
Combined Sarcoidosis and Idiopathic Pulmonary Fibrosis (CSIPF): A New Phenotype or a Fortuitous Overlap? Scoping Review and Case Series

 ,
,  , ,
, ,  , and
, and
Abstract
:1. Introduction
2. Methods
2.1. Eligibility Criteria
2.2. Information Sources and Search
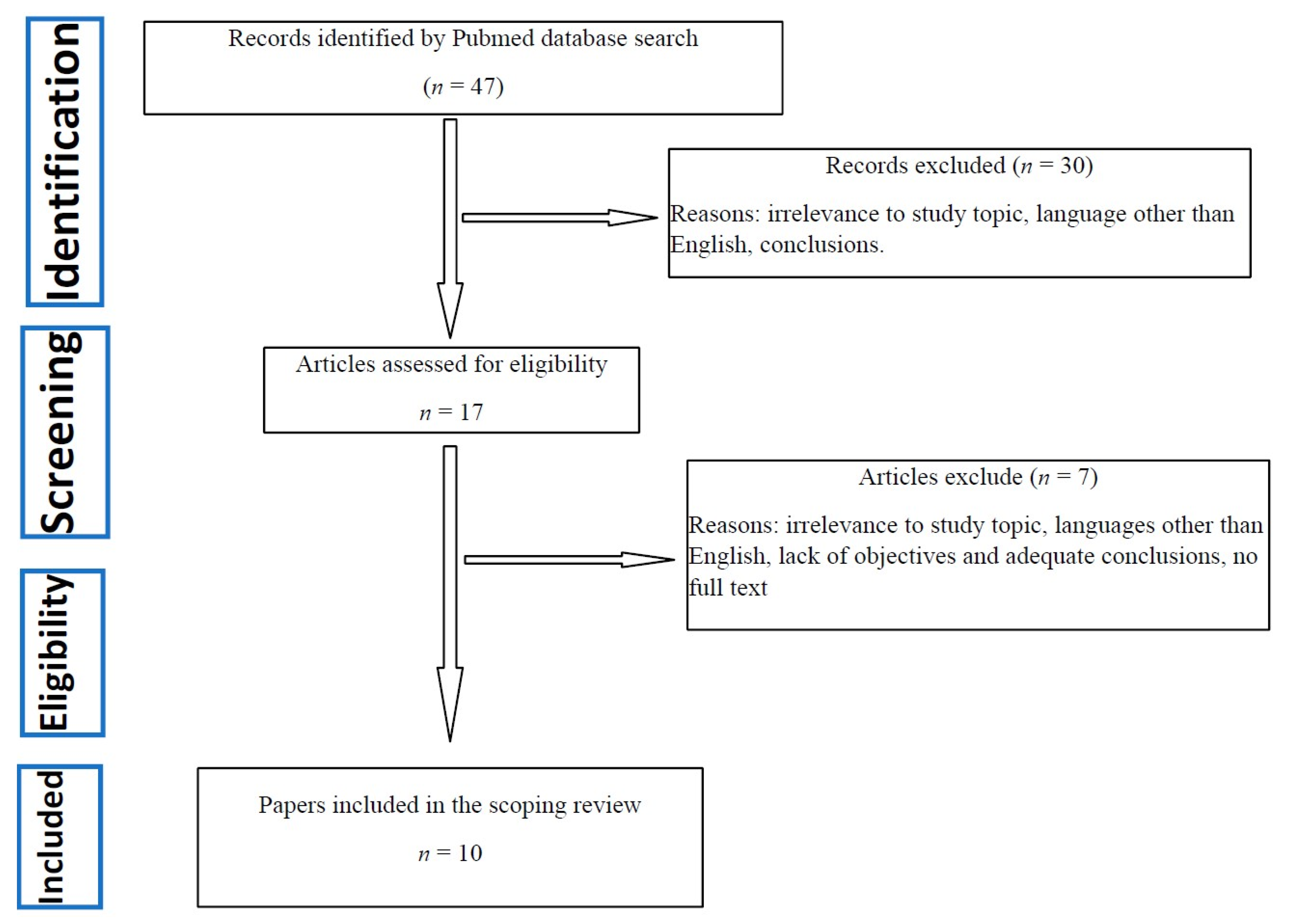
2.3. Selection Process
2.4. Data Charting and Items
2.5. Patient Cohorts
2.6. Statistical Analysis
3. Results
3.1. Descriptive Data and Quality Assessment after SANRA Assessment
3.2. Cohort Description
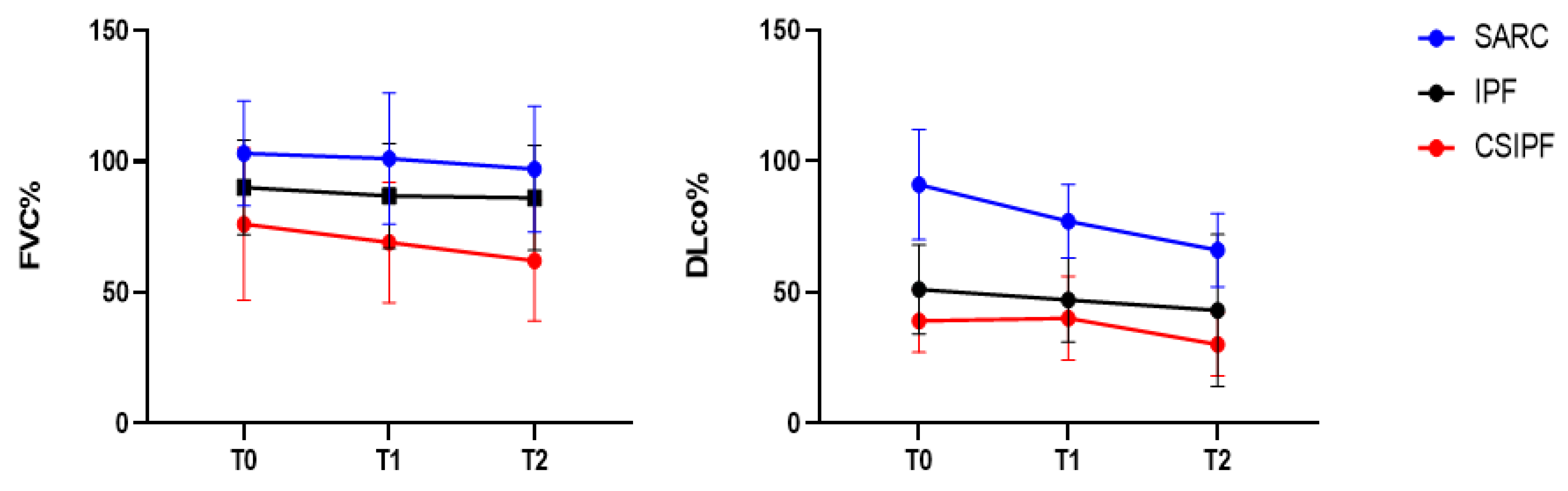
3.3. Lung Function Tests
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Ryerson, C.J.; Collard, H.R. Update on the diagnosis and classification of ILD. Curr. Opin. Pulm. Med. 2013, 19, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Vietri, L.; Cameli, P.; Perruzza, M.; Cekorja, B.; Bergantini, L.; D’Alessandro, M.; Refini, R.M.; Pieroni, M.; Fossi, A.; Bennett, D.; et al. Pirfenidone in idiopathic pulmonary fibrosis: Real-life experience in the referral centre of Siena. Ther. Adv. Respir. Dis. 2020, 14, 1753466620906326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergantini, L.; D’Alessandro, M.; Cameli, P.; Otranto, A.; Finco, T.; Curatola, G.; Sestini, P.; Bargagli, E. Prognostic role of NK cell percentages in bronchoalveolar lavage from patients with different fibrotic interstitial lung diseases. Clin. Immunol. 2021, 230, 108827. [Google Scholar] [CrossRef] [PubMed]
- Cameli, P.; Refini, R.M.; Bergantini, L.; D’Alessandro, M.; Alonzi, V.; Magnoni, C.; Rottoli, P.; Sestini, P.; Bargagli, E. Long-Term Follow-Up of Patients With Idiopathic Pulmonary Fibrosis Treated With Pirfenidone or Nintedanib: A Real-Life Comparison Study. Front. Mol. Biosci. 2020, 7, 581828. [Google Scholar] [CrossRef] [PubMed]
- Cottin, V.; Hirani, N.A.; Hotchkin, D.L.; Nambiar, A.M.; Ogura, T.; Otaola, M.; Skowasch, D.; Park, J.S.; Poonyagariyagorn, H.K.; Wuyts, W.; et al. Presentation, diagnosis and clinical course of the spectrum of progressive-fibrosing interstitial lung diseases. Eur. Respir. Rev. 2018, 27, 180076. [Google Scholar] [CrossRef] [Green Version]
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68. [Google Scholar] [CrossRef]
- Cameli, P.; Bargagli, E.; Bergantini, L.; Refini, R.M.; Pieroni, M.; Sestini, P.; Rottoli, P. Evaluation of multiple-flows exhaled nitric oxide in idiopathic and non-idiopathic interstitial lung disease. J. Breath Res. 2019, 13, 026008. [Google Scholar] [CrossRef]
- Vietri, L.; Bennett, D.; Cameli, P.; Bergantini, L.; Cillis, G.; Sestini, P.; Bargagli, E.; Rottoli, P. Serum amyloid A in patients with idiopathic pulmonary fibrosis. Respir. Investig. 2019, 57, 430–434. [Google Scholar] [CrossRef]
- Kärkkäinen, M.; Kettunen, H.-P.; Nurmi, H.; Selander, T.; Purokivi, M.; Kaarteenaho, R. Effect of smoking and comorbidities on survival in idiopathic pulmonary fibrosis. Respir. Res. 2017, 18, 160. [Google Scholar] [CrossRef] [Green Version]
- Cameli, P.; Bergantini, L.; Salvini, M.; Refini, R.M.; Pieroni, M.; Bargagli, E.; Sestini, P. Alveolar concentration of nitric oxide as a prognostic biomarker in idiopathic pulmonary fibrosis. Nitric Oxide 2019, 89, 41–45. [Google Scholar] [CrossRef]
- Landi, C.; Bergantini, L.; Cameli, P.; D’Alessandro, M.; Carleo, A.; Shaba, E.; Rottoli, P.; Bini, L.; Bargagli, E. Idiopathic Pulmonary Fibrosis Serum proteomic analysis before and after nintedanib therapy. Sci. Rep. 2020, 10, 9378. [Google Scholar] [CrossRef] [PubMed]
- Hunninghake, G.W.; Zimmerman, M.B.; Schwartz, D.A.; King, T.E.; Lynch, J.; Hegele, R.; Waldron, J.; Colby, T.; Müller, N.; Lynch, D.; et al. Utility of a Lung Biopsy for the Diagnosis of Idiopathic Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2001, 164, 193–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raghu, G.; Mageto, Y.N.; Lockhart, D.; Schmidt, R.A.; Wood, D.E.; Godwin, J.D. The accuracy of the clinical diagnosis of new-onset idiopathic pulmonary fibrosis and other interstitial lung disease: A prospective study. Chest 1999, 116, 1168–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souza, C.A.; Müller, N.L.; Lee, K.S.; Johkoh, T.; Mitsuhiro, H.; Chong, S. Idiopathic Interstitial Pneumonias: Prevalence of Mediastinal Lymph Node Enlargement in 206 Patients. Am. J. Roentgenol. 2006, 186, 995–999. [Google Scholar] [CrossRef] [PubMed]
- Salah, S.; Abad, S.; Monnet, D.; Brézin, A.P. Sarcoidosis. J. Fr. Ophtalmol. 2018, 41, e451–e467. [Google Scholar] [CrossRef]
- Cameli, P.; Gonnelli, S.; Bargagli, E.; D’Alessandro, M.; Bergantini, L.; Favetta, V.; Pitinca, M.D.T.; Lisi, E.; Refini, R.M.; Pieroni, M.; et al. The Role of Urinary Calcium and Chitotriosidase in a Cohort of Chronic Sarcoidosis Patients. Respiration 2020, 99, 207–212. [Google Scholar] [CrossRef]
- d’Alessandro, M.; Bergantini, L.; Cameli, P.; Mezzasalma, F.; Refini, R.M.; Pieroni, M.; Sestini, P.; Bargagli, E. Adaptive immune system in pulmonary sarcoidosis-Comparison of peripheral and alveolar biomarkers. Clin. Exp. Immunol. 2021, 205, 406–416. [Google Scholar] [CrossRef]
- d’Alessandro, M.; Bergantini, L.; Perrone, A.; Cameli, P.; Cameli, M.; Prasse, A.; Plataroti, D.; Sestini, P.; Bargagli, E. Serial investigation of Angiotensin-Converting Enzyme in sarcoidosis patients treated with Angiotensin-Converting Enzyme Inhibitor. Eur. J. Intern. Med. 2020, 78, 58–62. [Google Scholar] [CrossRef]
- Bargagli, E.; Prasse, A. Sarcoidosis: A review for the internist. Intern. Emerg. Med. 2018, 13, 325–331. [Google Scholar] [CrossRef]
- Rottoli, P.; Bargagli, E. Is bronchoalveolar lavage obsolete in the diagnosis of interstitial lung disease? Curr. Opin. Pulm. Med. 2003, 9, 418–425. [Google Scholar] [CrossRef]
- Silva, M.; Nunes, H.; Valeyre, D.; Sverzellati, N. Imaging of Sarcoidosis. Clin. Rev. Allergy Immunol. 2015, 49, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Ellinghaus, D.; Nutsua, M.; Hofmann, S.; Montgomery, C.G.; Iannuzzi, M.C.; Rybicki, B.A.; Petrek, M.; Mrazek, F.; Pabst, S.; et al. Identification of Immune-Relevant Factors Conferring Sarcoidosis Genetic Risk. Am. J. Respir. Crit. Care Med. 2015, 192, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, S.; Franke, A.; Fischer, A.; Jacobs, G.; Nothnagel, M.; Gaede, K.I.; Schürmann, M.; Müller-Quernheim, J.; Krawczak, M.; Rosenstiel, P.; et al. Genome-wide association study identifies ANXA11 as a new susceptibility locus for sarcoidosis. Nat. Genet. 2008, 40, 1103–1106. [Google Scholar] [CrossRef] [PubMed]
- Munn, Z.; Peters, M.D.J.; Stern, C.; Tufanaru, C.; McArthur, A.; Aromataris, E. Systematic review or scoping review? Guidance for authors when choosing between a systematic or scoping review approach. BMC Med. Res. Methodol. 2018, 18, 143. [Google Scholar] [CrossRef] [PubMed]
- Baethge, C.; Goldbeck-Wood, S.; Mertens, S. SANRA—A scale for the quality assessment of narrative review articles. Res. Integr. Peer Rev. 2019, 4, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teirstein, A.T.; Morgenthau, A.S. “End-stage” Pulmonary Fibrosis in Sarcoidosis. Mount Sinai J. Med. J. Transl. Personal. Med. 2009, 76, 30–36. [Google Scholar] [CrossRef]
- Shigemitsu, H.; Azuma, A. Sarcoidosis and interstitial pulmonary fibrosis; two distinct disorders or two ends of the same spectrum. Curr. Opin. Pulm. Med. 2011, 17, 303–307. [Google Scholar] [CrossRef]
- Patterson, K.C.; Strek, M.E. Pulmonary Fibrosis in Sarcoidosis. Clinical Features and Outcomes. Ann. Am. Thorac. Soc. 2013, 10, 362–370. [Google Scholar] [CrossRef]
- Nobata, K.; Kasai, T.; Fujimura, M.; Mizuguchi, M.; Nishi, K.; Ishiura, Y.; Yasui, M.; Nakao, S. Pulmonary Sarcoidosis with Usual Interstitial Pneumonia Distributed Predominantly in the Lower Lung Fields. Intern. Med. 2006, 45, 359–362. [Google Scholar] [CrossRef] [Green Version]
- Tachibana, K.; Arai, T.; Kagawa, T.; Minomo, S.; Akira, M.; Kitaichi, M.; Inoue, Y. A Case of Combined Sarcoidosis and Usual Interstitial Pneumonia. Intern. Med. 2012, 51, 1893–1897. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Kligerman, S.; Burke, A. End-stage Sarcoid Lung Disease Is Distinct From Usual Interstitial Pneumonia. Am. J. Surg. Pathol. 2013, 37, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Collins, B.F.; McClelland, R.L.; Ho, L.A.; Mikacenic, C.R.; Hayes, J.; Spada, C.; Raghu, G. Sarcoidosis and IPF in the same patient-a coincidence, an association or a phenotype? Respir. Med. 2018, 144, S20–S27. [Google Scholar] [CrossRef] [PubMed]
- Collins, B.F.; Raghu, G. Sarcoidosis and idiopathic pulmonary fibrosis: The same tale or a tale of two diseases in one. Respir. Med. 2019, 160, 105668. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, F.; Piccioli, C.; Rosi, E.; Carobene, L.; Spina, D.; Mazzei, M.A.; Bartolucci, M.; Moroni, C.; Novelli, L.; Rottoli, P.; et al. Combined sarcoidosis and idiopathic pulmonary fibrosis (CSIPF): A novel disease phenotype? Respir. Med. 2019, 160, 105650. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| No. | Title and Authors | Justification of the Article’s Importance for the Readership | Statement of Concrete Aims or Formulation of Questions | Description of the Literature Search | Referencing | Scientific Reasoning | Appropriate Presentation of Data | Total Score |
|---|---|---|---|---|---|---|---|---|
| 1 | Bianchi, F., Piccioli, C., Rosi, E., Carobene, L., Spina, D., Mazzei, M. A., Bartolucci, M., Moroni, C., Novelli, L., Rottoli, P., & Bargagli, E. (2019). Combined sarcoidosis and idiopathic pulmonary fibrosis (CSIPF): A novel disease phenotype? Respiratory medicine, 160, 105650. | 2 | 1 | 2 | 2 | 1 | 2 | 10 |
| 2 | Morgenthau A. S. (2018). Combined sarcoidosis and idiopathic pulmonary fibrosis (CSIPF): Genuine disease entity, obscure clinical phenotype or diagnostic red herring? Respiratory medicine, 144S, S3–S4. | 2 | 2 | 2 | 2 | 2 | 2 | 12 |
| 3 | Collins, B. F., McClelland, R. L., Ho, L. A., Mikacenic, C. R., Hayes, J., Spada, C., & Raghu, G. (2018). Sarcoidosis and IPF in the same patient-a coincidence, an association or a phenotype? Respiratory medicine, 144S, S20–S27. | 2 | 2 | 2 | 2 | 2 | 2 | 12 |
| 4 | Tachibana, K., Arai, T., Kagawa, T., Minomo, S., Akira, M., Kitaichi, M., & Inoue, Y. (2012). A case of combined sarcoidosis and usual interstitial pneumonia. Internal medicine (Tokyo, Japan), 51(14), 1893–1897 | 2 | 2 | 1 | 2 | 1 | 1 | 9 |
| 5 | Collins, B. F., & Raghu, G. (2019). Sarcoidosis and idiopathic pulmonary fibrosis: The same tale or a tale of two diseases in one. Respiratory medicine, 160, 105668. | 2 | 2 | 2 | 2 | 2 | 2 | 12 |
| 6 | Shigemitsu, H., & Azuma, A. (2011). Sarcoidosis and interstitial pulmonary fibrosis; two distinct disorders or two ends of the same spectrum. Current opinion in pulmonary medicine, 17(5), 303–307 | 2 | 1 | 1 | 2 | 2 | 2 | 8 |
| 7 | Patterson, K. C., & Strek, M. E. (2013). Pulmonary fibrosis in sarcoidosis. Clinical features and outcomes. Annals of the American Thoracic Society, 10(4), 362–370. | 1 | 2 | 1 | 1 | 1 | 2 | 8 |
| 8 | Teirstein, A. T., & Morgenthau, A. S. (2009). “End-stage” pulmonary fibrosis in sarcoidosis. The Mount Sinai journal of medicine, New York, 76(1), 30–36. | 2 | 1 | 1 | 1 | 1 | 2 | 8 |
| 9 | Xu, L., Kligerman, S., & Burke, A. (2013). End-stage sarcoid lung disease is distinct from usual interstitial pneumonia. The American journal of surgical pathology, 37(4), 593–600. | 2 | 1 | 1 | 1 | 1 | 2 | 8 |
| 10 | Nobata, K., Kasai, T., Fujimura, M., Mizuguchi, M., Nishi, K., Ishiura, Y., Yasui, M., & Nakao, S. (2006). Pulmonary sarcoidosis with usual interstitial pneumonia distributed predominantly in the lower lung fields. Internal medicine (Tokyo, Japan), 45(6), 359–362 | 2 | 2 | 1 | 1 | 1 | 2 | 9 |
| Characteristics | CSIPF (n = 9) | Lone-IPF (n = 19) | Stage 4 Sarcoidosis (n = 26) | p-Values |
|---|---|---|---|---|
| Sex (m/f) | 8/1 | (15/4) | (7/19) | 0.0001 |
| Caucasian (n) | 8 | 19 | 25 | ns |
| Smoking (current/never/former) | 0/2/7 | 0/4/15 | 1/16/9 | 0.01 |
| Age at sarcoidosis diagnosis (CSIPF diagnosis of granulomatous disease) (mean ± S.D) | 69.5 ± 8.7 | na | 42.8 ± 12.4 | 0.0001 |
| Age at IPF diagnosis (mean ± S.D) | 62.9 ± 10.4 | 72 ± 7.1 | na | 0.06 |
| Family history (n) | 7 | 2 | 0 | 0.0001 |
| Occupational exposure (yes/no) | 2/7 | 14/5 | 15/11 | 0.01 |
| Main comorbidities | na | |||
| ● GERD | 2 | 5 | 0 | |
| ● PH | 1 | 1 | 2 | |
| ● AH | 3 | 3 | 2 | |
| ● Infectious diseases | 2 | 1 | 3 | |
| ● Other lung disorders | 1 | 2 | 7 | |
| Extrapulmonary localization (yes/no) | 2/7 | na | 10/16 | 0.03 |
| Antifibrotic Therapy | na | na | ||
| ● Pirfenidone | 6 | 10 | ||
| ● Nintedanib | 3 | 9 | ||
| HRCT evidence of UIP | na | |||
| ● Consistent | 6 | 16 | 0 | |
| ● Inconsistent | 0 | 0 | 26 | |
| ● Possible | 2 | 3 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bergantini, L.; Nardelli, G.; d’Alessandro, M.; Montuori, G.; Piccioli, C.; Rosi, E.; Gangi, S.; Cavallaro, D.; Cameli, P.; Bargagli, E. Combined Sarcoidosis and Idiopathic Pulmonary Fibrosis (CSIPF): A New Phenotype or a Fortuitous Overlap? Scoping Review and Case Series. J. Clin. Med. 2022, 11, 2065. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm11072065
Bergantini L, Nardelli G, d’Alessandro M, Montuori G, Piccioli C, Rosi E, Gangi S, Cavallaro D, Cameli P, Bargagli E. Combined Sarcoidosis and Idiopathic Pulmonary Fibrosis (CSIPF): A New Phenotype or a Fortuitous Overlap? Scoping Review and Case Series. Journal of Clinical Medicine. 2022; 11(7):2065. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm11072065
Chicago/Turabian StyleBergantini, Laura, Gabriele Nardelli, Miriana d’Alessandro, Giusy Montuori, Caterina Piccioli, Elisabetta Rosi, Sara Gangi, Dalila Cavallaro, Paolo Cameli, and Elena Bargagli. 2022. "Combined Sarcoidosis and Idiopathic Pulmonary Fibrosis (CSIPF): A New Phenotype or a Fortuitous Overlap? Scoping Review and Case Series" Journal of Clinical Medicine 11, no. 7: 2065. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm11072065