
The Importance of Arterial Stiffness Assessment in Patients with Familial Hypercholesterolemia

 and
and
Abstract
:1. Introduction
2. Genotype and Phenotype of Familial Hypercholesterolemia
2.1. Heterozygous FH
2.2. Homozygous FH
3. Treatments of Familial Hypercholesterolemia
3.1. Pharmacological Treatment
3.2. Selective LDL Apheresis Treatment
4. Significance of Cardiovascular Risk Assessment
5. Arterial Stiffness
6. Relationship between Cholesterol and Arterial Stiffness
7. Assessing Arterial Stiffness in Familial Hypercholesterolemia
8. Effect of Traditional Oral Lipid-Lowering Treatment on Arterial Stiffness
9. Changes in Arterial Stiffness and PCSK9-Inhibitor Monoclonal Antibody Treatment
10. The Impact of LDL Apheresis Treatment on Vascular Parameters
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ABPM | ambulatory blood pressure monitoring |
| Aix | augmentation index |
| ApoB100 | apolipoprotein B100 |
| CAD | coronary artery disease |
| CRP | C-reactive protein |
| CVD | cardiovascular disease |
| DALI | direct adsorption of lipoproteins |
| FH | familial hypercholesterolemia |
| HDL-C | high-density lipoprotein cholesterol |
| LDL | low-density lipoprotein |
| LDL-C | low-density lipoprotein cholesterol |
| LDLR | LDL receptor |
| LDLRAP1 | LDL receptor adaptor protein 1 |
| Lp (a) | lipoprotein (a) |
| MMP-9 | matrix metalloprotease-9 |
| oxLDL | oxidized LDL |
| PAD | peripheral artery disease |
| PCSK9 | proprotein convertase subtilisin/kexin type 9 |
| siRNA | small interfering ribonucleic acid |
| TG | triglycerides |
| PWV | pulse wave velocity |
| STAP1 | signal transducing adaptor family member 1 |
| VLDL | very low-density lipoprotein |
References
- Goldstein, J.L.; Hazzard, W.R.; Schrott, H.G.; Bierman, E.L.; Motulsky, A.G. Hyperlipidemia in coronary heart disease. I. Lipid levels in 500 survivors of myocardial infarction. J. Clin. Investig. 1973, 52, 1533–1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ang, T.W.; ten Have, H.; Solbakk, J.H.; Nys, H. UNESCO Global Ethics Observatory: Database on ethics related legislation and guidelines. J. Med. Ethics 2008, 34, 738–741. [Google Scholar] [CrossRef] [PubMed]
- Catapano, A.L.; Wiklund, O.; Society, E.A. Think Again About Cholesterol Survey. Atheroscler. Suppl. 2015, 20, 1–5. [Google Scholar] [CrossRef]
- Paththinige, C.S.; Sirisena, N.D.; Dissanayake, V. Genetic determinants of inherited susceptibility to hypercholesterolemia-a comprehensive literature review. Lipids Health Dis. 2017, 16, 103. [Google Scholar] [CrossRef]
- Di Taranto, M.D.; Giacobbe, C.; Fortunato, G. Familial hypercholesterolemia: A complex genetic disease with variable phenotypes. Eur. J. Med. Genet. 2019, 63, 103831. [Google Scholar] [CrossRef] [PubMed]
- Fouchier, S.W.; Dallinga-Thie, G.M.; Meijers, J.C.; Zelcer, N.; Kastelein, J.J.; Defesche, J.C.; Hovingh, G.K. Mutations in STAP1 are associated with autosomal dominant hypercholesterolemia. Circ. Res. 2014, 115, 552–555. [Google Scholar] [CrossRef] [Green Version]
- Loaiza, N.; Hartgers, M.L.; Reeskamp, L.F.; Balder, J.W.; Rimbert, A.; Bazioti, V.; Wolters, J.C.; Winkelmeijer, M.; Jansen, H.P.G.; Dallinga-Thie, G.M.; et al. Taking One Step Back in Familial Hypercholesterolemia: STAP1 Does Not Alter Plasma LDL (Low-Density Lipoprotein) Cholesterol in Mice and Humans. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 973–985. [Google Scholar] [CrossRef]
- Kanuri, B.; Fong, V.; Haller, A.; Hui, D.Y.; Patel, S.B. Mice lacking global Stap1 expression do not manifest hypercholesterolemia. BMC Med. Genet. 2020, 21, 234. [Google Scholar] [CrossRef]
- Hegele, R.A.; Knowles, J.W.; Horton, J.D. Delisting. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 847–849. [Google Scholar] [CrossRef]
- Tada, H.; Kawashiri, M.A.; Ohtani, R.; Noguchi, T.; Nakanishi, C.; Konno, T.; Hayashi, K.; Nohara, A.; Inazu, A.; Kobayashi, J.; et al. A novel type of familial hypercholesterolemia: Double heterozygous mutations in LDL receptor and LDL receptor adaptor protein 1 gene. Atherosclerosis 2011, 219, 663–666. [Google Scholar] [CrossRef] [Green Version]
- Olmastroni, E.; Gazzotti, M.; Arca, M.; Averna, M.; Pirillo, A.; Catapano, A.L.; Casula, M.; Bertolini, S.; Calandra, S.; Tarugi, P.; et al. Twelve Variants Polygenic Score for Low-Density Lipoprotein Cholesterol Distribution in a Large Cohort of Patients With Clinically Diagnosed Familial Hypercholesterolemia With or Without Causative Mutations. J. Am. Heart Assoc. 2022, 11, e023668. [Google Scholar] [CrossRef] [PubMed]
- Trinder, M.; Francis, G.A.; Brunham, L.R. Association of Monogenic vs Polygenic Hypercholesterolemia With Risk of Atherosclerotic Cardiovascular Disease. JAMA Cardiol. 2020, 5, 390–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowman, F.L.; Molster, C.M.; Lister, K.J.; Bauskis, A.T.; Garton-Smith, J.; Vickery, A.W.; Watts, G.F.; Martin, A.C. Identifying Perceptions and Preferences of the General Public Concerning Universal Screening of Children for Familial Hypercholesterolaemia. Public Health Genom. 2019, 22, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Paragh, G.; Harangi, M.; Karányi, Z.; Daróczy, B.; Németh, Á.; Fülöp, P. Identifying patients with familial hypercholesterolemia using data mining methods in the Northern Great Plain region of Hungary. Atherosclerosis 2018, 277, 262–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiegman, A. Lipid Screening, Action, and Follow-up in Children and Adolescents. Curr. Cardiol. Rep. 2018, 20, 80. [Google Scholar] [CrossRef] [PubMed]
- Al-Rasadi, K.; Al-Waili, K.; Al-Sabti, H.A.; Al-Hinai, A.; Al-Hashmi, K.; Al-Zakwani, I.; Banerjee, Y. Criteria for Diagnosis of Familial Hypercholesterolemia: A Comprehensive Analysis of the Different Guidelines, Appraising their Suitability in the Omani Arab Population. Oman Med. J. 2014, 29, 85–91. [Google Scholar] [CrossRef]
- Bajnok, L. Newer evidences and recommendations in lipidology. Orvosi Hetil. 2018, 159, 1303–1309. [Google Scholar] [CrossRef] [Green Version]
- Wright, R.S.; Ray, K.K.; Raal, F.J.; Kallend, D.G.; Jaros, M.; Koenig, W.; Leiter, L.A.; Landmesser, U.; Schwartz, G.G.; Friedman, A.; et al. Pooled Patient-Level Analysis of Inclisiran Trials in Patients With Familial Hypercholesterolemia or Atherosclerosis. J. Am. Coll. Cardiol. 2021, 77, 1182–1193. [Google Scholar] [CrossRef]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2019, 294, 80–82. [Google Scholar] [CrossRef] [Green Version]
- Sjouke, B.; Kusters, D.M.; Kindt, I.; Besseling, J.; Defesche, J.C.; Sijbrands, E.J.; Roeters van Lennep, J.E.; Stalenhoef, A.F.; Wiegman, A.; de Graaf, J.; et al. Homozygous autosomal dominant hypercholesterolaemia in the Netherlands: Prevalence, genotype-phenotype relationship, and clinical outcome. Eur. Heart J. 2015, 36, 560–565. [Google Scholar] [CrossRef] [Green Version]
- Cuchel, M.; Meagher, E.A.; du Toit Theron, H.; Blom, D.J.; Marais, A.D.; Hegele, R.A.; Averna, M.R.; Sirtori, C.R.; Shah, P.K.; Gaudet, D.; et al. Efficacy and safety of a microsomal triglyceride transfer protein inhibitor in patients with homozygous familial hypercholesterolaemia: A single-arm, open-label, phase 3 study. Lancet 2013, 381, 40–46. [Google Scholar] [CrossRef] [Green Version]
- Akdim, F.; Stroes, E.S.; Sijbrands, E.J.; Tribble, D.L.; Trip, M.D.; Jukema, J.W.; Flaim, J.D.; Su, J.; Yu, R.; Baker, B.F.; et al. Efficacy and safety of mipomersen, an antisense inhibitor of apolipoprotein B, in hypercholesterolemic subjects receiving stable statin therapy. J. Am. Coll. Cardiol. 2010, 55, 1611–1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nordestgaard, B.G.; Chapman, M.J.; Humphries, S.E.; Ginsberg, H.N.; Masana, L.; Descamps, O.S.; Wiklund, O.; Hegele, R.A.; Raal, F.J.; Defesche, J.C.; et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: Guidance for clinicians to prevent coronary heart disease: Consensus statement of the European Atherosclerosis Society. Eur. Heart J. 2013, 34, 3478–3490. [Google Scholar] [CrossRef] [Green Version]
- Németh, Á.; Daróczy, B.; Juhász, L.; Fülöp, P.; Harangi, M.; Paragh, G. Assessment of Associations Between Serum Lipoprotein (a) Levels and Atherosclerotic Vascular Diseases in Hungarian Patients With Familial Hypercholesterolemia Using Data Mining and Machine Learning. Front. Genet. 2022, 13, 849197. [Google Scholar] [CrossRef] [PubMed]
- Alonso, R.; Andres, E.; Mata, N.; Fuentes-Jiménez, F.; Badimón, L.; López-Miranda, J.; Padró, T.; Muñiz, O.; Díaz-Díaz, J.L.; Mauri, M.; et al. Lipoprotein(a) levels in familial hypercholesterolemia: An important predictor of cardiovascular disease independent of the type of LDL receptor mutation. J. Am. Coll. Cardiol. 2014, 63, 1982–1989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoffel, W.; Demant, T. Selective removal of apolipoprotein B-containing serum lipoproteins from blood plasma. Proc. Natl. Acad. Sci. USA 1981, 78, 611–615. [Google Scholar] [CrossRef] [Green Version]
- Varga, V.E.; Lőrincz, H.; Zsíros, N.; Fülöp, P.; Seres, I.; Paragh, G.; Balla, J.; Harangi, M. Impact of selective LDL apheresis on serum chemerin levels in patients with hypercholesterolemia. Lipids Health Dis. 2016, 15, 182. [Google Scholar] [CrossRef] [Green Version]
- Varga, V.E.; Lőrincz, H.; Szentpéteri, A.; Juhász, L.; Seres, I.; Paragh, G.; Balla, J.; Harangi, M. Changes in serum afamin and vitamin E levels after selective LDL apheresis. J. Clin. Apher. 2018, 33, 569–575. [Google Scholar] [CrossRef]
- Raal, F.; Scott, R.; Somaratne, R.; Bridges, I.; Li, G.; Wasserman, S.M.; Stein, E.A. Low-density lipoprotein cholesterol-lowering effects of AMG 145, a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 serine protease in patients with heterozygous familial hypercholesterolemia: The Reduction of LDL-C with PCSK9 Inhibition in Heterozygous Familial Hypercholesterolemia Disorder (RUTHERFORD) randomized trial. Circulation 2012, 126, 2408–2417. [Google Scholar] [CrossRef] [Green Version]
- Stefanutti, C.; Morozzi, C.; Petta, A. Lipid and low-density-lipoprotein apheresis. Effects on plasma inflammatory profile and on cytokine pattern in patients with severe dyslipidemia. Cytokine 2011, 56, 842–849. [Google Scholar] [CrossRef]
- Harangi, M.; Juhász, L.; Nádró, B.; Paragh, G. Az LDL-aferezis helye a dyslipidaemia kezelésében a 2017-ben érvényes magyar és európai irányelvek alapján. Metabolizmus 2017, 15, 79–84. [Google Scholar]
- Paquette, M.; Baass, A. Predicting cardiovascular disease in familial hypercholesterolemia. Curr. Opin. Lipidol. 2018, 29, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Mata, P.; Alonso, R.; Pérez de Isla, L. Atherosclerotic cardiovascular disease risk assessment in familial hypercholesterolemia: Does one size fit all? Curr. Opin. Lipidol. 2018, 29, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Paquette, M.; Dufour, R.; Baass, A. The Montreal-FH-SCORE: A new score to predict cardiovascular events in familial hypercholesterolemia. J. Clin. Lipidol. 2017, 11, 80–86. [Google Scholar] [CrossRef]
- Riggio, S.; Mandraffino, G.; Sardo, M.A.; Iudicello, R.; Camarda, N.; Imbalzano, E.; Alibrandi, A.; Saitta, C.; Carerj, S.; Arrigo, T.; et al. Pulse wave velocity and augmentation index, but not intima-media thickness, are early indicators of vascular damage in hypercholesterolemic children. Eur. J. Clin. Investig. 2010, 40, 250–257. [Google Scholar] [CrossRef] [PubMed]
- Kavey, R.E.; Allada, V.; Daniels, S.R.; Hayman, L.L.; McCrindle, B.W.; Newburger, J.W.; Parekh, R.S.; Steinberger, J. Cardiovascular risk reduction in high-risk pediatric patients: A scientific statement from the American Heart Association Expert Panel on Population and Prevention Science; the Councils on Cardiovascular Disease in the Young, Epidemiology and Prevention, Nutrition, Physical Activity and Metabolism, High Blood Pressure Research, Cardiovascular Nursing, and the Kidney in Heart Disease; and the Interdisciplinary Working Group on Quality of Care and Outcomes Research: Endorsed by the American Academy of Pediatrics. Circulation 2006, 114, 2710–2738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Townsend, R.R. Arterial Stiffness: Recommendations and Standardization. Pulse 2017, 4, 3–7. [Google Scholar] [CrossRef] [Green Version]
- Nemcsik, J.; Cseprekál, O.; Tislér, A. Measurement of Arterial Stiffness: A Novel Tool of Risk Stratification in Hypertension. Adv. Exp. Med. Biol. 2017, 956, 475–488. [Google Scholar] [CrossRef]
- Williams, B.; Mancia, G.; Spiering, W.; Agabiti Rosei, E.; Azizi, M.; Burnier, M.; Clement, D.L.; Coca, A.; de Simone, G.; Dominiczak, A.; et al. 2018 ESC/ESH Guidelines for the management of arterial hypertension. Eur. Heart J. 2018, 39, 3021–3104. [Google Scholar] [CrossRef]
- Omboni, S.; Posokhov, I.N.; Kotovskaya, Y.V.; Protogerou, A.D.; Blacher, J. Twenty-Four-Hour Ambulatory Pulse Wave Analysis in Hypertension Management: Current Evidence and Perspectives. Curr. Hypertens. Rep. 2016, 18, 72. [Google Scholar] [CrossRef]
- Rajendran, P.; Jayakumar, T.; Nishigaki, I.; Ekambaram, G.; Nishigaki, Y.; Vetriselvi, J.; Sakthisekaran, D. Immunomodulatory Effect of Mangiferin in Experimental Animals with Benzo(a)Pyrene-induced Lung Carcinogenesis. Int. J. Biomed. Sci. 2013, 9, 68–74. [Google Scholar] [PubMed]
- Wilkinson, I.; Cockcroft, J.R. Cholesterol, lipids and arterial stiffness. In Atherosclerosis, Large Arteries and Cardiovascular Risk; Safar, M., Frohlich, E., Eds.; Karger: Basel, Switzerland, 2007; Volume 44, pp. 261–277. [Google Scholar]
- Paik, D.C.; Ramey, W.G.; Dillon, J.; Tilson, M.D. The nitrite/elastin reaction: Implications for in vivo degenerative effects. Connect. Tissue Res. 1997, 36, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Yasmin; McEniery, C.M.; Wallace, S.; Mackenzie, I.S.; Cockcroft, J.R.; Wilkinson, I.B. C-reactive protein is associated with arterial stiffness in apparently healthy individuals. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 969–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasmin; McEniery, C.M.; Wallace, S.; Dakham, Z.; Pulsalkar, P.; Pusalkar, P.; Maki-Petaja, K.; Ashby, M.J.; Cockcroft, J.R.; Wilkinson, I.B. Matrix metalloproteinase-9 (MMP-9), MMP-2, and serum elastase activity are associated with systolic hypertension and arterial stiffness. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 372. [Google Scholar] [CrossRef]
- Niederhoffer, N.; Lartaud-Idjouadiene, I.; Giummelly, P.; Duvivier, C.; Peslin, R.; Atkinson, J. Calcification of medial elastic fibers and aortic elasticity. Hypertension 1997, 29, 999–1006. [Google Scholar] [CrossRef]
- Chowienczyk, P.J.; Watts, G.F.; Cockcroft, J.R.; Ritter, J.M. Impaired endothelium-dependent vasodilation of forearm resistance vessels in hypercholesterolaemia. Lancet 1992, 340, 1430–1432. [Google Scholar] [CrossRef]
- Sakurai, K.; Cominacini, L.; Garbin, U.; Fratta Pasini, A.; Sasaki, N.; Takuwa, Y.; Masaki, T.; Sawamura, T. Induction of endothelin-1 production in endothelial cells via co-operative action between CD40 and lectin-like oxidized LDL receptor (LOX-1). J. Cardiovasc. Pharmacol. 2004, 44 (Suppl. S1), S173–S180. [Google Scholar] [CrossRef]
- Dart, A.M.; Lacombe, F.; Yeoh, J.K.; Cameron, J.D.; Jennings, G.L.; Laufer, E.; Esmore, D.S. Aortic distensibility in patients with isolated hypercholesterolaemia, coronary artery disease, or cardiac transplant. Lancet 1991, 338, 270–273. [Google Scholar] [CrossRef]
- Hopkins, K.D.; Lehmann, E.D.; Gosling, R.G.; Parker, J.R.; Sönksen, P.H. Biochemical correlates of aortic distensibility in vivo in normal subjects. Clin. Sci. 1993, 84, 593–597. [Google Scholar] [CrossRef]
- Cameron, J.D.; Jennings, G.L.; Dart, A.M. The relationship between arterial compliance, age, blood pressure and serum lipid levels. J. Hypertens. 1995, 13, 1718–1723. [Google Scholar] [CrossRef]
- Giannattasio, C.; Mangoni, A.A.; Failla, M.; Stella, M.L.; Carugo, S.; Bombelli, M.; Sega, R.; Mancia, G. Combined effects of hypertension and hypercholesterolemia on radial artery function. Hypertension 1997, 29, 583–586. [Google Scholar] [CrossRef] [PubMed]
- Márk, L.; Harangi, M.; Paragh, G. The labyrinth of residual risk: Reduction of the remaining lipid and inflammation risk in the prevention of atherosclerosis. Rvosi Hetil. 2018, 159, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Mundal, L.; Igland, J.; Ose, L.; Holven, K.B.; Veierød, M.B.; Leren, T.P.; Retterstøl, K. Cardiovascular disease mortality in patients with genetically verified familial hypercholesterolemia in Norway during 1992–2013. Eur. J. Prev. Cardiol. 2017, 24, 137–144. [Google Scholar] [CrossRef]
- Cheng, H.M.; Ye, Z.X.; Chiou, K.R.; Lin, S.J.; Charng, M.J. Vascular stiffness in familial hypercholesterolaemia is associated with C-reactive protein and cholesterol burden. Eur. J. Clin. Investig. 2007, 37, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Ellins, E.A.; New, K.J.; Datta, D.B.; Watkins, S.; Haralambos, K.; Rees, A.; Aled Rees, D.; Halcox, J.P. Validation of a new method for non-invasive assessment of vasomotor function. Eur. J. Prev. Cardiol. 2016, 23, 577–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewandowski, P.; Romanowska-Kocejko, M.; Węgrzyn, A.; Chmara, M.; Żuk, M.; Limon, J.; Wasąg, B.; Rynkiewicz, A.; Gruchała, M. Noninvasive assessment of endothelial function and vascular parameters in patients with familial and nonfamilial hypercholesterolemia. Pol. Arch. Med. Wewn. 2014, 124, 516–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plana, N.; Ferré, R.; Merino, J.; Aragonès, G.; Girona, J.; Heras, M.; Masana, L. Heterozygous familial hypercholesterolaemic patients have increased arterial stiffness, as determined using the augmentation index. J. Atheroscler. Thromb. 2011, 18, 1110–1116. [Google Scholar] [CrossRef] [Green Version]
- Ershova, A.I.; Meshkov, A.N.; Rozhkova, T.A.; Kalinina, M.V.; Deev, A.D.; Rogoza, A.N.; Balakhonova, T.V.; Boytsov, S.A. Carotid and Aortic Stiffness in Patients with Heterozygous Familial Hypercholesterolemia. PLoS ONE 2016, 11, e0158964. [Google Scholar] [CrossRef]
- Tada, H.; Kawashiri, M.A.; Nohara, A.; Inazu, A.; Mabuchi, H.; Yamagishi, M. Assessment of arterial stiffness in patients with familial hypercholesterolemia. J. Clin. Lipidol. 2018, 12, 397–402.e392. [Google Scholar] [CrossRef]
- Tran, A.; Burkhardt, B.; Tandon, A.; Blumenschein, S.; van Engelen, A.; Cecelja, M.; Zhang, S.; Uribe, S.; Mura, J.; Greil, G.; et al. Pediatric heterozygous familial hypercholesterolemia patients have locally increased aortic pulse wave velocity and wall thickness at the aortic root. Int. J. Cardiovasc. Imaging 2019, 35, 1903–1911. [Google Scholar] [CrossRef]
- Vlahos, A.P.; Naka, K.K.; Bechlioulis, A.; Theoharis, P.; Vakalis, K.; Moutzouri, E.; Miltiadous, G.; Michalis, L.K.; Siamopoulou-Mavridou, A.; Elisaf, M.; et al. Endothelial dysfunction, but not structural atherosclerosis, is evident early in children with heterozygous familial hypercholesterolemia. Pediatr. Cardiol. 2014, 35, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Waluś-Miarka, M.; Wojciechowska, W.; Miarka, P.; Kloch-Badełek, M.; Woźniakiewicz, E.; Czarnecka, D.; Sanak, M.; Małecki, M.; Idzior-Waluś, B. Intima-media thickness correlates with features of metabolic syndrome in young people with a clinical diagnosis of familial hypercholesterolaemia. Kardiol. Pol. 2013, 71, 566–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiner, Ž.; Simental-Mendía, L.E.; Ruscica, M.; Katsiki, N.; Banach, M.; Al Rasadi, K.; Jamialahmadi, T.; Sahebkar, A. Pulse wave velocity as a measure of arterial stiffness in patients with familial hypercholesterolemia: A systematic review and meta-analysis. Arch. Med. Sci. 2019, 15, 1365–1374. [Google Scholar] [CrossRef] [PubMed]
- Bjelakovic, B.; Stefanutti, C.; Reiner, Ž.; Watts, G.F.; Moriarty, P.; Marais, D.; Widhalm, K.; Cohen, H.; Harada-Shiba, M.; Banach, M. Risk Assessment and Clinical Management of Children and Adolescents with Heterozygous Familial Hypercholesterolaemia. A Position Paper of the Associations of Preventive Pediatrics of Serbia, Mighty Medic and International Lipid Expert Panel. J. Clin. Med. 2021, 10, 4930. [Google Scholar] [CrossRef] [PubMed]
- Martinez, L.R.; Miname, M.H.; Bortolotto, L.A.; Chacra, A.P.; Rochitte, C.E.; Sposito, A.C.; Santos, R.D. No correlation and low agreement of imaging and inflammatory atherosclerosis’ markers in familial hypercholesterolemia. Atherosclerosis 2008, 200, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Bedi, O.; Dhawan, V.; Sharma, P.L.; Kumar, P. Pleiotropic effects of statins: New therapeutic targets in drug design. Naunyn Schmiedebergs Arch. Pharmacol. 2016, 389, 695–712. [Google Scholar] [CrossRef] [PubMed]
- Oesterle, A.; Laufs, U.; Liao, J.K. Pleiotropic Effects of Statins on the Cardiovascular System. Circ. Res. 2017, 120, 229–243. [Google Scholar] [CrossRef] [Green Version]
- Kool, M.; Lustermans, F.; Kragten, H.; Struijker Boudier, H.; Hoeks, A.; Reneman, R.; Rila, H.; Hoogendam, I.; Van Bortel, L. Does lowering of cholesterol levels influence functional properties of large arteries? Eur. J. Clin. Pharmacol. 1995, 48, 217–223. [Google Scholar] [CrossRef]
- Raison, J.; Rudnichi, A.; Safar, M.E. Effects of atorvastatin on aortic pulse wave velocity in patients with hypertension and hypercholesterolaemia: A preliminary study. J. Hum. Hypertens. 2002, 16, 705–710. [Google Scholar] [CrossRef] [Green Version]
- Kontopoulos, A.G.; Athyros, V.G.; Pehlivanidis, A.N.; Demitriadis, D.S.; Papageorgiou, A.A.; Boudoulas, H. Long-term treatment effect of atorvastatin on aortic stiffness in hypercholesterolaemic patients. Curr. Med. Res. Opin. 2003, 19, 22–27. [Google Scholar] [CrossRef]
- Ferrier, K.E.; Muhlmann, M.H.; Baguet, J.P.; Cameron, J.D.; Jennings, G.L.; Dart, A.M.; Kingwell, B.A. Intensive cholesterol reduction lowers blood pressure and large artery stiffness in isolated systolic hypertension. J. Am. Coll. Cardiol. 2002, 39, 1020–1025. [Google Scholar] [CrossRef] [Green Version]
- Matsuo, T.; Iwade, K.; Hirata, N.; Yamashita, M.; Ikegami, H.; Tanaka, N.; Aosaki, M.; Kasanuki, H. Improvement of arterial stiffness by the antioxidant and anti-inflammatory effects of short-term statin therapy in patients with hypercholesterolemia. Heart Vessel. 2005, 20, 8–12. [Google Scholar] [CrossRef] [PubMed]
- Muramatsu, J.; Kobayashi, A.; Hasegawa, N.; Yokouchi, S. Hemodynamic changes associated with reduction in total cholesterol by treatment with the HMG-CoA reductase inhibitor pravastatin. Atherosclerosis 1997, 130, 179–182. [Google Scholar] [CrossRef]
- Poulter, N.R.; Wedel, H.; Dahlöf, B.; Sever, P.S.; Beevers, D.G.; Caulfield, M.; Kjeldsen, S.E.; Kristinsson, A.; McInnes, G.T.; Mehlsen, J.; et al. Role of blood pressure and other variables in the differential cardiovascular event rates noted in the Anglo-Scandinavian Cardiac Outcomes Trial-Blood Pressure Lowering Arm (ASCOT-BPLA). Lancet 2005, 366, 907–913. [Google Scholar] [CrossRef]
- Toyama, K.; Sugiyama, S.; Oka, H.; Iwasaki, Y.; Sumida, H.; Tanaka, T.; Tayama, S.; Jinnouchi, H.; Ogawa, H. Combination treatment of rosuvastatin or atorvastatin, with regular exercise improves arterial wall stiffness in patients with coronary artery disease. PLoS ONE 2012, 7, e41369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, B.; Che, W.; Yan, H.; Zhu, W.; Wang, H. Effects of rosuvastatin vs. simvastatin/ezetimibe on arterial wall stiffness in patients with coronary artery disease. Intern. Med. 2013, 52, 2715–2719. [Google Scholar] [CrossRef] [Green Version]
- Toscano, A.; Cinquegrani, M.; Scuruchi, M.; Di Pino, A.; Piro, S.; Ferrara, V.; Morace, C.; Lo Gullo, A.; Imbalzano, E.; Purrello, F.; et al. PCSK9 Plasma Levels Are Associated with Mechanical Vascular Impairment in Familial Hypercholesterolemia Subjects without a History of Atherosclerotic Cardiovascular Disease: Results of Six-Month Add-On PCSK9 Inhibitor Therapy. Biomolecules 2022, 12, 562. [Google Scholar] [CrossRef]
- Ruscica, M.; Tokgözoğlu, L.; Corsini, A.; Sirtori, C.R. PCSK9 inhibition and inflammation: A narrative review. Atherosclerosis 2019, 288, 146–155. [Google Scholar] [CrossRef] [Green Version]
- Sahebkar, A.; Simental-Mendía, L.E.; Guerrero-Romero, F.; Golledge, J.; Watts, G.F. Effect of statin therapy on plasma proprotein convertase subtilisin kexin 9 (PCSK9) concentrations: A systematic review and meta-analysis of clinical trials. Diabetes Obes. Metab. 2015, 17, 1042–1055. [Google Scholar] [CrossRef]
- Bambauer, R.; Bambauer, C.; Lehmann, B.; Latza, R.; Schiel, R. LDL-apheresis: Technical and clinical aspects. Sci. World J. 2012, 2012, 314283. [Google Scholar] [CrossRef] [Green Version]
- Mandraffino, G.; Scicali, R.; Rodríguez-Carrio, J.; Savarino, F.; Mamone, F.; Scuruchi, M.; Cinquegrani, M.; Imbalzano, E.; Di Pino, A.; Piro, S.; et al. Arterial stiffness improvement after adding on PCSK9 inhibitors or ezetimibe to high-intensity statins in patients with familial hypercholesterolemia: A Two-Lipid Center Real-World Experience. J. Clin. Lipidol. 2020, 14, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Sinzinger, H.; Steiner, S.; Derfler, K. Pleiotropic effects of regular lipoprotein-apheresis. Atheroscler. Suppl. 2017, 30, 122–127. [Google Scholar] [CrossRef] [PubMed]


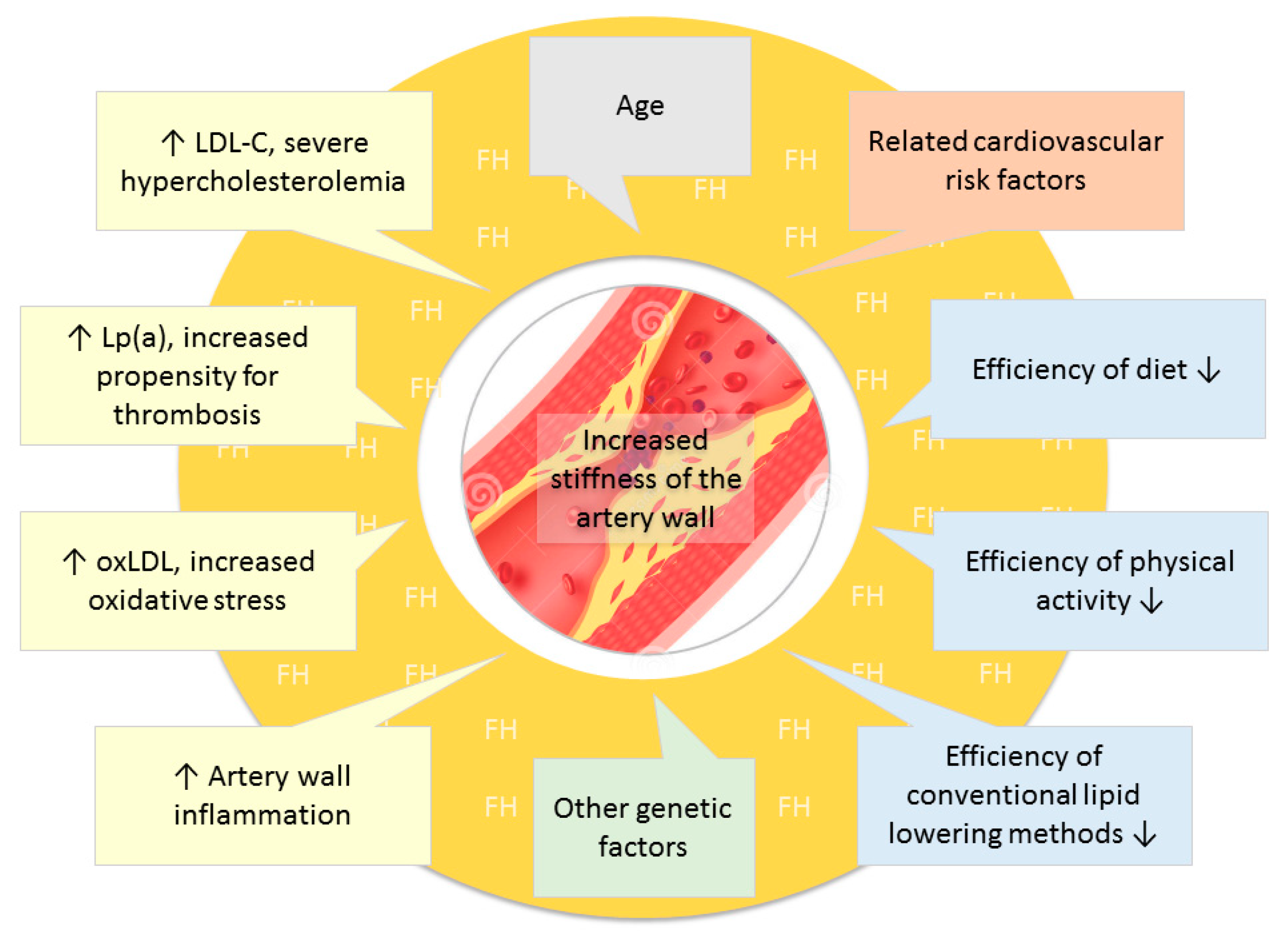{kind=link}
| Inheritance | Chromosome | Gene | Name | Prevalence |
|---|---|---|---|---|
| Autosomal dominant | 19p13 | LDLR | Familial hypercholesterolemia (FH) | 60–80% |
| 2p24–p23 | ApoB100 | Familial defective ApoB syndrome (FDB)/(FH2) | 1–5% | |
| 1p32 | PCSK9 | PCSK9 gain-of-function (FH3) | 0–3% | |
| several genes | Polygenic forms | 20–40% | ||
| Autosomal recessive | 1p35 | LDLRAP1 | Autosomal recessive hypercholesterolemia | rare |
| Family History | Earl-Onset CAD or PAD First-Degree Relative | 1 point |
| Presence of Xanthomata or Corneal Arcus in First-Degree Relative | 2 points | |
| Clinical history | CAD in women under 60, in men under 55 | 2 points |
| stroke or PAD in women under 60, in men under 55 | 1 point | |
| Physical examination | presence of tendonous xanthomata at any age | 6 points |
| presence of corneal arcus under 45 | 4 points | |
| Laboratory tests | LDL > 8.5 mmol/L | 8 points |
| LDL 6.5–8.4 mmol/L | 5 points | |
| LDL 5.0–6.4 mmol/L | 3 points | |
| LDL 4.0–4.9 mmol/L | 1 point | |
| note: HDL and TG levels norm. | ||
| DNA analysis | detectable mutation in the LDL receptor gene | 8 points |
| The diagnosis is verified if score is higher than 8 points | ||
| The diagnosis is probable: 6–8 points | ||
| The diagnosis is possible: 3–5 points | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kovács, B.; Cseprekál, O.; Diószegi, Á.; Lengyel, S.; Maroda, L.; Paragh, G.; Harangi, M.; Páll, D. The Importance of Arterial Stiffness Assessment in Patients with Familial Hypercholesterolemia. J. Clin. Med. 2022, 11, 2872. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm11102872
Kovács B, Cseprekál O, Diószegi Á, Lengyel S, Maroda L, Paragh G, Harangi M, Páll D. The Importance of Arterial Stiffness Assessment in Patients with Familial Hypercholesterolemia. Journal of Clinical Medicine. 2022; 11(10):2872. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm11102872
Chicago/Turabian StyleKovács, Beáta, Orsolya Cseprekál, Ágnes Diószegi, Szabolcs Lengyel, László Maroda, György Paragh, Mariann Harangi, and Dénes Páll. 2022. "The Importance of Arterial Stiffness Assessment in Patients with Familial Hypercholesterolemia" Journal of Clinical Medicine 11, no. 10: 2872. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm11102872