
Lymphocyte Medium-Chain Acyl-CoA Dehydrogenase Activity and Its Potential as a Diagnostic Confirmation Tool in Newborn Screening Cases

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Biochemical Analysis
2.3. MCAD Enzyme Activity in Lymphocytes
2.4. Genetic Analysis
2.5. Statistical Analysis
3. Results
3.1. Newborn Screening
3.2. Confirmatory Testing
3.2.1. Biochemical Confirmatory Analysis
3.2.2. Genetic Studies
3.2.3. Lymphocyte Enzyme Activity
3.3. Clinical and Biochemical Outcomes and Treatment
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Rocha, H.; Castiñeiras, D.; Delgado, C.; Egea, J.; Yahyaoui, R.; González, Y.; Conde, M.; González, I.; Rueda, I.; Rello, L.; et al. Birth Prevalence of Fatty Acid β-Oxidation Disorders in Iberia. In JIMD Reports; Springer: Berlin/Heidelberg, Germany, 2014; Volume 16, pp. 89–94. [Google Scholar] [CrossRef] [Green Version]
- Hoflack, M.; Caruba, C.; Pitelet, G.; Haas, H.; Mas, J.-C.; Paquis, V.; Berard, E. Coma du nourrisson aux urgences: 2 cas de déficit en MCAD. Arch. De Pédiatrie 2010, 17, 1074–1077. [Google Scholar] [CrossRef] [PubMed]
- Feillet, F.; Ogier, H.; Cheillan, D.; Aquaviva, C.; Labarthe, F.; Baruteau, J.; Chabrol, B.; de Lonlay, P.; Valayanopoulos, V.; Garnotel, R.; et al. Déficit en acyl-CoA-déshydrogénase des acides gras à chaîne moyenne (MCAD): Consensus français pour le dépistage, le diagnostic, et la prise en charge. Arch. De Pédiatrie 2012, 19, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Jager, E.A.; Kuijpers, M.M.; Bosch, A.M.; Mulder, M.F.; Gozalbo, E.R.; Visser, G.; Vries, M.; Williams, M.; Waterham, H.R.; Spronsen, F.J.; et al. A Nationwide Retrospective Observational Study of Population Newborn Screening for Medium-chain Acyl-CoA Dehydrogenase (MCAD) Deficiency in the Netherlands. J. Inherit. Metab. Dis. 2019, 42, 890–897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilcken, B.; Haas, M.; Joy, P.; Wiley, V.; Chaplin, M.; Black, C.; Fletcher, J.; McGill, J.; Boneh, A. Outcome of Neonatal Screening for Medium-Chain Acyl-CoA Dehydrogenase Deficiency in Australia: A Cohort Study. Lancet 2007, 369, 37–42. [Google Scholar] [CrossRef]
- Schatz, U.A.; Ensenauer, R. The Clinical Manifestation of MCAD Deficiency: Challenges towards Adulthood in the Screened Population. J. Inherit. Metab. Dis. 2010, 33, 513–520. [Google Scholar] [CrossRef]
- Grosse, S.D.; Khoury, M.J.; Greene, C.L.; Crider, K.S.; Pollitt, R.J. The Epidemiology of Medium Chain Acyl-CoA Dehydrogenase Deficiency: An Update. Genet. Med. 2006, 8, 205–212. [Google Scholar] [CrossRef] [Green Version]
- Gramer, G.; Haege, G.; Fang-Hoffmann, J.; Hoffmann, G.F.; Bartram, C.R.; Hinderhofer, K.; Burgard, P.; Lindner, M. Medium-Chain Acyl-CoA Dehydrogenase Deficiency: Evaluation of Genotype-Phenotype Correlation in Patients Detected by Newborn Screening. In JIMD Reports; Springer: Berlin/Heidelberg, Germany, 2015; Volume 23, pp. 101–112. [Google Scholar] [CrossRef] [Green Version]
- Merritt, J.L.; Chang, I.J. Medium-Chain Acyl-Coenzyme A Dehydrogenase Deficiency. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Ter Veld, F.; Mueller, M.; Kramer, S.; Haussmann, U.; Herebian, D.; Mayatepek, E.; Laryea, M.D.; Primassin, S.; Spiekerkoetter, U. A Novel Tandem Mass Spectrometry Method for Rapid Confirmation of Medium- and Very Long-Chain Acyl-CoA Dehydrogenase Deficiency in Newborns. PLoS ONE 2009, 4, e6449. [Google Scholar] [CrossRef]
- Vianey-Saban, C.; Divry, P.; Brivet, M.; Nada, M.; Zabot, M.T.; Mathieu, M.; Roe, C. Mitochondrial Very-Long-Chain Acyl-Coenzyme A Dehydrogenase Deficiency: Clinical Characteristics and Diagnostic Considerations in 30 Patients. Clin. Chim. Acta 1998, 269, 43–62. [Google Scholar] [CrossRef]
- Touw, C.M.L.; Smit, G.P.A.; de Vries, M.; de Klerk, J.B.C.; Bosch, A.M.; Visser, G.; Mulder, M.F.; Rubio-Gozalbo, M.; Elvers, B.; Niezen-Koning, K.E.; et al. Risk Stratification by Residual Enzyme Activity after Newborn Screening for Medium-Chain Acyl-CoA Dehyrogenase Deficiency: Data from a Cohort Study. Orphanet J. Rare Dis. 2012, 7, 30. [Google Scholar] [CrossRef] [Green Version]
- Derks, T.G.J.; Boer, T.S.; van Assen, A.; Bos, T.; Ruiter, J.; Waterham, H.R.; Niezen-Koning, K.E.; Wanders, R.J.A.; Rondeel, J.M.M.; Loeber, J.G.; et al. Neonatal Screening for Medium-Chain Acyl-CoA Dehydrogenase (MCAD) Deficiency in The Netherlands: The Importance of Enzyme Analysis to Ascertain True MCAD Deficiency. J. Inherit. Metab. Dis. 2008, 31, 88–96. [Google Scholar] [CrossRef]
- Sturm, M.; Herebian, D.; Mueller, M.; Laryea, M.D.; Spiekerkoetter, U. Functional Effects of Different Medium-Chain Acyl-CoA Dehydrogenase Genotypes and Identification of Asymptomatic Variants. PLoS ONE 2012, 7, e45110. [Google Scholar] [CrossRef] [PubMed]
- Touw, C.M.L.; Smit, G.P.A.; Niezen-Koning, K.E.; Bosgraaf-de Boer, C.; Gerding, A.; Reijngoud, D.-J.; Derks, T.G.J. In Vitro and in Vivo Consequences of Variant Medium-Chain Acyl-CoA Dehydrogenase Genotypes. Orphanet J. Rare Dis. 2013, 8, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tucci, S.; Wagner, C.; Grünert, S.C.; Matysiak, U.; Weinhold, N.; Klein, J.; Porta, F.; Spada, M.; Bordugo, A.; Rodella, G.; et al. Genotype and Residual Enzyme Activity in Medium-chain acyl-CoA Dehydrogenase (MCAD) Deficiency: Are Predictions Possible? J. Inherit. Metab. Dis. 2021, 44, 916–925. [Google Scholar] [CrossRef] [PubMed]
- Tajima, G.; Sakura, N.; Shirao, K.; Okada, S.; Tsumura, M.; Nishimura, Y.; Ono, H.; Hasegawa, Y.; Hata, I.; Naito, E.; et al. Development of a New Enzymatic Diagnosis Method for Very-Long-Chain Acyl-CoA Dehydrogenase Deficiency by Detecting 2-Hexadecenoyl-CoA Production and Its Application in Tandem Mass Spectrometry-Based Selective Screening and Newborn Screening in Japan. Pediatr. Res. 2008, 64, 667–672. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, I.; Ruiz-Sala, P.; Vicente, Y.; Merinero, B.; Pérez-Cerdá, C.; Ugarte, M. Separation and Identification of Plasma Short-Chain Acylcarnitine Isomers by HPLC/MS/MS for the Differential Diagnosis of Fatty Acid Oxidation Defects and Organic Acidemias. J. Chromatogr. B 2007, 860, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Merinero, B.; Alcaide, P.; Martín-Hernández, E.; Morais, A.; García-Silva, M.T.; Quijada-Fraile, P.; Pedrón-Giner, C.; Dulin, E.; Yahyaoui, R.; Egea, J.M.; et al. Four Years’ Experience in the Diagnosis of Very Long-Chain Acyl-CoA Dehydrogenase Deficiency in Infants Detected in Three Spanish Newborn Screening Centers. In JIMD Reports; Morava, E., Baumgartner, M., Patterson, M., Rahman, S., Zschocke, J., Peters, V., Eds.; Springer: Berlin/Heidelberg, Germany, 2017; Volume 39, pp. 63–74. ISBN 978-3-662-57576-5. [Google Scholar]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Tajima, G.; Hara, K.; Tsumura, M.; Kagawa, R.; Okada, S.; Sakura, N.; Hata, I.; Shigematsu, Y.; Kobayashi, M. Screening of MCAD Deficiency in Japan: 16years’ Experience of Enzymatic and Genetic Evaluation. Mol. Genet. Metab. 2016, 119, 322–328. [Google Scholar] [CrossRef]
- Navarrete, R.; Leal, F.; Vega, A.I.; Morais-López, A.; Garcia-Silva, M.T.; Martín-Hernández, E.; Quijada-Fraile, P.; Bergua, A.; Vives, I.; García-Jiménez, I.; et al. Value of Genetic Analysis for Confirming Inborn Errors of Metabolism Detected through the Spanish Neonatal Screening Program. Eur. J. Hum. Genet. 2019, 27, 556–562. [Google Scholar] [CrossRef] [Green Version]
- Blau, N.; Duran, M.; Gibson, K.M.; Dionisi Vici, C. (Eds.) Physician’s Guide to the Diagnosis, Treatment, and Follow-Up of Inherited Metabolic Diseases; Springer: Berlin/Heidelberg, Germany, 2014; ISBN 978-3-642-40336-1. [Google Scholar]
- Couce, M.L.; Sánchez-Pintos, P.; Diogo, L.; Leão-Teles, E.; Martins, E.; Santos, H.; Bueno, M.A.; Delgado-Pecellín, C.; Castiñeiras, D.E.; Cocho, J.A.; et al. Newborn Screening for Medium-Chain Acyl-CoA Dehydrogenase Deficiency: Regional Experience and High Incidence of Carnitine Deficiency. Orphanet J. Rare Dis. 2013, 8, 102. [Google Scholar] [CrossRef] [Green Version]
- Wilcken, B.; Haas, M.; Joy, P.; Wiley, V.; Bowling, F.; Carpenter, K.; Christodoulou, J.; Cowley, D.; Ellaway, C.; Fletcher, J.; et al. Expanded Newborn Screening: Outcome in Screened and Unscreened Patients at Age 6 Years. Pediatrics 2009, 124, e241–e248. [Google Scholar] [CrossRef]
- Yusuf, K.; Jirapradittha, J.; Amin, H.J.; Yu, W.; Hasan, S.U. Neonatal Ventricular Tachyarrhythmias in Medium Chain Acyl-CoA Dehydrogenase Deficiency. Neonatology 2010, 98, 260–264. [Google Scholar] [CrossRef] [PubMed]
- Cyriac, J.; Venkatesh, V.; Gupta, C. A Fatal Neonatal Presentation of Medium-Chain Acyl Coenzyme a Dehydrogenase Deficiency. J. Int. Med. Res. 2008, 36, 609–610. [Google Scholar] [CrossRef] [Green Version]
- Maier, E.M.; Liebl, B.; Röschinger, W.; Nennstiel-Ratzel, U.; Fingerhut, R.; Olgemöller, B.; Busch, U.; Krone, N.; Kries, R.V.; Roscher, A.A. Population Spectrum of ACADM Genotypes Correlated to Biochemical Phenotypes in Newborn Screening for Medium-Chain Acyl-CoA Dehydrogenase Deficiency. Hum. Mutat. 2005, 25, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Bentler, K.; Zhai, S.; Elsbecker, S.A.; Arnold, G.L.; Burton, B.K.; Vockley, J.; Cameron, C.A.; Hiner, S.J.; Edick, M.J.; Berry, S.A.; et al. 221 Newborn-Screened Neonates with Medium-Chain Acyl-Coenzyme A Dehydrogenase Deficiency: Findings from the Inborn Errors of Metabolism Collaborative. Mol. Genet. Metab. 2016, 119, 75–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnold, G.L.; Saavedra-Matiz, C.A.; Galvin-Parton, P.A.; Erbe, R.; DeVincentis, E.; Kronn, D.; Mofidi, S.; Wasserstein, M.; Pellegrino, J.E.; Levy, P.A.; et al. Lack of Genotype–Phenotype Correlations and Outcome in MCAD Deficiency Diagnosed by Newborn Screening in New York State. Mol. Genet. Metab. 2010, 99, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.-W.; Zytkovicz, T.H.; Comeau, A.M.; Strauss, A.W.; Marsden, D.; Shih, V.E.; Grady, G.F.; Eaton, R.B. Spectrum of Medium-Chain Acyl-CoA Dehydrogenase Deficiency Detected by Newborn Screening. Pediatrics 2008, 121, e1108–e1114. [Google Scholar] [CrossRef] [PubMed]
- Waddell, L.; Wiley, V.; Carpenter, K.; Bennetts, B.; Angel, L.; Andresen, B.S.; Wilcken, B. Medium-Chain Acyl-CoA Dehydrogenase Deficiency: Genotype–Biochemical Phenotype Correlations. Mol. Genet. Metab. 2006, 87, 32–39. [Google Scholar] [CrossRef]
- Anderson, D.R.; Viau, K.; Botto, L.D.; Pasquali, M.; Longo, N. Clinical and Biochemical Outcomes of Patients with Medium-Chain Acyl-CoA Dehydrogenase Deficiency. Mol. Genet. Metab. 2020, 129, 13–19. [Google Scholar] [CrossRef]


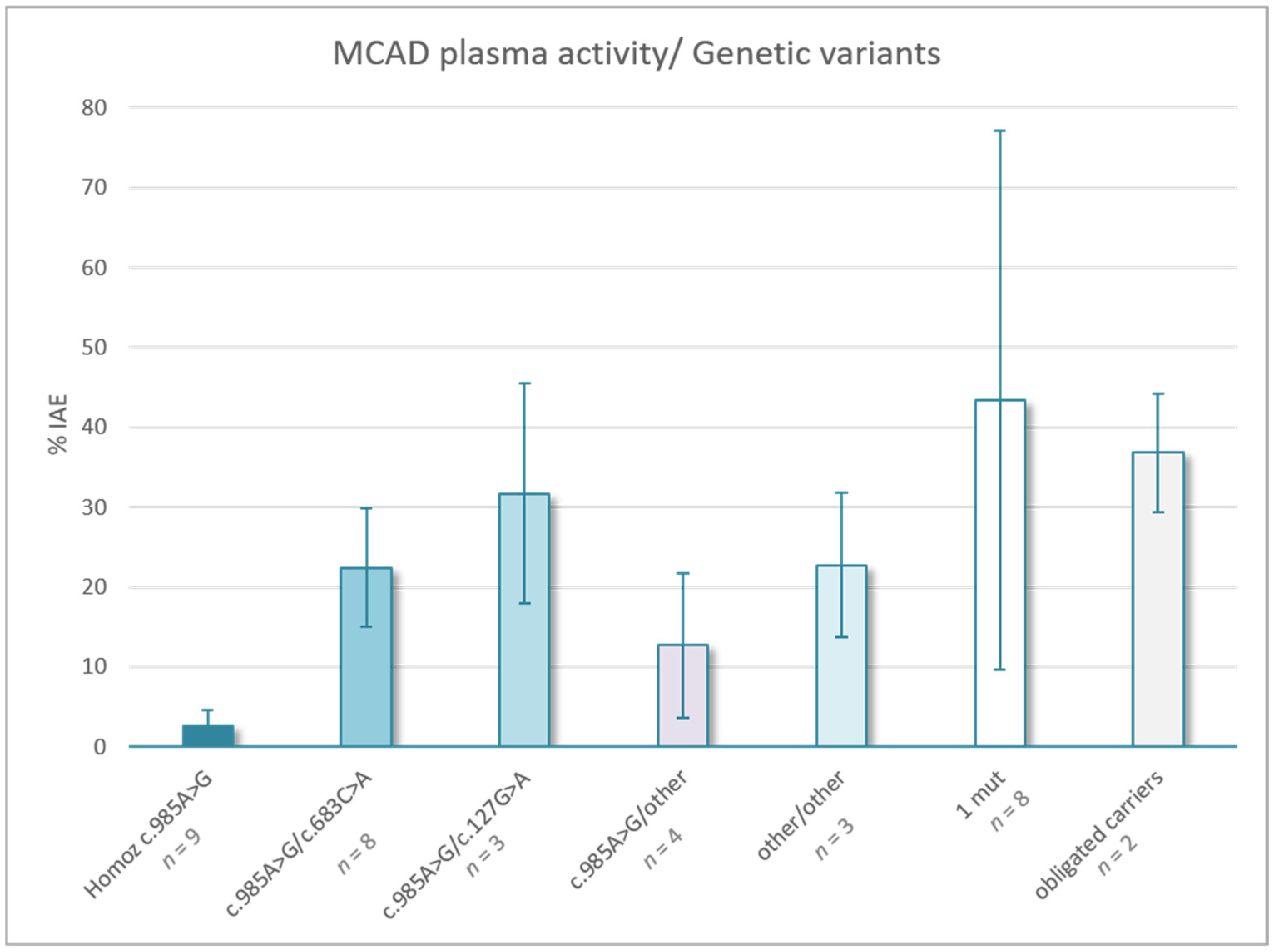{kind=link}
| C8 (DBS) NBS 48 h | C8 Confirmatory Test CEDEM (pl) | Nucleotide Variation | Protein Effect | Act | Act (Lf) | Therapy | Clinical Outcome | |
|---|---|---|---|---|---|---|---|---|
| NR: 0.16 µmol/L | mean (range) NR < 0.25 µmol/L | nmol/min/mg prot | % IAC | |||||
| P1 | 8.4 | 5.73 (4–8.9) | c.985A > G/c.985A > G | p.Lys329Glu/p.Lys329Glu | 0.06 | 5.9 | C, AF | CD |
| P2 | 10.6 | 5.43 (4.1–6.7) | c.985A > G/c.985A > G | p.Lys329Glu/p.Lys329Glu | 0.04 | 5.8 | C, AF | AS |
| P3 | 15.2 | 5.85 (1.7–12.8) | c.985A > G/c.985A > G | p.Lys329Glu/p.Lys329Glu | 0.05 | 4.5 | C, AF | AS |
| P4 | 7.2 | 2.89 (0.87–5.2) | c.985A > G/c.985A > G | p.Lys329Glu/p.Lys329Glu | 0.06 | 4.2 | C, AF | AB, LR, MC |
| P5 | 11.4 | 5.9 (2–11) | c.985A > G/c.985A > G | p.Lys329Glu/p.Lys329Glu | 0.16 | 2.6 | C, AF | FI, V |
| P67 | 6.6 | 9.6 (8.1–12.8) | c.985A > G/c.985A > G | p.Lys329Glu/p.Lys329Glu | 0.09 | 1.7 | C, AF | FH |
| P76 | 7.2 | 3.8 (3.4–4) | c.985A > G/c.985A > G | p.Lys329Glu/p.Lys329Glu | 0.09 | 1.7 | C, AF | AS |
| P8 | NP | 10.88 (7.2–17.9) | c.985A > G/c.985A > G | p.Lys329Glu/p.Lys329Glu | 0.03 | 1.2 | C, AF | CSE, FH, CD (d16 m) |
| P9 | 13.7 | 7(2.4–12.5) | c.985A > G/c.985A > G | p.Lys329Glu/p.Lys329Glu | 0.09 | 1.3 | PC | AB (d2 y) |
| P1011 | 20 | 9.78 (9.1–10) | c.985A > G/c.985A > G | p.Lys329Glu/p.Lys329Glu | 0.08 | 1.1 | C, AF | AS |
| P1110 | 7.9 | 6.72 (5.7–8.2) | c.985A > G/c.985A > G | p.Lys329Glu/p.Lys329Glu | 0.05 | 0.7 | C, AF | AS |
| P12 | 8.1 | 9.9 | c.985A > G/c.985A > G | p.Lys329Glu/p.Lys329Glu | 0.05 | 0.7 | C, AF | AS |
| P1318 | 0.39 | 0.35 (0.35–0.36) | c.985A > G/c.683C > A | p.Lys329Glu/p.Thr228Asn | 0.34 | 30.6 | AF | H, FI |
| P14 | 0.65 | 0.32 (0.17–0.6) | c.985A > G/c.683C > A | p.Lys329Glu/p.Thr228Asn | 1.10 | 28.7 | C, AF | AS |
| P15 | 0.93 | 0.45 (0.28–0.65) | c.985A > G/c.683C > A | p.Lys329Glu/p.Thr228Asn | 0.36 | 28.8 | C, AF | AS |
| P16 | 1.19 | 0.33 (0.2–0.4) | c.985A > G/c.683C > A | p.Lys329Glu/p.Thr228Asn | 0.62 | 26.7 | AF | LGW, CK |
| P1720 | 0.37 | 0.25 (0.11–0.33) | c.985A > G/c.683C > A | p.Lys329Glu/p.Thr228Asn | 0.24 | 21.6 | AF | J |
| P1813 | 1.56 | 0.35 (0.21–0.46) | c.985A > G/c.683C > A | p.Lys329Glu/p.Thr228Asn | 0.24 | 16.9 | AF | AS |
| P19 | 0.67 | 0.49 (0.34–0.64) | c.985A > G/c.683C > A | p.Lys329Glu/p.Thr228Asn | 0.54 | 15 | AF | AS |
| P2017 | NP | 0.31 | c.985A > G/c.683C > A | p.Lys329Glu/p.Thr228Asn | 0.38 | 11 | AF | AS |
| P21 | 0.61 | 0.57 (0.32–0.82) | c.985A > G/c.127G > A | p.Lys329Glu/p.Glu43Lys | 0.45 | 40.5 | AF | AS |
| P22 | 0.38 | 0.23 (0.16–0.3) | c.985A > G/c.127G > A | p.Lys329Glu/p.Glu43Lys | 1.50 | 38.8 | AF | AS |
| P23 | 0.47 | 0.43 (0.33–0.52) | c.985A > G/c.127G > A | p.Lys329Glu/p.Glu43Lys | 0.16 | 15.8 | C, AF | LWB, AS |
| P2435 | 1.24 | 0.46 | c.985A > G/c.599+3A > G | p.Lys329Glu/p.? | 0.59 | 22 | AF | AS |
| P25 | 3.89 | 2 (1.3–2.7) | c.985A > G/G626C > T | p.Lys329Glu/p.Pro209Leu | 0.18 | 18.3 | C, AF | AS |
| P26 | 3.43 | 5.17 (2.5–8.9) | c.985A > G/c.250C > T | p.Lys329Glu/p.Leu84Phe | 0.46 | 8.2 | C, AF | AS |
| P27 | 6.0 | 1.5 | c.985A > G/c.346T > G | p.Lys329Glu/p.Cys116Gly | 0.03 | 2.4 | C, AF | AS |
| P28 | 1.03 | 0.3 (0.21–0.46) | c.683C > A/c.999_1011 dup13 | p.Thr228Asn/p.Gln338Term | 0.56 | 31 | C, AF | AS |
| P29 | 0.48 | 0.2 (0.08–0.48) | c.351A > C/c.503A > C | p.Thr117Thr/p.Asp168Ala | 0.45 | 24 | AF | AS |
| P30 | 2.31 | 1.0 (0.34–1.5) | c.338C > A/c.940G > C | p.Ala113Asp/p.Val314Leu | 0.53 | 13.1 | C, AF | AS |
| P31 | 0.5 | 0.12 | c.985A > G | p.Lys329Glu | 1.42 | 100 | No | AS |
| P32 | 0.5 | 0.79 | c.253G > C | p.Gly85Arg | 1.10 | 9.5 | No | FH |
| P33 | 0.49 | 0.34 (0.08–0.73) | c.449_452delCTGA | p.Thr150Argfs*4 | 1.27 | 37 | AF | LC, Ds |
| P34 | 0.17 | 0.57 | c.985A > G | p.Lys329Glu | 0.81 | 34 | No | AS |
| P3524 | 0.12 | 0.27 | c.985A > G | p.Lys329Glu | 0.79 | 30 | No | AS |
| P36 | 0.43 | 0.22 | c.985A > G | p.Lys329Glu | 0.51 | 22.3 | No | AS |
| P37 | 0.42 | 0.11 | c.985A > G | p.Lys329Glu | 0.28 | 16 | No | AS |
| P38 | NP | 0.27 (0.11–0.53) | c.985A > G | p.Lys329Glu | 0.48 | 14.7 | AF | CV, AP, D, P, TE (d10 y) |
| P39m3 | NP | NP | c.985A > G | p.Lys329Glu | 1.72 | 42 | No | / |
| P40f35 | NP | NP | c.985A > G | p.Lys329Glu | 0.73 | 32 | No | / |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alcaide, P.; Ferrer-López, I.; Gutierrez, L.; Leal, F.; Martín-Hernández, E.; Quijada-Fraile, P.; Bellusci, M.; Moráis, A.; Pedrón-Giner, C.; Rausell, D.; et al. Lymphocyte Medium-Chain Acyl-CoA Dehydrogenase Activity and Its Potential as a Diagnostic Confirmation Tool in Newborn Screening Cases. J. Clin. Med. 2022, 11, 2933. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm11102933
Alcaide P, Ferrer-López I, Gutierrez L, Leal F, Martín-Hernández E, Quijada-Fraile P, Bellusci M, Moráis A, Pedrón-Giner C, Rausell D, et al. Lymphocyte Medium-Chain Acyl-CoA Dehydrogenase Activity and Its Potential as a Diagnostic Confirmation Tool in Newborn Screening Cases. Journal of Clinical Medicine. 2022; 11(10):2933. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm11102933
Chicago/Turabian StyleAlcaide, Patricia, Isaac Ferrer-López, Leticia Gutierrez, Fatima Leal, Elena Martín-Hernández, Pilar Quijada-Fraile, Marcello Bellusci, Ana Moráis, Consuelo Pedrón-Giner, Dolores Rausell, and et al. 2022. "Lymphocyte Medium-Chain Acyl-CoA Dehydrogenase Activity and Its Potential as a Diagnostic Confirmation Tool in Newborn Screening Cases" Journal of Clinical Medicine 11, no. 10: 2933. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm11102933