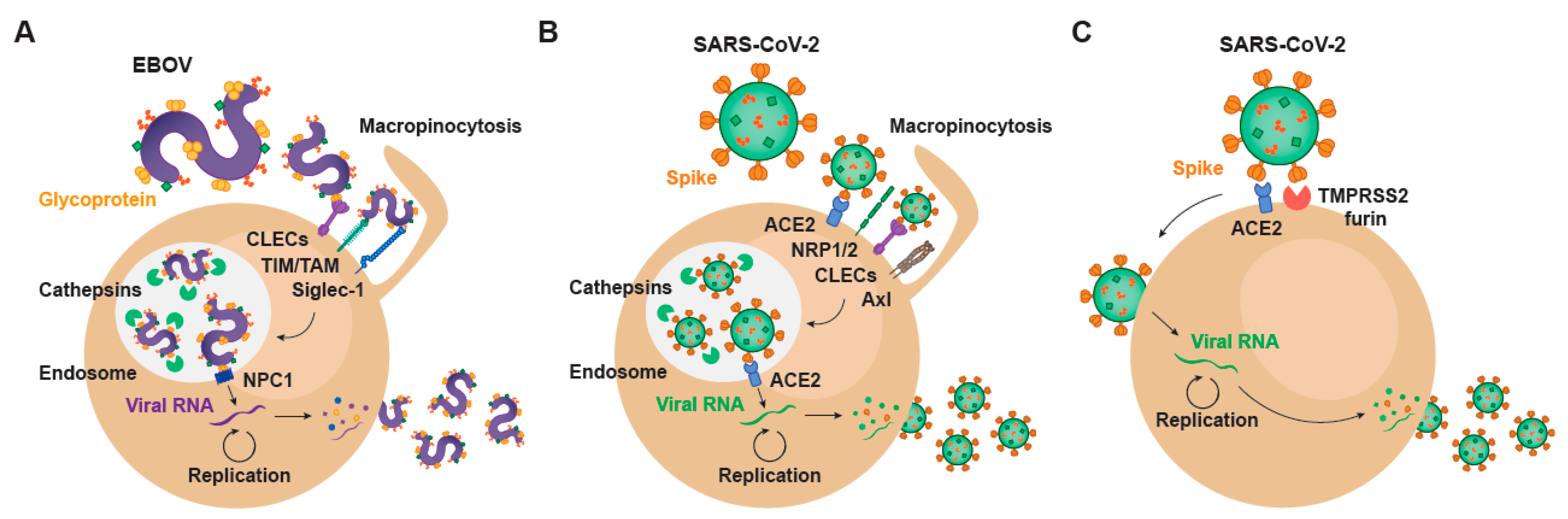{kind=link}
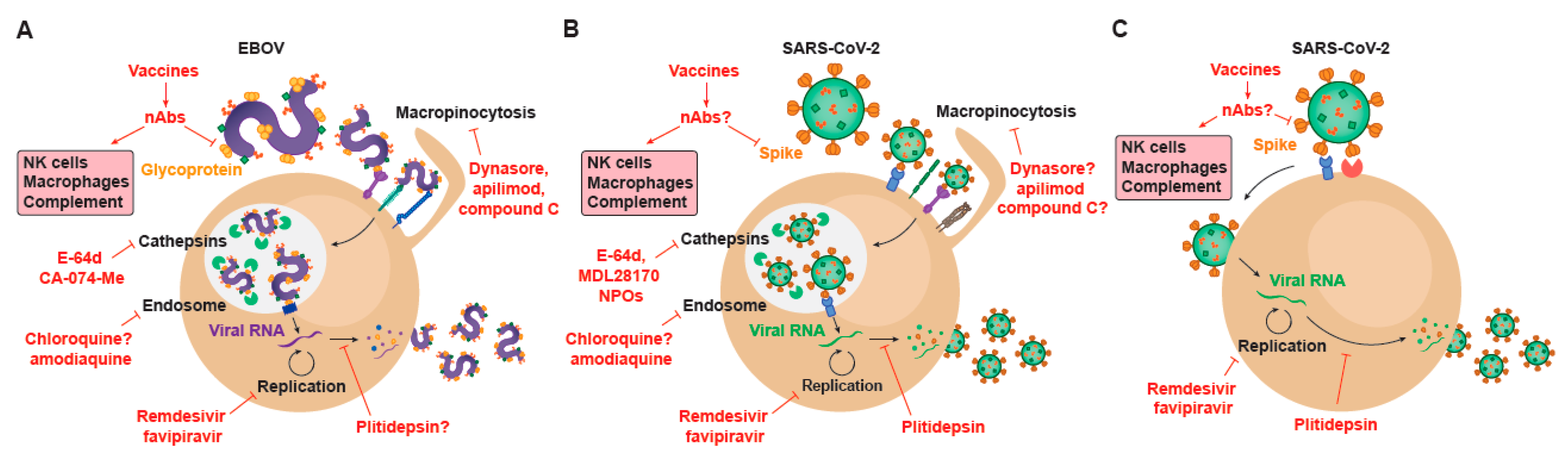{kind=link}
Abstract
:1. Introduction
2. Setting the Stage for Infection: Viral Binding and Host Attachment Receptors for EBOV and SARS-CoV-2
3. EBOV Entry Converges with the Endosomal Route of SARS-CoV-2
4. Transcription and Replication of EBOV and SARS-CoV-2
5. EBOV Egress and Common Gateways to SARS-CoV-2
6. Repurposing Drugs against EBOV and SARS-CoV-2: A Shared Strategy to Find Effective Antivirals
7. How the Development of EBOV Treatments Based on Monoclonal Antibodies Targeting the Virus Guides the Design of Novel Therapies for SARS-CoV-2
8. Vaccines for EBOV and for SARS-CoV-2 Infection
9. Conclusions and Future Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Moller-Tank, S.; Maury, W. Phosphatidylserine Receptors: Enhancers of Enveloped Virus Entry and Infection. Virology 2014, 468, 565–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moller-Tank, S.; Albritton, L.M.; Rennert, P.D.; Maury, W. Characterizing Functional Domains for TIM-Mediated Enveloped Virus Entry. J. Virol. 2014, 88, 6702–6713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, M.E.; Adhikary, S.; Kolokoltsov, A.A.; Davey, R.A. Ebolavirus Requires Acid Sphingomyelinase Activity and Plasma Membrane Sphingomyelin for Infection. J. Virol. 2012, 86, 7473–7483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Zsolt, D.; Erkizia, I.; Pino, M.; García-Gallo, M.; Martin, M.T.; Benet, S.; Chojnacki, J.; Fernández-Figueras, M.T.; Guerrero, D.; Urrea, V.; et al. Anti-Siglec-1 Antibodies Block Ebola Viral Uptake and Decrease Cytoplasmic Viral Entry. Nat. Microbiol. 2019, 4, 1558–1570. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Structural Basis for the Recognition of SARS-CoV-2 by Full-Length Human ACE2. Science 2020, 367, 2444–2447. [Google Scholar]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 1–10. [Google Scholar] [CrossRef]
- Clausen, T.M.; Sandoval, D.R.; Spliid, C.B.; Pihl, J.; Perrett, H.R.; Painter, C.D.; Narayanan, A.; Majowicz, S.A.; Kwong, E.M.; McVicar, R.N.; et al. SARS-CoV-2 Infection Depends on Cellular Heparan Sulfate and ACE2. Cell 2020, 183, 1043–1057. [Google Scholar] [CrossRef]
- Sigrist, C.J.; Bridge, A.; Le Mercier, P. A Potential Role for Integrins in Host Cell Entry by SARS-CoV-2. Antivir. Res. 2020, 177, 104759. [Google Scholar] [CrossRef]
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; van der Meer, F.; Kallio, K.; Kaya, T.; Anastasina, M.; et al. Neuropilin-1 Facilitates SARS-CoV-2 Cell Entry and Infectivity. Science 2020, 370, 856–860. [Google Scholar] [CrossRef]
- Daly, J.L.; Simonetti, B.; Klein, K.; Chen, K.-E.; Williamson, M.K.; Antón-Plágaro, C.; Shoemark, D.K.; Simón-Gracia, L.; Bauer, M.; Hollandi, R.; et al. Neuropilin-1 Is a Host Factor for SARS-CoV-2 Infection. Science 2020, 370, 861–865. [Google Scholar] [CrossRef]
- Wang, K.; Chen, W.; Zhou, Y.-S.; Lian, J.-Q.; Zhang, Z.; Du, P.; Gong, L.; Zhang, Y.; Cui, H.Y.; Geng, J.-J.; et al. SARS-CoV-2 Invades Host Cells via a Novel Route: CD147-Spike Protein. BioRxiv 2020, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Thepaut, M.; Luczkowiak, J.; Vives, C.; Labiod, N.; Bally, I.; Lasala, F.; Grimoire, Y.; Fenel, D.; Sattin, S.; Thielens, N.; et al. DC/L-SIGN Recognition of Spike Glycoprotein Promotes SARS-CoV-2 Trans-Infection and Can Be Inhibited by a Glycomimetic Antagonist. Biorxiv 2020. [Google Scholar] [CrossRef]
- Wang, S.; Qiu, Z.; Hou, Y.; Deng, X.; Zheng, T.; Yan, R.; Wu, P.; Xie, S.; Zhou, Q.; Huang, J.; et al. AXL Promotes SARS-CoV-2 Infection of Pulmonary and Bronchial Epithelial Cells. BioRxiv 2020. [Google Scholar] [CrossRef]
- Schloer, S.; Brunotte, L.; Goretzko, J.; Mecate-Zambrano, A.; Korthals, N.; Gerke, V.; Ludwig, S.; Rescher, U. Targeting the Endolysosomal Host-SARS-CoV-2 Interface by Clinically Licensed Functional Inhibitors of Acid Sphingomyelinase (FIASMA) Including the Antidepressant Fluoxetine. Emerg. Microbes Infect. 2020, 9, 2245–2255. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 180, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Belouzard, S.; Chu, V.C.; Whittaker, G.R. Activation of the SARS Coronavirus Spike Protein via Sequential Proteolytic Cleavage at Two Distinct Sites. Proc. Natl. Acad. Sci. USA 2009, 106, 5871–5876. [Google Scholar] [CrossRef] [Green Version]
- Walls, A.C.; Tortorici, M.A.; Snijder, J.; Xiong, X.; Bosch, B.J.; Rey, F.A.; Veesler, D. Tectonic Conformational Changes of a Coronavirus Spike Glycoprotein Promote Membrane Fusion. Proc. Natl. Acad. Sci. USA 2017, 114, 11157–11162. [Google Scholar] [CrossRef] [Green Version]
- Coutard, B.; Valle, C.; de Lamballerie, X.; Canard, B.; Seidah, N.G.; Decroly, E. The Spike Glycoprotein of the New Coronavirus 2019-NCoV Contains a Furin-like Cleavage Site Absent in CoV of the Same Clade. Antivir. Res. 2020, 176, 104742. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 Spike Receptor-Binding Domain Bound to the ACE2 Receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor Recognition by the Novel Coronavirus from Wuhan: An Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [Green Version]
- Jaimes, J.A.; Millet, J.K.; Whittaker, G.R. Proteolytic Cleavage of the SARS-CoV-2 Spike Protein and the Role of the Novel S1/S2 Site. iScience 2020, 23, 101212. [Google Scholar] [CrossRef] [PubMed]
- Madu, I.G.; Roth, S.L.; Belouzard, S.; Whittaker, G.R. Characterization of a Highly Conserved Domain within the Severe Acute Respiratory Syndrome Coronavirus Spike Protein S2 Domain with Characteristics of a Viral Fusion Peptide. J. Virol. 2009, 83, 7411–7421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zang, R.; Gomez Castro, M.F.; McCune, B.T.; Zeng, Q.; Rothlauf, P.W.; Sonnek, N.M.; Liu, Z.; Brulois, K.F.; Wang, X.; Greenberg, H.B.; et al. TMPRSS2 and TMPRSS4 Promote SARS-CoV-2 Infection of Human Small Intestinal Enterocytes. Sci. Immunol. 2020, 5, eabc3582. [Google Scholar] [CrossRef] [PubMed]
- Nanbo, A.; Imai, M.; Watanabe, S.; Noda, T.; Takahashi, K.; Neumann, G.; Halfmann, P.; Kawaoka, Y. Ebolavirus Is Internalized into Host Cells via Macropinocytosis in a Viral Glycoprotein-Dependent Manner. PLoS Pathog. 2010, 6, e1001121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saeed, M.F.; Kolokoltsov, A.A.; Albrecht, T.; Davey, R.A. Cellular Entry of Ebola Virus Involves Uptake by a Macropinocytosis-like Mechanism and Subsequent Trafficking through Early and Late Endosomes. PLoS Pathog. 2010, 6, e1001110. [Google Scholar] [CrossRef] [Green Version]
- Volchkov, V.E.; Feldmann, H.; Volchkova, V.A.; Klenk, H.D. Processing of the Ebola Virus Glycoprotein by the Proprotein Convertase Furin. Proc. Natl. Acad. Sci. USA 1998, 95, 5762–5767. [Google Scholar] [CrossRef] [Green Version]
- Brecher, M.; Schornberg, K.L.; Delos, S.E.; Fusco, M.L.; Saphire, E.O.; White, J.M. Cathepsin Cleavage Potentiates the Ebola Virus Glycoprotein To Undergo a Subsequent Fusion-Relevant Conformational Change. J. Virol. 2012, 86, 364–372. [Google Scholar] [CrossRef] [Green Version]
- Chandran, K.; Sullivan, N.J.; Felbor, U.; Whelan, S.P.; Cunningham, J.M. Virology: Endosomal Proteolysis of the Ebola Virus Glycoprotein Is Necessary for Infection. Science 2005, 308, 1643–1646. [Google Scholar] [CrossRef] [Green Version]
- Mingo, R.M.; Simmons, J.A.; Shoemaker, C.J.; Nelson, E.A.; Schornberg, K.L.; D’Souza, R.S.; Casanova, J.E.; White, J.M. Ebola Virus and Severe Acute Respiratory Syndrome Coronavirus Display Late Cell Entry Kinetics: Evidence That Transport to NPC1+ Endolysosomes Is a Rate-Defining Step. J. Virol. 2015, 89, 2931–2943. [Google Scholar] [CrossRef] [Green Version]
- Carette, J.E.; Raaben, M.; Wong, A.C.; Herbert, A.S.; Obernosterer, G.; Mulherkar, N.; Kuehne, A.I.; Kranzusch, P.J.; Griffin, A.M.; Ruthel, G.; et al. Ebola Virus Entry Requires the Cholesterol Transporter Niemann-Pick C1. Nature 2011, 477, 340–343. [Google Scholar] [CrossRef] [Green Version]
- Côté, M.; Misasi, J.; Ren, T.; Bruchez, A.; Lee, K.; Filone, C.M.; Hensley, L.; Li, Q.; Ory, D.; Chandran, K.; et al. Small Molecule Inhibitors Reveal Niemann-Pick C1 Is Essential for Ebola Virus Infection. Nature 2011, 477, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, Y.; Kolokoltsov, A.A.; Chen, C.C.; Tidwell, M.W.; Bauta, W.E.; Klugbauer, N.; Grimm, C.; Wahl-Schott, C.; Biel, M.; Davey, R.A. Two-Pore Channels Control Ebola Virus Host Cell Entry and Are Drug Targets for Disease Treatment. Science 2015, 347, 995–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM Structure of the 2019-NCoV Spike in the Prefusion Conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banadyga, L.; Hoenen, T.; Ambroggio, X.; Dunham, E.; Groseth, A.; Ebihara, H. Ebola Virus VP24 Interacts with NP to Facilitate Nucleocapsid Assembly and Genome Packaging. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Watt, A.; Moukambi, F.; Banadyga, L.; Groseth, A.; Callison, J.; Herwig, A.; Ebihara, H.; Feldmann, H.; Hoenen, T. A Novel Life Cycle Modeling System for Ebola Virus Shows a Genome Length-Dependent Role of VP24 in Virus Infectivity. J. Virol. 2014, 88, 10511–10524. [Google Scholar] [CrossRef] [Green Version]
- Mühlberger, E.; Weik, M.; Volchkov, V.E.; Klenk, H.-D.; Becker, S. Comparison of the Transcription and Replication Strategies of Marburg Virus and Ebola Virus by Using Artificial Replication Systems. J. Virol. 1999, 73, 2333–2342. [Google Scholar] [CrossRef] [Green Version]
- Finkel, Y.; Mizrahi, O.; Nachshon, A.; Weingarten-Gabbay, S.; Morgenstern, D.; Yahalom-Ronen, Y.; Tamir, H.; Achdout, H.; Stein, D.; Israeli, O.; et al. The Coding Capacity of SARS-CoV-2. Nature 2020, 589, 125–130. [Google Scholar] [CrossRef]
- Sims, A.C.; Ostermann, J.; Denison, M.R. Mouse Hepatitis Virus Replicase Proteins Associate with Two Distinct Populations of Intracellular Membranes. J. Virol. 2000, 74, 5647–5654. [Google Scholar] [CrossRef] [Green Version]
- Snijder, E.J.; Decroly, E.; Ziebuhr, J. The Nonstructural Proteins Directing Oronavirus RNA Synthesis and Processing. Adv. Virus Res. 2016, 96, 58–126. [Google Scholar]
- Philip, V.; Markus, G.; Jenna, K.; Stephanie, P.; Nadine, E.; Sophie, B.L.; Cedric, S.; Jasmine, P.; Hanspeter, S.; Véronique, G.; et al. Determination of Host Cell Proteins Constituting the Molecular Microenvironment of Coronavirus Replicase Complexes by Proximity-Labeling. eLife 2019, 8, e42037. [Google Scholar]
- Schubert, K.; Karousis, E.D.; Jomaa, A.; Scaiola, A.; Echeverria, B.; Gurzeler, L.A.; Leibundgut, M.; Thiel, V.; Mühlemann, O.; Ban, N. SARS-CoV-2 Nsp1 Binds the Ribosomal MRNA Channel to Inhibit Translation. Nat. Struct. Mol. Biol. 2020, 27, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Thoms, M.; Buschauer, R.; Ameismeier, M.; Koepke, L.; Denk, T.; Hirschenberger, M.; Kratzat, H.; Hayn, M.; MacKens-Kiani, T.; Cheng, J.; et al. Structural Basis for Translational Shutdown and Immune Evasion by the Nsp1 Protein of SARS-CoV-2. Science 2020, 369, 1249–1256. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Gupta, S.P. Fundamentals of Viruses and Their Proteases. In Viral Proteases and Their Inhibitors; Elsevier: Amsterdam, The Netherlands, 2017; pp. 1–24. ISBN 978-0-12-809712-0. [Google Scholar]
- Amin, S.A.; Banerjee, S.; Ghosh, K.; Gayen, S.; Jha, T. Protease Targeted COVID-19 Drug Discovery and Its Challenges: Insight into Viral Main Protease (Mpro) and Papain-like Protease (PLpro) Inhibitors. Bioorg. Med. Chem. 2020, 115860. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Yan, L.; Huang, Y.; Liu, F.; Zhao, Y.; Cao, L.; Wang, T.; Sun, Q.; Ming, Z.; Zhang, L.; et al. Structure of the RNA-Dependent RNA Polymerase from COVID-19 Virus. Science 2020, 368, 779–782. [Google Scholar] [CrossRef] [Green Version]
- Fang, J.; Pietzsch, C.; Ramanathan, P.; Santos, R.I.; Ilinykh, P.A.; Garcia-Blanco, M.A.; Bukreyev, A.; Bradrick, S.S. Staufen1 Interacts with Multiple Components of the Ebola Virus Ribonucleoprotein and Enhances Viral RNA Synthesis. MBio 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Halfmann, P.; Oyama, M.; Kozuka-Hata, H.; Noda, T.; Kawaoka, Y. DNA Topoisomerase 1 Facilitates the Transcription and Replication of the Ebola Virus Genome. J. Virol. 2013, 87, 8862–8869. [Google Scholar] [CrossRef] [Green Version]
- Martin, S.; Chiramel, A.I.; Schmidt, M.L.; Chen, Y.C.; Whitt, N.; Watt, A.; Dunham, E.C.; Shifflett, K.; Traeger, S.; Leske, A.; et al. A Genome-Wide SiRNA Screen Identifies a Druggable Host Pathway Essential for the Ebola Virus Life Cycle. Genome Med. 2018, 10, 58. [Google Scholar] [CrossRef]
- Ammosova, T.; Pietzsch, C.A.; Saygideǧer, Y.; Ilatovsky, A.; Lin, X.; Ivanov, A.; Kumari, N.; Jerebtsova, M.; Kulkarni, A.; Petukhov, M.; et al. Protein Phosphatase 1-Targeting Small-Molecule C31 Inhibits Ebola Virus Replication. J. Infect. Dis. 2018, 218, S627–S635. [Google Scholar] [CrossRef] [Green Version]
- Modrof, J.; Mühlberger, E.; Klenk, H.D.; Becker, S. Phosphorylation of VP30 Impairs Ebola Virus Transcription. J. Biol. Chem. 2002, 277, 33099–33104. [Google Scholar] [CrossRef] [Green Version]
- Batra, J.; Hultquist, J.F.; Liu, D.; Shtanko, O.; Von Dollen, J.; Satkamp, L.; Jang, G.M.; Luthra, P.; Schwarz, T.M.; Gabriel, I.; et al. Protein Interactio Mapping Identifies RBBP6 as a Negative Regulator of Ebola Virus Replication. Cell 2019, 175, 1917–1930. [Google Scholar] [CrossRef] [Green Version]
- Mehedi, M.; Falzarano, D.; Seebach, J.; Hu, X.; Carpenter, M.S.; Schnittler, H.-J.; Feldmann, H. A New Ebola Virus Nonstructural Glycoprotein Expressed through RNA Editing. J. Virol. 2011, 85, 5406–5414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Wilde, A.H.; Snijder, E.J.; Kikkert, M.; van Hemert, M.J. Host Factors in Coronavirus Replication. Curr. Top. Microbiol. Immunol. 2018, 419, 1–42. [Google Scholar] [PubMed] [Green Version]
- Gabriel, G.; Feldmann, F.; Reimer, R.; Thiele, S.; Fischer, M.; Hartmann, E.; Bader, M.; Ebihara, H.; Hoenen, T.; Feldmann, H. Importin-Α7 Is Involved in the Formation of Ebola Virus Inclusion Bodies but Is Not Essential for Pathogenicity in Mice. J. Infect. Dis. 2015, 212, S316–S321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelini, M.M.; Akhlaghpour, M.; Neuman, B.W.; Buchmeier, M.J. Severe Acute Respiratory Syndrome Coronavirus Nonstructural Proteins 3, 4, and 6 Induce Double-Membrane Vesicles. MBio 2013, 4, e00524-13. [Google Scholar] [CrossRef] [Green Version]
- Lundin, A.; Dijkman, R.; Bergström, T.; Kann, N.; Adamiak, B.; Hannoun, C.; Kindler, E.; Jónsdóttir, H.R.; Muth, D.; Kint, J.; et al. Targeting Membrane-Bound Viral RNA Synthesis Reveals Potent Inhibition of Diverse Coronaviruses Including the Middle East Respiratory Syndrome Virus. PLoS Pathog. 2014, 10, e1004166. [Google Scholar] [CrossRef] [Green Version]
- Oudshoorn, D.; Rijs, K.; Limpens, R.W.A.L.; Groen, K.; Koster, A.J.; Snijder, E.J.; Kikkert, M.; Bárcena, M. Expression and Cleavage of Middle East Respiratory Syndrome Coronavirus Nsp3-4 Polyprotein Induce the Formation of Double-Membrane Vesicles That Mimic Those Associated with Coronaviral RNA Replication. MBio 2017, 8, e01658-17. [Google Scholar] [CrossRef] [Green Version]
- Wolff, G.; Limpens, R.W.A.L.; Zevenhoven-Dobbe, J.C.; Laugks, U.; Zheng, S.; de Jong, A.W.M.; Koning, R.I.; Agard, D.A.; Grünewald, K.; Koster, A.J.; et al. A Molecular Pore Spans the Double Membrane of the Coronavirus Replication Organelle. Science 2020, 369, 1395–1398. [Google Scholar] [CrossRef]
- Schudt, G.; Dolnik, O.; Kolesnikova, L.; Biedenkopf, N.; Herwig, A.; Becker, S. Transport of Ebolavirus Nucleocapsids Is Dependent on Actin Polymerization: Live-Cell Imaging Analysis of Ebolavirus-Infected Cells. J. Infect. Dis. 2015, 212, S160–S166. [Google Scholar] [CrossRef]
- Takamatsu, Y.; Kolesnikova, L.; Becker, S. Ebola Virus Proteins NP, VP35, and VP24 Are Essential and Sufficient to Mediate Nucleocapsid Transport. Proc. Natl. Acad. Sci. USA 2018, 115, 1075–1080. [Google Scholar] [CrossRef] [Green Version]
- Noda, T.; Sagara, H.; Suzuki, E.; Takada, A.; Kida, H.; Kawaoka, Y. Ebola Virus VP40 Drives the Formation of Virus-Like Filamentous Particles Along with GP. J. Virol. 2002, 76, 4855–4865. [Google Scholar] [CrossRef] [Green Version]
- Adu-Gyamfi, E.; Digman, M.A.; Gratton, E.; Stahelin, R.V. Single-Particle Tracking Demonstrates That Actin Coordinates the Movement of the Ebola Virus Matrix Protein. Biophys. J. 2012, 103, L41–L43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Z.; Harty, R.N. Packaging of Actin into Ebola Virus VLPs. Virol. J. 2005, 2, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruthel, G.; Demmin, G.L.; Kallstrom, G.; Javid, M.P.; Badie, S.S.; Will, A.B.; Nelle, T.; Schokman, R.; Nguyen, T.L.; Carra, J.H.; et al. Association of Ebola Virus Matrix Protein VP40 with Microtubules. J. Virol. 2005, 79, 4709–4719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamayoshi, S.; Noda, T.; Ebihara, H.; Goto, H.; Morikawa, Y.; Lukashevich, I.S.; Neumann, G.; Feldmann, H.; Kawaoka, Y. Ebola Virus Matrix Protein VP40 Uses the COPII Transport System for Its Intracellular Transport. Cell Host Microbe 2008, 3, 168–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, G.; Feldmann, H.; Watanabe, S.; Lukashevich, I.; Kawaoka, Y. Reverse Genetics Demonstrates That Proteolytic Processing of the Ebola Virus Glycoprotein Is Not Essential for Replication in Cell Culture. J. Virol. 2002, 76, 406–410. [Google Scholar] [CrossRef] [Green Version]
- Wool-Lewis, R.J.; Bates, P. Endoproteolytic Processing of the Ebola Virus Envelope Glycoprotein: Cleavage Is Not Required for Function. J. Virol. 1999, 73, 1419–1426. [Google Scholar] [CrossRef] [Green Version]
- Licata, J.M.; Simpson-Holley, M.; Wright, N.T.; Han, Z.; Paragas, J.; Harty, R.N. Overlapping Motifs (PTAP and PPEY) within the Ebola Virus VP40 Protein Function Independently as Late Budding Domains: Involvement of Host Proteins TSG101 and VPS-4. J. Virol. 2003, 77, 1812–1819. [Google Scholar] [CrossRef] [Green Version]
- Bavari, S.; Bosio, C.M.; Wiegand, E.; Ruthel, G.; Will, A.B.; Geisbert, T.W.; Hevey, M.; Schmaljohn, C.; Schmaljohn, A.; Aman, M.J. Lipid Raft Microdomains: A Gateway for Compartmentalized Trafficking of Ebola and Marburg Viruses. J. Exp. Med. 2002, 195, 593–602. [Google Scholar] [CrossRef]
- Nanbo, A.; Maruyama, J.; Imai, M.; Ujie, M.; Fujioka, Y.; Nishide, S.; Takada, A.; Ohba, Y.; Kawaoka, Y. Ebola Virus Requires a Host Scramblase for Externalization of Phosphatidylserine on the Surface of Viral Particles. PLoS Pathog. 2018, 14, e1006848. [Google Scholar] [CrossRef]
- Moller-Tank, S.; Maury, W. Ebola Virus Entry: A Curious and Complex Series of Events. PLoS Pathog. 2015, 11, e1004731. [Google Scholar] [CrossRef]
- De Haan, C.A.M.; Rottier, P.J.M. Molecular Interactions in the Assembly of Coronaviruses. Adv. Virus Res. 2005, 64, 165–230. [Google Scholar] [PubMed]
- Stertz, S.; Reichelt, M.; Spiegel, M.; Kuri, T.; Martínez-Sobrido, L.; García-Sastre, A.; Weber, F.; Kochs, G. The Intracellular Sites of Early Replication and Budding of SARS-Coronavirus. Virology 2007, 361, 304–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, S.; Cortese, M.; Winter, S.L.; Wachsmuth-Melm, M.; Neufeldt, C.J.; Cerikan, B.; Stanifer, M.L.; Boulant, S.; Bartenschlager, R.; Chlanda, P. SARS-CoV-2 Structure and Replication Characterized by in Situ Cryo-Electron Tomography. Nat. Commun. 2020, 11, 5885. [Google Scholar] [CrossRef] [PubMed]
- Mihelič, M.; Doberšek, A.; Gunčar, G.; Turk, D. Inhibitory Fragment from the P41 Form of Invariant Chain Can Regulate Activity of Cysteine Cathepsins in Antigen Presentation. J. Biol. Chem. 2008, 283, 14453–14460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruchez, A.; Sha, K.; Johnson, J.; Chen, L.; Stefani, C.; McConnell, H.; Gaucherand, L.; Prins, R.; Matreyek, K.A.; Hume, A.J.; et al. MHC Class II Transactivator CIITA Induces Cell Resistance to Ebola Virus and SARS-like Coronaviruses. Science 2020, 370, 241–247. [Google Scholar] [CrossRef]
- Ghosh, S.; Dellibovi-Ragheb, T.A.; Kerviel, A.; Pak, E.; Qiu, Q.; Fisher, M.; Takvorian, P.M.; Bleck, C.; Hsu, V.W.; Fehr, A.R.; et al. β-Coronaviruses Use Lysosomes for Egress Instead of the Biosynthetic Secretory Pathway. Cell 2020, 183, 1520–1535. [Google Scholar] [CrossRef] [PubMed]
- Riva, L.; Yuan, S.; Yin, X.; Martin-Sancho, L.; Matsunaga, N.; Pache, L.; Burgstaller-Muehlbacher, S.; De Jesus, P.D.; Teriete, P.; Hull, M.V.; et al. Discovery of SARS-CoV-2 Antiviral Drugs through Large-Scale Compound Repurposing. Nature 2020, 586, 113–119. [Google Scholar] [CrossRef]
- Cai, X.; Xu, Y.; Cheung, A.K.; Tomlinson, R.C.; Alcázar-Román, A.; Murphy, L.; Billich, A.; Zhang, B.; Feng, Y.; Klumpp, M.; et al. PIKfyve, a Class III PI Kinase, Is the Target of the Small Molecular IL-12/IL-23 Inhibitor Apilimod and a Player in Toll-like Receptor Signaling. Chem. Biol. 2013, 20, 912–921. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.-L.; Chou, Y.; Rothlauf, P.W.; Liu, Z.; Soh, T.K.; Cureton, D.; Brett Case, J.; Chen, R.E.; Diamond, M.S.; Whelan, S.P.J.; et al. Inhibition of PIKfyve Kinase Prevents Infection by Zaire Ebolavirus and SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 20803–20813. [Google Scholar] [CrossRef]
- Madrid, P.B.; Chopra, S.; Manger, I.D.; Gilfillan, L.; Keepers, T.R.; Shurtleff, A.C.; Green, C.E.; Iyer, L.V.; Dilks, H.H.; Davey, R.A.; et al. A Systematic Screen of FDA-Approved Drugs for Inhibitors of Biological Threat Agents. PLoS ONE 2013, 8, e60579. [Google Scholar] [CrossRef] [Green Version]
- Dowall, S.D.; Bosworth, A.; Watson, R.; Bewley, K.; Taylor, I.; Rayner, E.; Hunter, L.; Pearson, G.; Easterbrook, L.; Pitman, J.; et al. Chloroquine Inhibited Ebola Virus Replication in Vitro but Failed to Protect against Infection and Disease in the in Vivo Guinea Pig Model. J. Gen. Virol. 2015, 96, 3484–3492. [Google Scholar] [CrossRef] [PubMed]
- Falzarano, D.; Safronetz, D.; Prescott, J.; Marzi, A.; Feldmann, F.; Feldmann, H. Lack of Protection against Ebola Virus from Chloroquine in Mice and Hamsters. Emerg. Infect. Dis. 2015, 21, 1065–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madrid, P.B.; Panchal, R.G.; Warren, T.K.; Shurtleff, A.C.; Endsley, A.N.; Green, C.E.; Kolokoltsov, A.; Davey, R.; Manger, I.D.; Gilfillan, L.; et al. Evaluation of Ebola Virus Inhibitors for Drug Repurposing. ACS Infect. Dis. 2015, 1, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Gignoux, E.; Azman, A.S.; de Smet, M.; Azuma, P.; Massaquoi, M.; Job, D.; Tiffany, A.; Petrucci, R.; Sterk, E.; Potet, J.; et al. Effect of Artesunate–Amodiaquine on Mortality Related to Ebola Virus Disease. N. Engl. J. Med. 2016, 374, 23–34. [Google Scholar] [CrossRef]
- Hu, T.Y.; Frieman, M.; Wolfram, J. Insights from Nanomedicine into Chloroquine Efficacy against COVID-19. Nat. Nanotechnol. 2020, 15, 247–249. [Google Scholar] [CrossRef] [Green Version]
- Fantini, J.; Di Scala, C.; Chahinian, H.; Yahi, N. Structural and Molecular Modelling Studies Reveal a New Mechanism of Action of Chloroquine and Hydroxychloroquine against SARS-CoV-2 Infection. Int. J. Antimicrob. Agents 2020, 55, 105960. [Google Scholar] [CrossRef]
- Lu, Y.; Liu, D.X.; Tam, J.P. Lipid Rafts Are Involved in SARS-CoV Entry into Vero E6 Cells. Biochem. Biophys. Res. Commun. 2008, 369, 344–349. [Google Scholar] [CrossRef]
- Liu, J.; Cao, R.; Xu, M.; Wang, X.; Zhang, H.; Hu, H.; Li, Y.; Hu, Z.; Zhong, W.; Wang, M. Hydroxychloroquine, a Less Toxic Derivative of Chloroquine, Is Effective in Inhibiting SARS-CoV-2 Infection in Vitro. Cell Discov. 2020, 6, 16. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and Chloroquine Effectively Inhibit the Recently Emerged Novel Coronavirus (2019-NCoV) in Vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef]
- Martinez, O.; Johnson, J.; Manicassamy, B.; Rong, L.; Olinger, G.G.; Hensley, L.E.; Basler, C.F. Zaire Ebola Virus Entry into Human Dendritic Cells Is Insensitive to Cathepsin L Inhibition. Cell. Microbiol. 2010, 12, 148–157. [Google Scholar] [CrossRef] [Green Version]
- Rodon, J.; Muñoz-Basagoiti, J.; Perez-Zsolt, D.; Noguera-Julian, M.; Paredes, R.; Mateu, L.; Quiñones, C.; Erkizia, I.; Blanco, I.; Valencia, A.; et al. Search for SARS-CoV-2 Inhibitors in Currently Approved Drugs to Tackle COVID-19 Pandemia. BioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Maisonnasse, P.; Guedj, J.; Contreras, V.; Behillil, S.; Solas, C.; Marlin, R.; Naninck, T.; Pizzorno, A.; Lemaitre, J.; Gonçalves, A.; et al. Hydroxychloroquine Use against SARS-CoV-2 Infection in Non-Human Primates. Nature 2020, 585, 584–587. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; Peto, R.; Henao-Restrepo, A.-M.; Preziosi, M.-P.; Sathiyamoorthy, V.; Abdool Karim, Q.; Alejandria, M.M.; Hernández García, C.; Kieny, M.-P.; Malekzadeh, R.; et al. Repurposed Antiviral Drugs for Covid-19—Interim WHO Solidarity Trial Results. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Boulware, D.R.; Pullen, M.F.; Bangdiwala, A.S.; Pastick, K.A.; Lofgren, S.M.; Okafor, E.C.; Skipper, C.P.; Nascene, A.A.; Nicol, M.R.; Abassi, M.; et al. A Randomized Trial of Hydroxychloroquine as Postexposure Prophylaxis for Covid-19. N. Engl. J. Med. 2020, 383, 517–525. [Google Scholar] [CrossRef]
- Cavalcanti, A.B.; Zampieri, F.G.; Rosa, R.G.; Azevedo, L.C.P.; Veiga, V.C.; Avezum, A.; Damiani, L.P.; Marcadenti, A.; Kawano-Dourado, L.; Lisboa, T.; et al. Hydroxychloroquine with or without Azithromycin in Mild-to-Moderate Covid-19. N. Engl. J. Med. 2020, 383, 2041–2052. [Google Scholar] [CrossRef]
- Hanley, B.; Naresh, K.N.; Roufosse, C.; Nicholson, A.G.; Weir, J.; Cooke, G.S.; Thursz, M.; Manousou, P.; Corbett, R.; Goldin, R.; et al. Histopathological Findings and Viral Tropism in UK Patients with Severe Fatal COVID-19: A Post-Mortem Study. Lancet Microbe 2020, 1, e245–e253. [Google Scholar] [CrossRef]
- Ou, T.; Mou, H.; Zhang, L.; Ojha, A.; Choe, H.; Farzan, M. Hydroxychloroquine-Mediated Inhibition of SARS-CoV-2 Entry Is Attenuated by TMPRSS2. Biorxiv 2020. [Google Scholar] [CrossRef]
- Mulangu, S.; Dodd, L.E.; Davey, R.T.; Tshiani Mbaya, O.; Proschan, M.; Mukadi, D.; Lusakibanza Manzo, M.; Nzolo, D.; Tshomba Oloma, A.; Ibanda, A.; et al. A Randomized, Controlled Trial of Ebola Virus Disease Therapeutics. N. Engl. J. Med. 2019, 381, 2293–2303. [Google Scholar] [CrossRef]
- Warren, T.K.; Jordan, R.; Lo, M.K.; Ray, A.S.; Mackman, R.L.; Soloveva, V.; Siegel, D.; Perron, M.; Bannister, R.; Hui, H.C.; et al. Therapeutic Efficacy of the Small Molecule GS-5734 against Ebola Virus in Rhesus Monkeys. Nature 2016, 531, 381–385. [Google Scholar] [CrossRef]
- Sheahan, T.P.; Sims, A.C.; Graham, R.L.; Menachery, V.D.; Gralinski, L.E.; Case, J.B.; Leist, S.R.; Pyrc, K.; Feng, J.Y.; Trantcheva, I.; et al. Broad-Spectrum Antiviral GS-5734 Inhibits Both Epidemic and Zoonotic Coronaviruses. Sci. Transl. Med. 2017, 9, eaal3653. [Google Scholar] [CrossRef] [Green Version]
- Williamson, B.N.; Feldmann, F.; Schwarz, B.; Meade-White, K.; Porter, D.P.; Schulz, J.; van Doremalen, N.; Leighton, I.; Yinda, C.K.; Pérez-Pérez, L.; et al. Clinical Benefit of Remdesivir in Rhesus Macaques Infected with SARS-CoV-2. Nature 2020, 585, 273–276. [Google Scholar] [CrossRef] [PubMed]
- WHO “Solitdarity” Clinical Trial for COVID-19 Treatments. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019/global-research-on-novel-coronavirus-2019-ncov/solidarity-clinical-trial-for-covid-19-treatments (accessed on 17 January 2021).
- Gillenwater, S.; Rahaghi, F.; Hadeh, A. Remdesivir for the Treatment of Covid-19—Preliminary Report. N. Engl. J. Med. 2020, 383, 992–994. [Google Scholar] [PubMed]
- Grein, J.; Ohmagari, N.; Shin, D.; Diaz, G.; Asperges, E.; Castagna, A.; Feldt, T.; Green, G.; Green, M.L.; Lescure, F.-X.; et al. Compassionate Use of Remdesivir for Patients with Severe Covid-19. N. Engl. J. Med. 2020, 382, 2327–2336. [Google Scholar] [CrossRef] [PubMed]
- Kaptein, K.; Jacobs, S.; Langendries, L.; Seldeslachts, L.; ter Horst, S.; Liesenborghs, L.; Hens, B.; Vergote, V.; Heylen, E.; Barthelemy, K.; et al. Favipiravir at High Doses Has Potent Antiviral Activity in SARS-CoV-2−infected Hamsters, Whereas Hydroxychloroquine Lacks Activity. Proc. Natl. Acad. Sci. USA 2020, 117, 26955–26965. [Google Scholar] [CrossRef] [PubMed]
- Guedj, J.; Piorkowski, G.; Jacquot, F.; Madelain, V.; Nguyen, T.H.T.; Rodallec, A.; Gunther, S.; Carbonnelle, C.; Mentré, F.; Raoul, H.; et al. Antiviral Efficacy of Favipiravir against Ebola Virus: A Translational Study in Cynomolgus Macaques. PLoS Med. 2018, 15, e1002535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Losada, A.; Muñoz-Alonso, M.J.; García, C.; Sánchez-Murcia, P.A.; Martínez-Leal, J.F.; Domínguez, J.M.; Lillo, M.P.; Gago, F.; Galmarini, C.M. Translation Elongation Factor EEF1A2 Is a Novel Anticancer Target for the Marine Natural Product Plitidepsin. Sci. Rep. 2016, 6, 35100. [Google Scholar] [CrossRef] [PubMed]
- Lou, B.; Li, T.D.; Zheng, S.F.; Su, Y.Y.; Li, Z.Y.; Liu, W.; Yu, F.; Ge, S.X.; Zou, Q.D.; Yuan, Q.; et al. Serology Characteristics of SARS-CoV-2 Infection since Exposure and Post Symptom Onset. Eur. Respir. J. 2020, 56. [Google Scholar] [CrossRef] [PubMed]
- Jefferis, R. Isotype and Glycoform Selection for Antibody Therapeutics. Arch. Biochem. Biophys. 2012, 526, 159–166. [Google Scholar] [CrossRef]
- Jefferis, R.; Lund, J.; Pound, J.D. IgG-Fc-Mediated Effector Functions: Molecular Definition of Interaction Sites for Effector Ligands and the Role of Glycosylation. Immunol. Rev. 1998, 163, 59–76. [Google Scholar] [CrossRef]
- Ito, H.; Watanabe, S.; Takada, A.; Kawaoka, Y. Ebola Virus Glycoprotein: Proteolytic Processing, Acylation, Cell Tropism, and Detection of Neutralizing Antibodies. J. Virol. 2001, 75, 1576–1580. [Google Scholar] [CrossRef] [Green Version]
- Yuan, M.; Wu, N.C.; Zhu, X.; Lee, C.-C.D.; So, R.T.Y.; Lv, H.; Mok, C.K.P.; Wilson, I.A. A Highly Conserved Cryptic Epitope in the Receptor Binding Domains of SARS-CoV-2 and SARS-CoV. Science 2020, 368, 630–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Wang, P.; Nair, M.S.; Yu, J.; Rapp, M.; Wang, Q.; Luo, Y.; Chan, J.F.W.; Sahi, V.; Figueroa, A.; et al. Potent Neutralizing Antibodies against Multiple Epitopes on SARS-CoV-2 Spike. Nature 2020, 584, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Pelegrin, M.; Naranjo-Gomez, M.; Piechaczyk, M. Antiviral Monoclonal Antibodies: Can They Be More Than Simple Neutralizing Agents? Trends Microbiol. 2015, 23, 653–656. [Google Scholar] [CrossRef] [PubMed]
- Winau, F.; Winau, R. Emil von Behring and Serum Therapy. Microbes Infect. 2002, 4, 185–188. [Google Scholar] [CrossRef]
- Hsu, J.L.; Safdar, N. Polyclonal Immunoglobulins and Hyperimmune Globulins in Prevention and Management of Infectious Diseases. Infect. Dis. Clin. N. Am. 2011, 25, 773–788. [Google Scholar] [CrossRef] [PubMed]
- Köhler, G.; Milstein, C. Continuous Cultures of Fused Cells Secreting Antibody of Predefined Specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef]
- Li, F.; Vijayasankaran, N.; Shen, A.; Kiss, R.; Amanullah, A. Cell Culture Processes for Monoclonal Antibody Production. MAbs 2010, 2, 466–479. [Google Scholar] [CrossRef] [Green Version]
- Rosman, Z.; Shoenfeld, Y.; Zandman-Goddard, G. Biologic Therapy for Autoimmune Diseases: An Update. BMC Med. 2013, 11, 88. [Google Scholar] [CrossRef] [Green Version]
- Scott, A.M.; Wolchok, J.D.; Old, L.J. Antibody Therapy of Cancer. Nat. Rev. Cancer 2012, 12, 278–287. [Google Scholar] [CrossRef]
- Shadman, K.A.; Wald, E.R. A Review of Palivizumab and Emerging Therapies for Respiratory Syncytial Virus. Expert Opin. Biol. Ther. 2011, 11, 1455–1467. [Google Scholar] [CrossRef]
- Carrillo, J.; Clotet, B.; Blanco, J. Antibodies and Antibody Derivatives: New Partners in HIV Eradication Strategies. Front. Immunol. 2018, 9, 2429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shanmugaraj, B.; Siriwattananon, K.; Wangkanont, K.; Phoolcharoen, W. Perspectives on Monoclonal Antibody Therapy as Potential Therapeutic Intervention for Coronavirus Disease-19 (COVID-19). Asian Pac. J. Allergy Immunol. 2020, 38, 10–18. [Google Scholar] [PubMed]
- Moekotte, A.L.; Huson, M.A.M.; van der Ende, A.J.; Agnandji, S.T.; Huizenga, E.; Goorhuis, A.; Grobusch, M.P. Monoclonal Antibodies for the Treatment of Ebola Virus Disease. Expert Opin. Investig. Drugs 2016, 25, 1325–1335. [Google Scholar] [CrossRef] [PubMed]
- Saphire, E.O.; Schendel, S.L.; Fusco, M.L.; Gangavarapu, K.; Gunn, B.M.; Wec, A.Z.; Halfmann, P.J.; Brannan, J.M.; Herbert, A.S.; Qiu, X.; et al. Systematic Analysis of Monoclonal Antibodies against Ebola Virus GP Defines Features That Contribute to Protection. Cell 2018, 174, 938–952. [Google Scholar] [CrossRef] [Green Version]
- Sanders, J.M.; Monogue, M.L.; Jodlowski, T.Z.; Cutrell, J.B. Pharmacologic Treatments for Coronavirus Disease 2019 (COVID-19): A Review. JAMA J. Am. Med. Assoc. 2020, 323, 1824–1836. [Google Scholar] [CrossRef] [PubMed]
- Ju, B.; Zhang, Q.; Ge, J.; Wang, R.; Sun, J.; Ge, X.; Yu, J.; Shan, S.; Zhou, B.; Song, S.; et al. Human Neutralizing Antibodies Elicited by SARS-CoV-2 Infection. Nature 2020, 584, 115–119. [Google Scholar] [CrossRef]
- Tortorici, M.A.; Beltramello, M.; Lempp, F.A.; Pinto, D.; Dang, H.V.; Rosen, L.E.; Mccallum, M.; Bowen, J.; Minola, A.; Jaconi, S.; et al. Ultrapotent Human Antibodies Protect against SARS-CoV-2 Challenge via Multiple Mechanisms. Science 2020, 3354, 29–31. [Google Scholar] [CrossRef]
- Rogers, T.F.; Zhao, F.; Huang, D.; Beutler, N.; Burns, A.; He, W.; Limbo, O.; Smith, C.; Song, G.; Woehl, J.; et al. Isolation of Potent SARS-CoV-2 Neutralizing Antibodies and Protection from Disease in a Small Animal Model. Sciences 2020, 369, 956–963. [Google Scholar] [CrossRef]
- Hassan, A.O.; Case, J.B.; Winkler, E.S.; Thackray, L.B.; Kafai, N.M.; Bailey, A.L.; McCune, B.T.; Fox, J.M.; Chen, R.E.; Alsoussi, W.B.; et al. A SARS-CoV-2 Infection Model in Mice Demonstrates Protection by Neutralizing Antibodies. Cell 2020, 182, 744–753. [Google Scholar] [CrossRef]
- Rajendran, K.; Krishnasamy, N.; Rangarajan, J.; Rathinam, J.; Natarajan, M.; Ramachandran, A. Convalescent Plasma Transfusion for the Treatment of COVID-19: Systematic Review. J. Med. Virol. 2020, 92, 1475–1483. [Google Scholar] [CrossRef]
- Rojas, M.; Rodríguez, Y.; Monsalve, D.M.; Acosta-Ampudia, Y.; Camacho, B.; Gallo, J.E.; Rojas-Villarraga, A.; Ramírez-Santana, C.; Díaz-Coronado, J.C.; Manrique, R.; et al. Convalescent Plasma in Covid-19: Possible Mechanisms of Action. Autoimmun. Rev. 2020, 19, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zost, S.J.; Gilchuk, P.; Case, J.B.; Binshtein, E.; Chen, R.E.; Nkolola, J.P.; Schäfer, A.; Reidy, J.X.; Trivette, A.; Nargi, R.S.; et al. Potently Neutralizing and Protective Human Antibodies against SARS-CoV-2. Nature 2020, 584, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Dolzhikova, I.V.; Zubkova, O.V.; Tukhvatulin, A.I.; Dzharullaeva, A.S.; Tukhvatulina, N.M.; Shcheblyakov, D.V.; Shmarov, M.M.; Tokarskaya, E.A.; Simakova, Y.V.; Egorova, D.A.; et al. Safety and Immunogenicity of GamEvac-Combi, a Heterologous VSV- and Ad5-Vectored Ebola Vaccine: An Open Phase I/II Trial in Healthy Adults in Russia. Hum. Vaccines Immunother. 2017, 13, 613–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, F.C.; Wurie, A.H.; Hou, L.H.; Liang, Q.; Li, Y.H.; Russell, J.B.W.; Wu, S.P.; Li, J.X.; Hu, Y.M.; Guo, Q.; et al. Safety and Immunogenicity of a Recombinant Adenovirus Type-5 Vector-Based Ebola Vaccine in Healthy Adults in Sierra Leone: A Single-Centre, Randomised, Double-Blind, Placebo-Controlled, Phase 2 Trial. Lancet 2017, 389, 621–628. [Google Scholar] [CrossRef] [Green Version]
- Tapia, M.D.; Sow, S.O.; Lyke, K.E.; Haidara, F.C.; Diallo, F.; Doumbia, M.; Traore, A.; Coulibaly, F.; Kodio, M.; Onwuchekwa, U.; et al. Use of ChAd3-EBO-Z Ebola Virus Vaccine in Malian and US Adults, and Boosting of Malian Adults with MVA-BN-Filo: A Phase 1, Single-Blind, Randomised Trial, a Phase 1b, Open-Label and Double-Blind, Dose-Escalation Trial, and a Nested, Randomised, Double-Blind, Placebo-Controlled Trial. Lancet Infect. Dis. 2016, 16, 31–42. [Google Scholar]
- Medaglini, D.; Santoro, F.; Siegrist, C.-A. Correlates of Vaccine-Induced Protective Immunity against Ebola Virus Disease. Semin. Immunol. 2018, 39, 65–72. [Google Scholar] [CrossRef]
- Sullivan, N.J.; Sanchez, A.; Rollin, P.E.; Yang, Z.Y.; Nabel, G.J. Development of a Preventive Vaccine for Ebola Virus Infection in Primates. Nature 2000, 408, 605–609. [Google Scholar] [CrossRef] [PubMed]
- Warfield, K.L.; Olinger, G.; Deal, E.M.; Swenson, D.L.; Bailey, M.; Negley, D.L.; Hart, M.K.; Bavari, S. Induction of Humoral and CD8 + T Cell Responses Are Required for Protection against Lethal Ebola Virus Infection. J. Immunol. 2005, 175, 1184–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, J.A.; Hart, M.K. Protection from Ebola Virus Mediated by Cytotoxic T Lymphocytes Specific for the Viral Nucleoprotein. J. Virol. 2001, 75, 2660–2664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thom, R.; Tipton, T.; Strecker, T.; Hall, Y.; Akoi Bore, J.; Maes, P.; Raymond Koundouno, F.; Fehling, S.K.; Krähling, V.; Steeds, K.; et al. Longitudinal Antibody and T Cell Responses in Ebola Virus Disease Survivors and Contacts: An Observational Cohort Study. Lancet Infect. Dis. 2020, 3099, 1–10. [Google Scholar] [CrossRef]
- Baize, S.; Leroy, E.M.; Georges-Courbot, M.C.; Capron, M.; Lansoud-Soukate, J.; Debré, P.; Fisher-Hoch, S.P.; McCormick, J.B.; Georges, A.J. Defective Humoral Responses and Extensive Intravascular Apoptosis Are Associated with Fatal Outcome in Ebola Virus-Infected Patients. Nat. Med. 1999, 5, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Ksiazek, T.G.; Rollin, P.E.; Williams, A.J.; Bressler, D.S.; Martin, M.L.; Swanepoel, R.; Burt, F.J.; Leman, P.A.; Khan, A.S.; Rowe, A.K.; et al. Clinical Virology of Ebola Hemorrhagic Fever (EHF): Virus, Virus Antigen, and IgG and IgM Antibody Findings among EHF Patients in Kikwit, Democratic Republic of the Congo, 1995. J. Infect. Dis. 1999, 179, S117–S187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A New Coronavirus Associated with Human Respiratory Disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Krammer, F. SARS-CoV-2 Vaccines in Development. Nature 2020, 586, 516–527. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Lu, J.; Wu, D.; Zhang, L.; Zhao, H.; Rao, B.; Yang, Z. False Negative Rate of COVID-19 Is Eliminated by Using Nasal Swab Test. Travel Med. Infect. Dis. 2020, 37, 10–12. [Google Scholar] [CrossRef]
- Keech, C.; Albert, G.; Cho, I.; Robertson, A.; Reed, P.; Neal, S.; Plested, J.S.; Zhu, M.; Cloney-Clark, S.; Zhou, H.; et al. Phase 1–2 Trial of a SARS-CoV-2 Recombinant Spike Protein Nanoparticle Vaccine. N. Engl. J. Med. 2020, 383, 2320–2332. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, F.; Shen, C.; Peng, W.; Li, D.; Zhao, C.; Li, Z.; Li, S.; Bi, Y.; Yang, Y.; et al. A Noncompeting Pair of Human Neutralizing Antibodies Block COVID-19 Virus Binding to Its Receptor ACE2. Science 2020, 368, 1274–1278. [Google Scholar] [CrossRef]
- Addetia, A.; Crawford, K.H.D.; Dingens, A.; Zhu, H.; Roychoudhury, P.; Huang, M.-L.; Jerome, K.R.; Bloom, J.D.; Greninger, A.L. Neutralizing Antibodies Correlate with Protection from SARS-CoV-2 in Humans during a Fishery Vessel Outbreak with a High Attack Rate. J. Clin. Microbiol. 2020, 58, e02107–e02120. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muñoz-Basagoiti, J.; Perez-Zsolt, D.; Carrillo, J.; Blanco, J.; Clotet, B.; Izquierdo-Useros, N. SARS-CoV-2 Cellular Infection and Therapeutic Opportunities: Lessons Learned from Ebola Virus. Membranes 2021, 11, 64. https://0-doi-org.brum.beds.ac.uk/10.3390/membranes11010064
Muñoz-Basagoiti J, Perez-Zsolt D, Carrillo J, Blanco J, Clotet B, Izquierdo-Useros N. SARS-CoV-2 Cellular Infection and Therapeutic Opportunities: Lessons Learned from Ebola Virus. Membranes. 2021; 11(1):64. https://0-doi-org.brum.beds.ac.uk/10.3390/membranes11010064
Chicago/Turabian StyleMuñoz-Basagoiti, Jordana, Daniel Perez-Zsolt, Jorge Carrillo, Julià Blanco, Bonaventura Clotet, and Nuria Izquierdo-Useros. 2021. "SARS-CoV-2 Cellular Infection and Therapeutic Opportunities: Lessons Learned from Ebola Virus" Membranes 11, no. 1: 64. https://0-doi-org.brum.beds.ac.uk/10.3390/membranes11010064