
HspB4/αA-Crystallin Modulates Neuroinflammation in the Retina via the Stress-Specific Inflammatory Pathways


Abstract
:1. Introduction
2. Methods and Materials
2.1. Cell Culture
2.2. Transfection and Experimental Protocol
2.3. Subcellular Fractionation
2.4. Immunoblot
2.5. qPCR
2.6. Statistics
2.7. Study Approval
3. Results
3.1. HspB4/αA-Crystallin Overexpression Dampens the Metabolic-Stress-Induced Pro-inflammatory Transition of Müller Glial Cells (MGCs)
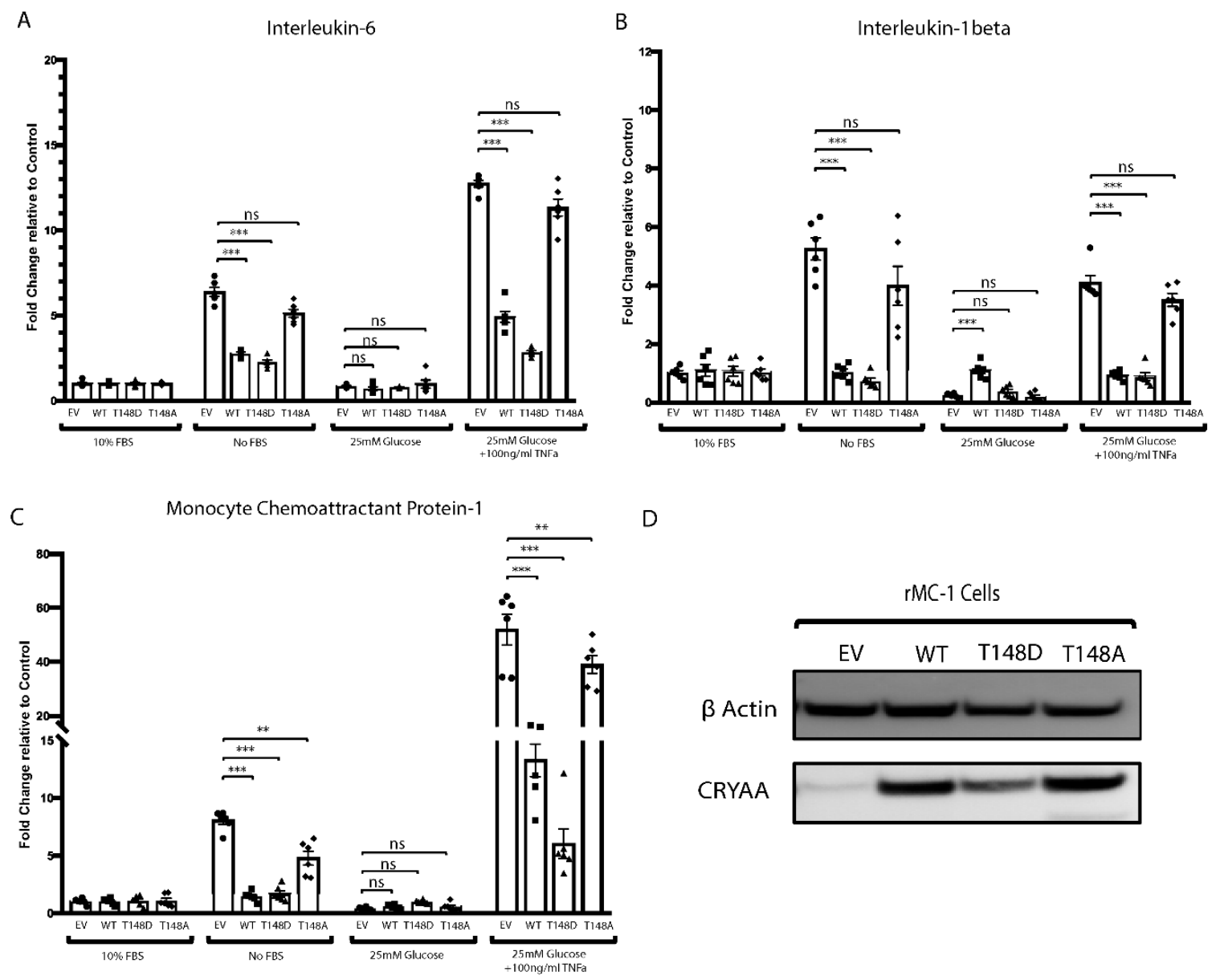
3.2. HspB4/αA-Crystallin Effect on the Metabolic-Stress-Induced Activation of Müller Glial Cells Is T148 Phosphorylation Dependent
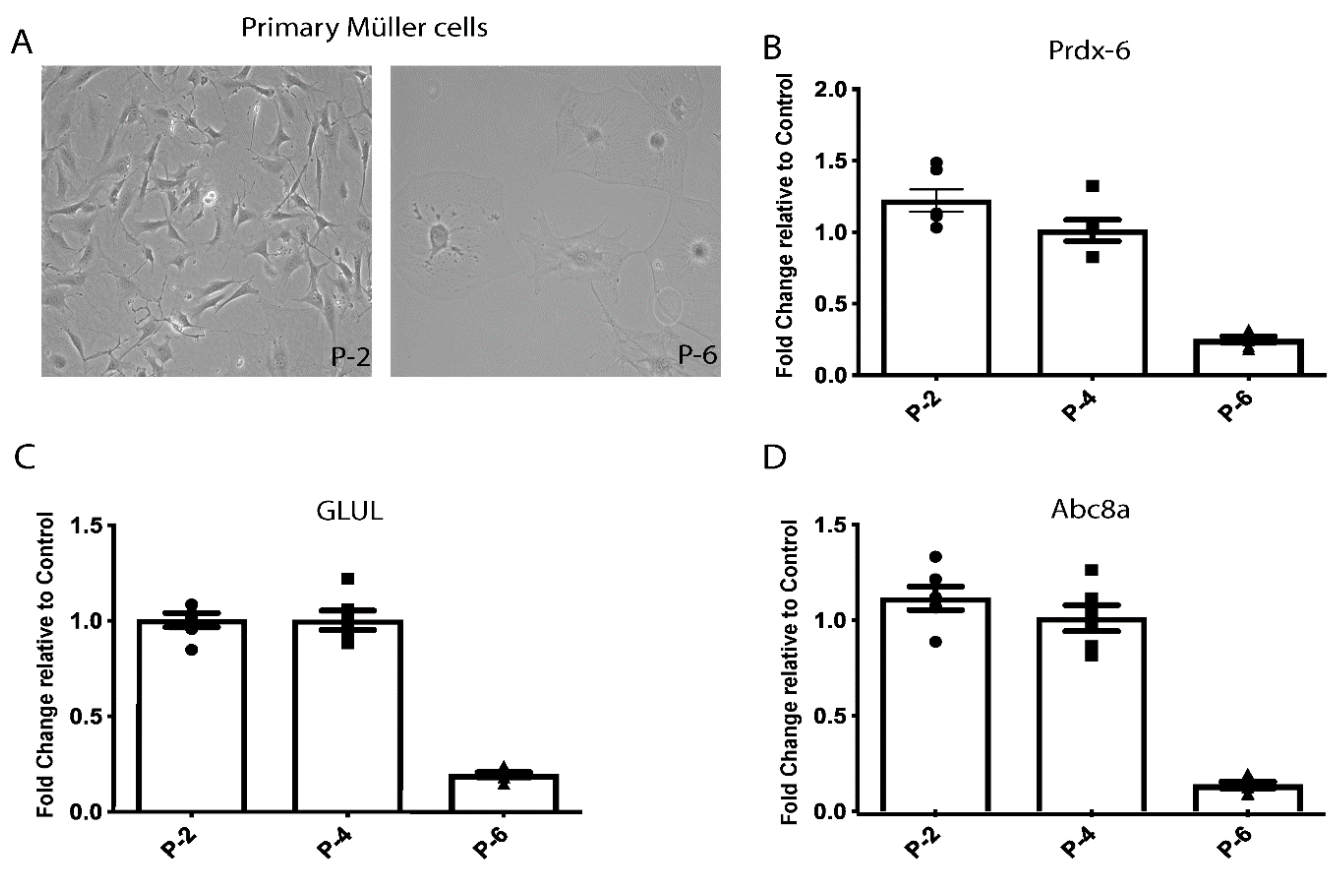
3.3. Primary Müller Cells Isolated from HspB4/αA-Crystallin Knockout Mice
3.4. The Metabolic-Stress-Induced Pro-Inflammatory Response of Primary MGCs (HspB4−/−) Is Dampened by HspB4/αA-Crystallin Expression
3.5. Phosphorylation of HspB4/αA-Crystallin on Residue 148 Controls the Neuroinflammatory Cascade in Metabolically Stressed Primary MGCs (HspB4−/−)
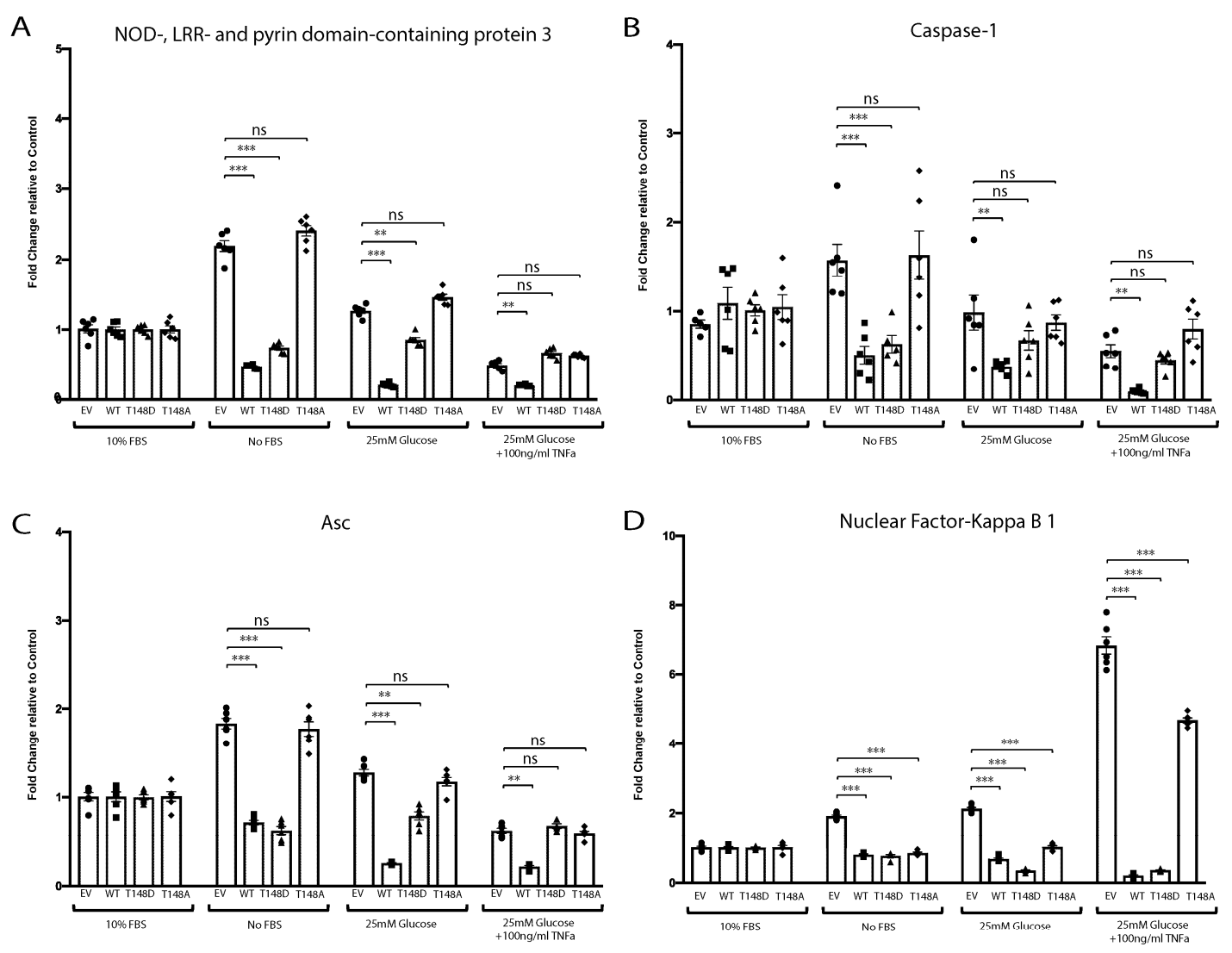
3.6. HspB4/αA-Crystallin Regulates MGCs Inflammatory Response through Stress-Specific Inflammatory Pathways
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Reichenbach, A.; Bringmann, A. Role of Purines in Muller Glia. J. Ocul. Pharmacol. Ther. 2016, 32, 518–533. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Shen, S.Q.; Jui, J.; Rupp, A.C.; Byrne, L.C.; Hattar, S.; Flannery, J.G.; Corbo, J.C.; Kefalov, V.J. CRALBP supports the mammalian retinal visual cycle and cone vision. J. Clin. Investig. 2015, 125, 727–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurley, J.B.; Lindsay, K.J.; Du, J. Glucose, lactate, and shuttling of metabolites in vertebrate retinas. J. Neurosci. Res. 2015, 93, 1079–1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindsay, K.J.; Du, J.; Sloat, S.R.; Contreras, L.; Linton, J.D.; Turner, S.J.; Sadilek, M.; Satrustegui, J.; Hurley, J.B. Pyruvate kinase and aspartate-glutamate carrier distributions reveal key metabolic links between neurons and glia in retina. Proc. Natl. Acad. Sci. USA 2014, 111, 15579–15584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subirada, P.V.; Paz, M.C.; Ridano, M.E.; Lorenc, V.E.; Vaglienti, M.V.; Barcelona, P.F.; Luna, J.D.; Sanchez, M.C. A journey into the retina: Muller glia commanding survival and death. Eur. J. Neurosci. 2018, 47, 1429–1443. [Google Scholar] [CrossRef] [PubMed]
- Yego, E.C.; Vincent, J.A.; Sarthy, V.; Busik, J.V.; Mohr, S. Differential regulation of high glucose-induced glyceraldehyde-3-phosphate dehydrogenase nuclear accumulation in Muller cells by IL-1beta and IL-6. Investig. Ophthalmol. Vis. Sci. 2009, 50, 1920–1928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, X.; Zhang, J.; Shen, J.; Hu, L.M.; Wu, Y.; Mou, L.; Xu, G.; Li, W.; Xu, G.T. EPO attenuates inflammatory cytokines by Muller cells in diabetic retinopathy. Front. Biosci. 2011, 3, 201–211. [Google Scholar]
- Abu el Asrar, A.M.; Maimone, D.; Morse, P.H.; Gregory, S.; Reder, A.T. Cytokines in the vitreous of patients with proliferative diabetic retinopathy. Am. J. Ophthalmol. 1992, 114, 731–736. [Google Scholar] [CrossRef]
- Blakytny, R.; Carver, J.A.; Harding, J.J.; Kilby, G.W.; Sheil, M.M. A spectroscopic study of glycated bovine alpha-crystallin: Investigation of flexibility of the C-terminal extension, chaperone activity and evidence for diglycation. Biochim. Biophys. Acta 1997, 1343, 299–315. [Google Scholar] [CrossRef]
- Deliyanti, D.; Alrashdi, S.F.; Tan, S.M.; Meyer, C.; Ward, K.W.; de Haan, J.B.; Wilkinson-Berka, J.L. Nrf2 Activation Is a Potential Therapeutic Approach to Attenuate Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2018, 59, 815–825. [Google Scholar] [CrossRef] [Green Version]
- Demircan, N.; Safran, B.G.; Soylu, M.; Ozcan, A.A.; Sizmaz, S. Determination of vitreous interleukin-1 (IL-1) and tumour necrosis factor (TNF) levels in proliferative diabetic retinopathy. Eye 2006, 20, 1366–1369. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, C.; Segura, R.M.; Fonollosa, A.; Carrasco, E.; Francisco, G.; Simo, R. Interleukin-8, monocyte chemoattractant protein-1 and IL-10 in the vitreous fluid of patients with proliferative diabetic retinopathy. Diabet. Med. 2005, 22, 719–722. [Google Scholar] [CrossRef]
- van Noort, J.M.; van Sechel, A.C.; Bajramovic, J.J.; el Ouagmiri, M.; Polman, C.H.; Lassmann, H.; Ravid, R. The small heat-shock protein alpha B-crystallin as candidate autoantigen in multiple sclerosis. Nature 1995, 375, 798–801. [Google Scholar] [CrossRef]
- Renkawek, K.; Stege, G.J.; Bosman, G.J. Dementia, gliosis and expression of the small heat shock proteins hsp27 and alpha B-crystallin in Parkinson’s disease. Neuroreport 1999, 10, 2273–2276. [Google Scholar] [CrossRef]
- Fort, P.E.; Freeman, W.M.; Losiewicz, M.K.; Singh, R.S.; Gardner, T.W. The retinal proteome in experimental diabetic retinopathy: Up-regulation of crystallins and reversal by systemic and periocular insulin. Mol. Cell. Proteom. 2009, 8, 767–779. [Google Scholar] [CrossRef] [Green Version]
- Ruebsam, A.; Dulle, J.E.; Myers, A.M.; Sakrikar, D.; Green, K.M.; Khan, N.W.; Schey, K.; Fort, P.E. A specific phosphorylation regulates the protective role of alphaA-crystallin in diabetes. JCI Insight 2018, 3, e97919. [Google Scholar] [CrossRef] [Green Version]
- Nagaraj, R.H.; Nahomi, R.B.; Mueller, N.H.; Raghavan, C.T.; Ammar, D.A.; Petrash, J.M. Therapeutic potential of alpha-crystallin. Biochim. Biophys. Acta 2016, 1860 Pt B, 252–257. [Google Scholar] [CrossRef] [Green Version]
- Nahomi, R.B.; Wang, B.; Raghavan, C.T.; Voss, O.; Doseff, A.I.; Santhoshkumar, P.; Nagaraj, R.H. Chaperone peptides of alpha-crystallin inhibit epithelial cell apoptosis, protein insolubilization, and opacification in experimental cataracts. J. Biol. Chem. 2013, 288, 13022–13035. [Google Scholar] [CrossRef] [Green Version]
- Masilamoni, J.G.; Jesudason, E.P.; Bharathi, S.N.; Jayakumar, R. The protective effect of alpha-crystallin against acute inflammation in mice. Biochim. Biophys. Acta 2005, 1740, 411–420. [Google Scholar] [CrossRef] [Green Version]
- Masilamoni, J.G.; Vignesh, S.; Kirubagaran, R.; Jesudason, E.P.; Jayakumar, R. The neuroprotective efficacy of alpha-crystallin against acute inflammation in mice. Brain Res. Bull. 2005, 67, 235–241. [Google Scholar] [CrossRef]
- Simirskii, V.N.; Panova, I.G.; Sologub, A.A.; Aleinikova, K.S. Localization of crystallins in Muellerian cells in the grass frog retina. Ontogenez 2003, 34, 365–370. [Google Scholar]
- Zayas-Santiago, A.; Rios, D.S.; Zueva, L.V.; Inyushin, M.Y. Localization of alphaA-Crystallin in Rat Retinal Muller Glial Cells and Photoreceptors. Microsc. Microanal. 2018, 24, 545–552. [Google Scholar] [CrossRef] [Green Version]
- Cherian, M.; Abraham, E.C. Decreased molecular chaperone property of alpha-crystallins due to posttranslational modifications. Biochem. Biophys. Res. Commun. 1995, 208, 675–679. [Google Scholar] [CrossRef]
- Cherian, M.; Smith, J.B.; Jiang, X.Y.; Abraham, E.C. Influence of protein-glutathione mixed disulfide on the chaperone-like function of alpha-crystallin. J. Biol. Chem. 1997, 272, 29099–29103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciano, M.; Allocca, S.; Ciardulli, M.C.; Della Volpe, L.; Bonatti, S.; D’Agostino, M. Differential phosphorylation-based regulation of alphaB-crystallin chaperone activity for multipass transmembrane proteins. Biochem. Biophys. Res. Commun. 2016, 479, 325–330. [Google Scholar] [CrossRef] [Green Version]
- Kamei, A.; Iwase, H.; Masuda, K. Cleavage of amino acid residue(s) from the N-terminal region of alpha A- and alpha B-crystallins in human crystalline lens during aging. Biochem. Biophys. Res. Commun. 1997, 231, 373–378. [Google Scholar] [CrossRef]
- Morrison, L.E.; Hoover, H.E.; Thuerauf, D.J.; Glembotski, C.C. Mimicking phosphorylation of alphaB-crystallin on serine-59 is necessary and sufficient to provide maximal protection of cardiac myocytes from apoptosis. Circ. Res. 2003, 92, 203–211. [Google Scholar] [CrossRef] [Green Version]
- Maddala, R.; Rao, V.P. alpha-Crystallin localizes to the leading edges of migrating lens epithelial cells. Exp. Cell Res. 2005, 306, 203–215. [Google Scholar] [CrossRef]
- Heise, E.A.; Marozas, L.M.; Grafton, S.A.; Green, K.M.; Kirwin, S.J.; Fort, P.E. Strain-independent increases of crystallin proteins in the retina of type 1 diabetic rats. PLoS ONE 2013, 8, e82520. [Google Scholar] [CrossRef]
- Kim, Y.H.; Choi, M.Y.; Kim, Y.S.; Han, J.M.; Lee, J.H.; Park, C.H.; Kang, S.S.; Choi, W.S.; Cho, G.J. Protein kinase C delta regulates anti-apoptotic alphaB-crystallin in the retina of type 2 diabetes. Neurobiol. Dis. 2007, 28, 293–303. [Google Scholar] [CrossRef]
- Rao, N.A.; Saraswathy, S.; Pararajasegaram, G.; Bhat, S.P. Small heat shock protein alphaA-crystallin prevents photoreceptor degeneration in experimental autoimmune uveitis. PLoS ONE 2012, 7, e33582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarthy, V.P.; Brodjian, S.J.; Dutt, K.; Kennedy, B.N.; French, R.P.; Crabb, J.W. Establishment and characterization of a retinal Muller cell line. Investig. Ophthalmol. Vis. Sci. 1998, 39, 212–216. [Google Scholar]
- Hicks, D.; Courtois, Y. The growth and behaviour of rat retinal Muller cells in vitro. 1. An improved method for isolation and culture. Exp. Eye Res. 1990, 51, 119–129. [Google Scholar] [CrossRef]
- Brady, J.P.; Garland, D.; Duglas-Tabor, Y.; Robison, W.G., Jr.; Groome, A.; Wawrousek, E.F. Targeted disruption of the mouse alpha A-crystallin gene induces cataract and cytoplasmic inclusion bodies containing the small heat shock protein alpha B-crystallin. Proc. Natl. Acad. Sci. USA 1997, 94, 884–889. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, K.; Bose, P.; Leong-Quong, R.Y.; Fujita, D.J.; Riabowol, K. REAP: A two minute cell fractionation method. BMC Res. Notes 2010, 3, 294. [Google Scholar] [CrossRef] [Green Version]
- Sambrook, J.; Russell, D.W. Purification of nucleic acids by extraction with phenol:chloroform. CSH Protoc. 2006, 2006, pdb.prot4455. [Google Scholar] [CrossRef]
- Yamagata, K.; Miyashita, A.; Matsufuji, H.; Chino, M. Dietary flavonoid apigenin inhibits high glucose and tumor necrosis factor alpha-induced adhesion molecule expression in human endothelial cells. J. Nutr. Biochem. 2010, 21, 116–124. [Google Scholar] [CrossRef]
- Sun, M.; Yang, J.; Wang, J.; Hao, T.; Jiang, D.; Bao, G.; Liu, G. TNF-alpha is upregulated in T2DM patients with fracture and promotes the apoptosis of osteoblast cells in vitro in the presence of high glucose. Cytokine 2016, 80, 35–42. [Google Scholar] [CrossRef]
- Christiansen, T.; Richelsen, B.; Bruun, J.M. Monocyte chemoattractant protein-1 is produced in isolated adipocytes, associated with adiposity and reduced after weight loss in morbid obese subjects. Int. J. Obes. 2005, 29, 146–150. [Google Scholar] [CrossRef] [Green Version]
- Ijima, R.; Kaneko, H.; Ye, F.; Nagasaka, Y.; Takayama, K.; Kataoka, K.; Kachi, S.; Iwase, T.; Terasaki, H. Interleukin-18 induces retinal pigment epithelium degeneration in mice. Investig. Ophthalmol. Vis. Sci. 2014, 55, 6673–6678. [Google Scholar] [CrossRef] [Green Version]
- Zhao, M.; Bai, Y.; Xie, W.; Shi, X.; Li, F.; Yang, F.; Sun, Y.; Huang, L.; Li, X. Interleukin-1beta Level Is Increased in Vitreous of Patients with Neovascular Age-Related Macular Degeneration (nAMD) and Polypoidal Choroidal Vasculopathy (PCV). PLoS ONE 2015, 10, e0125150. [Google Scholar]
- Coughlin, B.A.; Feenstra, D.J.; Mohr, S. Muller cells and diabetic retinopathy. Vision Res. 2017, 139, 93–100. [Google Scholar] [CrossRef]
- Mohammad, G.; AlSharif, H.M.; Siddiquei, M.M.; Ahmad, A.; Alam, K.; Abu El-Asrar, A.M. Rho-Associated Protein Kinase-1 Mediates the Regulation of Inflammatory Markers in Diabetic Retina and in Retinal Muller Cells. Ann. Clin. Lab. Sci. 2018, 48, 137–145. [Google Scholar]
- Sigurdardottir, S.; Zapadka, T.E.; Lindstrom, S.I.; Liu, H.; Taylor, B.E.; Lee, C.A.; Kern, T.S.; Taylor, P.R. Diabetes-mediated IL-17A enhances retinal inflammation, oxidative stress, and vascular permeability. Cell. Immunol. 2019, 341, 103921. [Google Scholar] [CrossRef]
- Zhou, T.; Che, D.; Lan, Y.; Fang, Z.; Xie, J.; Gong, H.; Li, C.; Feng, J.; Hong, H.; Qi, W.; et al. Mesenchymal marker expression is elevated in Muller cells exposed to high glucose and in animal models of diabetic retinopathy. Oncotarget 2017, 8, 4582–4594. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Yang, H.; Chen, X. Protective effects of sulforaphane on diabetic retinopathy: Activation of the Nrf2 pathway and inhibition of NLRP3 inflammasome formation. Exp. Anim. 2019, 68, 221–231. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Chen, C.; McLaughlin, T.; Wang, Y.; Le, Y.Z.; Wang, J.J.; Zhang, S.X. Loss of X-box binding protein 1 in Muller cells augments retinal inflammation in a mouse model of diabetes. Diabetologia 2019, 62, 531–543. [Google Scholar] [CrossRef] [Green Version]
- Hoover, H.E.; Thuerauf, D.J.; Martindale, J.J.; Glembotski, C.C. alpha B-crystallin gene induction and phosphorylation by MKK6-activated p38. A potential role for alpha B-crystallin as a target of the p38 branch of the cardiac stress response. J. Biol. Chem. 2000, 275, 23825–23833. [Google Scholar] [CrossRef] [Green Version]
- Piao, C.S.; Kim, S.W.; Kim, J.B.; Lee, J.K. Co-induction of alphaB-crystallin and MAPKAPK-2 in astrocytes in the penumbra after transient focal cerebral ischemia. Exp. Brain Res. 2005, 163, 421–429. [Google Scholar] [CrossRef]
- Li, R.; Zhu, Z.; Reiser, G. Specific phosphorylation of alphaA-crystallin is required for the alphaA-crystallin-induced protection of astrocytes against staurosporine and C2-ceramide toxicity. Neurochem. Int. 2012, 60, 652–658. [Google Scholar] [CrossRef]
- Webster, K.A. Serine phosphorylation and suppression of apoptosis by the small heat shock protein alphaB-crystallin. Circ. Res. 2003, 92, 130–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potilinski, M.C.; Lorenc, V.; Perisset, S.; Gallo, J.E. Mechanisms behind Retinal Ganglion Cell Loss in Diabetes and Therapeutic Approach. Int. J. Mol. Sci. 2020, 21, 2351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiesa, R.; Gawinowicz-Kolks, M.A.; Spector, A. The phosphorylation of the primary gene products of alpha-crystallin. J. Biol. Chem. 1987, 262, 1438–1441. [Google Scholar] [CrossRef]
- Kannan, R.; Sreekumar, P.G.; Hinton, D.R. Novel roles for alpha-crystallins in retinal function and disease. Prog. Retin. Eye Res. 2012, 31, 576–604. [Google Scholar] [CrossRef] [Green Version]
- Voorter, C.E.; Mulders, J.W.; Bloemendal, H.; de Jong, W.W. Some aspects of the phosphorylation of alpha-crystallin A. Eur. J. Biochem. 1986, 160, 203–210. [Google Scholar] [CrossRef]
- Harada, C.; Okumura, A.; Namekata, K.; Nakamura, K.; Mitamura, Y.; Ohguro, H.; Harada, T. Role of monocyte chemotactic protein-1 and nuclear factor kappa B in the pathogenesis of proliferative diabetic retinopathy. Diabetes Res. Clin. Pract. 2006, 74, 249–256. [Google Scholar] [CrossRef]
- Meleth, A.D.; Agron, E.; Chan, C.C.; Reed, G.F.; Arora, K.; Byrnes, G.; Csaky, K.G.; Ferris, F.L., 3rd; Chew, E.Y. Serum inflammatory markers in diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2005, 46, 4295–4301. [Google Scholar] [CrossRef]
- Kumar, A.; Pandey, R.K.; Miller, L.J.; Singh, P.K.; Kanwar, M. Muller glia in retinal innate immunity: A perspective on their roles in endophthalmitis. Crit. Rev. Immunol. 2013, 33, 119–135. [Google Scholar] [CrossRef]
- Kumar, A.; Shamsuddin, N. Retinal Muller glia initiate innate response to infectious stimuli via toll-like receptor signaling. PLoS ONE 2012, 7, e29830. [Google Scholar]
- Xiong, W.; Wu, D.M.; Xue, Y.; Wang, S.K.; Chung, M.J.; Ji, X.; Rana, P.; Zhao, S.R.; Mai, S.; Cepko, C.L. AAV cis-regulatory sequences are correlated with ocular toxicity. Proc. Natl. Acad. Sci. USA 2019, 116, 5785–5794. [Google Scholar] [CrossRef] [Green Version]
- Eastlake, K.; Banerjee, P.J.; Angbohang, A.; Charteris, D.G.; Khaw, P.T.; Limb, G.A. Muller glia as an important source of cytokines and inflammatory factors present in the gliotic retina during proliferative vitreoretinopathy. Glia 2016, 64, 495–506. [Google Scholar] [CrossRef] [Green Version]
- Funatsu, H.; Noma, H.; Mimura, T.; Eguchi, S.; Hori, S. Association of vitreous inflammatory factors with diabetic macular edema. Ophthalmology 2009, 116, 73–79. [Google Scholar] [CrossRef]
- Vujosevic, S.; Simo, R. Local and Systemic Inflammatory Biomarkers of Diabetic Retinopathy: An Integrative Approach. Investig. Ophthalmol. Vis. Sci. 2017, 58, BIO68–BIO75. [Google Scholar] [CrossRef]
- Chaurasia, S.S.; Lim, R.R.; Parikh, B.H.; Wey, Y.S.; Tun, B.B.; Wong, T.Y.; Luu, C.D.; Agrawal, R.; Ghosh, A.; Mortellaro, A.; et al. The NLRP3 Inflammasome May Contribute to Pathologic Neovascularization in the Advanced Stages of Diabetic Retinopathy. Sci. Rep. 2018, 8, 2847. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.; Santani, D. Role of NF-kappa B in the pathogenesis of diabetes and its associated complications. Pharmacol. Rep. 2009, 61, 595–603. [Google Scholar] [CrossRef]
- Suryavanshi, S.V.; Kulkarni, Y.A. NF-kappabeta: A Potential Target in the Management of Vascular Complications of Diabetes. Front. Pharmacol. 2017, 8, 798. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Sequence (5′-3′) | |
|---|---|---|
| β-actin | Forward | GGCTGTATTCCCCTCCATCG |
| Reverse | CCAGTTGGTAACAATGCCATGT | |
| IL-6 | Forward | TAGTCCTTCCTACCCCAATTTCC |
| Reverse | TTGGTCCTTAGCCACTCCTTC | |
| IL-1β | Forward | TTCAGGCAGGCAGTATCACTC |
| Reverse | GAAGGTCCACGGGAAAGACAC | |
| IL-18 | Forward | GACTCTTGCGTCAACTTCAAGG |
| Reverse | CAGGCTGTCTTTTGTCAACGA | |
| CCL2 | Forward | TTAAAAACCTGGATCGGAACCAA |
| Reverse | GCATTAGCTTCAGATTTACGGGT | |
| VEGF | Forward | GGCCTCCGAAACCATGAACTT |
| Reverse | TGGGACCACTTGGCATGGTG | |
| ICAM1 | Forward | GAGCCAATTTCTCATGCCGC |
| Reverse | GCTGGAAGATCGAAAGTCCG | |
| TNFa | Forward | CAGGCGGTGCCTATGTCTC |
| Reverse | CGATCACCCCGAAGTTCAGTAG | |
| Nf-kB1 | Forward | TCCACTGTCTGCCTCTCTCGTC |
| Reverse | GCCTTCAATAGGTCCTTCCTGC | |
| ASC | Forward | CAGCAACACTCCGGTCAG |
| Reverse | AGCTGGCTTTTCGTATATTGTG | |
| Casp1 | Forward | GGAAGCAATTTATCAACTCAGTG |
| Reverse | GCCTTGTCCATAGCAGTAATG | |
| Nlrp3 | Forward | ATTACCCGCCCGAGAAAGG |
| Reverse | TCGCAGCAAAGATCCACACAG | |
| Prdx6 | Forward | CGCCAGAGTTTGCCAAGAG |
| Reverse | TCCGTGGGTGTTTCACCATTG | |
| GLUL | Forward | TGAACAAAGGCATCAAGCAAATG |
| Reverse | CAGTCCAGGGTACGGGTCTT | |
| Abc8a | Forward | CGTGGGCCTTATTGTGCAAGA |
| Reverse | CAGGTCCACATCAGGCAGTG | |
| Rpe65 | Forward | ACCACTAACAGCTCATGTCACA |
| Reverse | ACAGGTGATAGAAAGGCTCAGAT | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nath, M.; Shan, Y.; Myers, A.M.; Fort, P.E. HspB4/αA-Crystallin Modulates Neuroinflammation in the Retina via the Stress-Specific Inflammatory Pathways. J. Clin. Med. 2021, 10, 2384. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm10112384
Nath M, Shan Y, Myers AM, Fort PE. HspB4/αA-Crystallin Modulates Neuroinflammation in the Retina via the Stress-Specific Inflammatory Pathways. Journal of Clinical Medicine. 2021; 10(11):2384. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm10112384
Chicago/Turabian StyleNath, Madhu, Yang Shan, Angela M. Myers, and Patrice Elie Fort. 2021. "HspB4/αA-Crystallin Modulates Neuroinflammation in the Retina via the Stress-Specific Inflammatory Pathways" Journal of Clinical Medicine 10, no. 11: 2384. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm10112384