Age-Related Macular Degeneration: A Disease of Systemic or Local Complement Dysregulation?

{kind=link}
Abstract
:1. Introduction
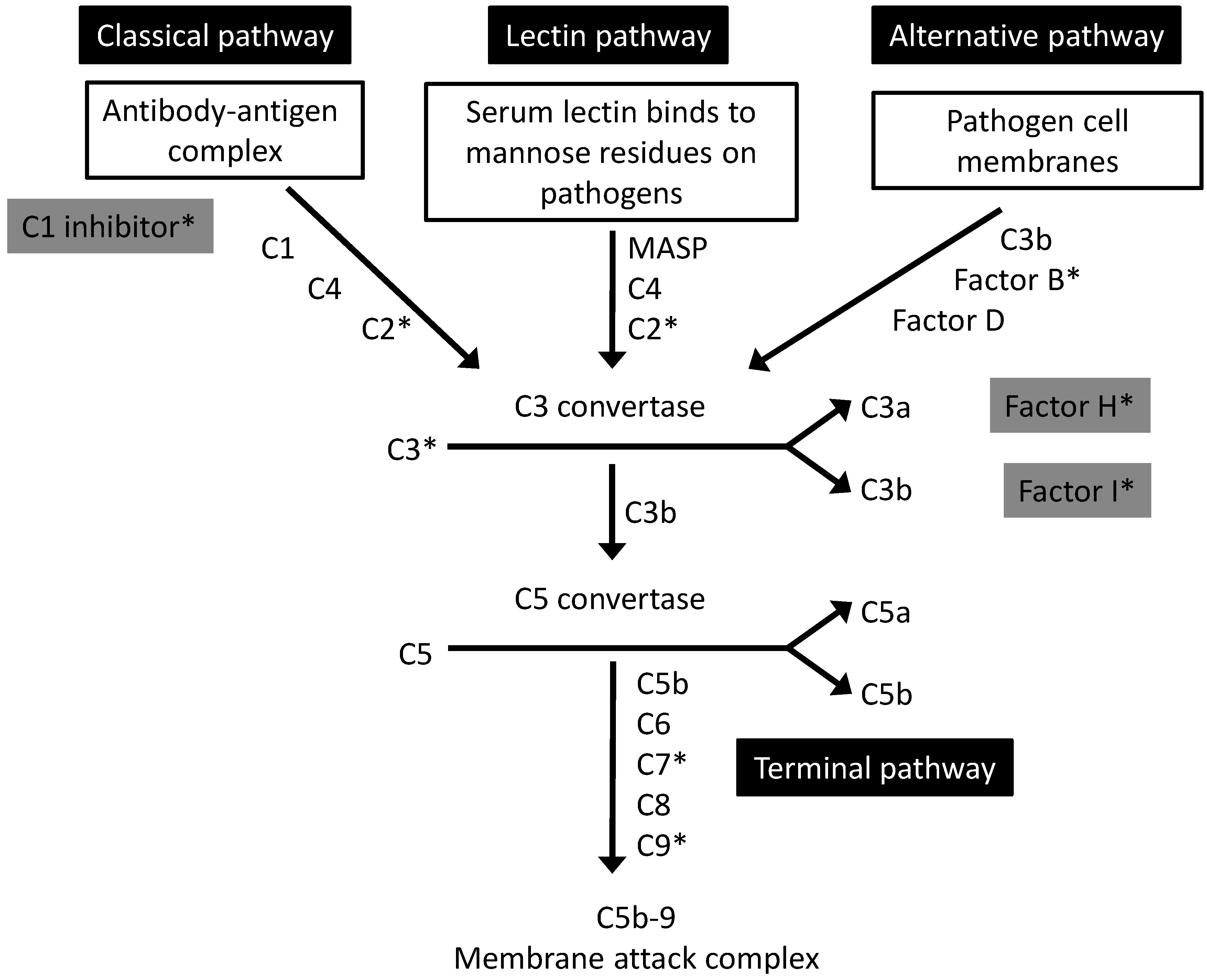
2. The Complement System: An Overview

3. Intraocular Complement and AMD
3.1. Complement Production in the Ageing Retina
3.2. Complement Production and Deposition in AMD
3.3. Complement Activation and AMD: Pathogenic Mechanisms
3.3.1. Functional Consequences of the CFH Y402H Genetic Variant
3.3.2. Potential Triggers for Complement Activation in AMD
3.3.2.1. Oxidative Stress
3.3.2.2. Pro-Inflammatory Agents Produced in the Retina
3.3.2.3. Amyloid Beta
4. Systemic Complement Activation and AMD
5. Systemic versus Local Manipulation of Complement
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Klein, R.; Peto, T.; Bird, A.; Vannewkirk, M.R. The epidemiology of age-related macular degeneration. Am. J. Ophthalmol. 2004, 137, 486–495. [Google Scholar] [CrossRef]
- Khandhadia, S.; Lotery, A. Oxidation and age-related macular degeneration: Insights from molecular biology. Expert. Rev. Mol. Med. 2010, 12. [Google Scholar] [CrossRef]
- Anderson, D.H.; Radeke, M.J.; Gallo, N.B.; Chapin, E.A.; Johnson, P.T.; Curletti, C.R.; Hancox, L.S.; Hu, J.; Elbright, J.N.; Malek, G.; et al. The pivotal role of the complement system in aging and age-related macular degeneration: Hypothesis re-visited. Prog. Retin. Eye Res. 2010, 29, 95–112. [Google Scholar] [CrossRef]
- Hageman, G.S.; Luthert, P.J.; Victor Chong, N.H.; Johnson, L.V.; Anderson, D.H.; Mullins, R.F. An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE-Bruch’s membrane interface in aging and age-related macular degeneration. Prog. Retin. Eye Res. 2001, 20, 705–732. [Google Scholar] [CrossRef]
- Johnson, L.V.; Leitner, W.P.; Staples, M.K.; Anderson, D.H. Complement activation and inflammatory processes in Drusen formation and age related macular degeneration. Exp. Eye Res. 2001, 73, 887–896. [Google Scholar] [CrossRef]
- Mullins, R.F.; Aptsiauri, N.; Hageman, G.S. Structure and composition of drusen associated with glomerulonephritis: Implications for the role of complement activation in drusen biogenesis. Eye (Lond) 2001, 15, 390–395. [Google Scholar] [CrossRef]
- Crabb, J.W.; Miyagi, M.; Gu, X.; Shadrach, K.; West, K.A.; Sakaguchi, H.; Kamei, M.; Hasan, A.; Yan, L.; Rayborn, M.E.; et al. Drusen proteome analysis: An approach to the etiology of age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2002, 99, 14682–14687. [Google Scholar] [CrossRef]
- Mullins, R.F.; Russell, S.R.; Anderson, D.H.; Hageman, G.S. Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. FASEB J. 2000, 14, 835–846. [Google Scholar]
- Hageman, G.S.; Anderson, D.H.; Johnson, L.V.; Hancox, L.S.; Taiber, A.J.; Hardisty, L.I.; Hageman, J.L.; Stockman, H.A.; Borchardt, J.D.; Gehrs, K.M.; et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2005, 102, 7227–7232. [Google Scholar] [CrossRef]
- Edwards, A.O.; Ritter, R., III; Abel, K.J.; Manning, A.; Panhuysen, C.; Farrer, L.A. Complement factor H polymorphism and age-related macular degeneration. Science 2005, 308, 421–424. [Google Scholar] [CrossRef]
- Haines, J.L.; Hauser, M.A.; Schmidt, S.; Scott, W.K.; Olson, L.M.; Gallins, P.; Spencer, K.L.; Kwan, S.Y.; Noureddine, M.; Gilbert, J.R.; et al. Complement factor H variant increases the risk of age-related macular degeneration. Science 2005, 308, 419–421. [Google Scholar] [CrossRef]
- Klein, R.J.; Zeiss, C.; Chew, E.Y.; Tsai, J.Y.; Sackler, R.S.; Haynes, C.; Henning, A.K.; SanGiovanni, J.P.; Mane, S.M.; Mayne, S.T.; et al. Complement factor H polymorphism in age-related macular degeneration. Science 2005, 308, 385–389. [Google Scholar] [CrossRef]
- Gold, B.; Merriam, J.E.; Zernant, J.; Hancox, L.S.; Taiber, A.J.; Gehrs, K.; Cramer, K.; Neel, J.; Bergeron, J.; Barile, G.R.; et al. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat. Genet. 2006, 38, 458–462. [Google Scholar] [CrossRef]
- Yates, J.R.; Sepp, T.; Matharu, B.K.; Khan, J.C.; Thurlby, D.A.; Shahid, H.; Clayton, D.G.; Hayward, C.; Morgan, J.; Wright, A.F.; et al. Complement C3 variant and the risk of age-related macular degeneration. N. Engl. J. Med. 2007, 357, 553–561. [Google Scholar]
- Dinu, V.; Miller, P.L.; Zhao, H. Evidence for association between multiple complement pathway genes and AMD. Genet. Epidemiol. 2007, 31, 224–237. [Google Scholar] [CrossRef]
- Seddon, J.M.; Yu, Y.; Miller, E.C.; Reynolds, R.; Tan, P.L.; Gowrisankar, S.; Goldstein, J.I.; Triebwasser, M.; Anderson, H.E.; Zerbib, J.; et al. Rare variants in CFI, C3 and C9 are associated with high risk of advanced age-related macular degeneration. Nat. Genet. 2013, 45, 1366–1370. [Google Scholar] [CrossRef]
- Fagerness, J.A.; Maller, J.B.; Neale, B.M.; Reynolds, R.C.; Daly, M.J.; Seddon, J.M. Variation near complement factor I is associated with risk of advanced AMD. Eur. J. Hum. Genet. 2009, 17, 100–104. [Google Scholar] [CrossRef]
- Ennis, S.; Jomary, C.; Mullins, R.; Cree, A.; Chen, X.; Macleod, A.; Jones, S.; Collins, A.; Stone, E.; Lotery, A. Association between the SERPING1 gene and age-related macular degeneration: A two-stage case-control study. Lancet 2008, 372, 1828–1834. [Google Scholar] [CrossRef]
- Allikmets, R.; Dean, M.; Hageman, G.S.; Baird, P.N.; Klaver, C.C.; Bergen, A.A.; Weber, B.H.; International AMD Genetics Consortium. The SERPING1 gene and age-related macular degeneration. Lancet 2009, 374, 875–876. [Google Scholar] [CrossRef]
- Coffey, P.J.; Gias, C.; McDermott, C.J.; Lundh, P.; Pickering, M.C.; Sethi, C.; Bird, A.; Fitzke, F.W.; Maass, A.; Chen, L.L.; et al. Complement factor H deficiency in aged mice causes retinal abnormalities and visual dysfunction. Proc. Natl. Acad. Sci. USA 2007, 104, 16651–16656. [Google Scholar] [CrossRef]
- Bora, P.S.; Sohn, J.H.; Cruz, J.M.; Jha, P.; Nishihori, H.; Wang, Y.; Kaliappan, S.; Kaplan, H.J.; Bora, N.S. Role of complement and complement membrane attack complex in laser-induced choroidal neovascularization. J. Immunol. 2005, 174, 491–497. [Google Scholar] [CrossRef]
- Bora, N.S.; Kaliappan, S.; Jha, P.; Xu, Q.; Sohn, J.H.; Dhaulakhandi, D.B.; Kaplan, H.J.; Bora, P.S. Complement activation via alternative pathway is critical in the development of laser-induced choroidal neovascularization: Role of factor B and factor H. J. Immunol. 2006, 177, 1872–1878. [Google Scholar] [CrossRef]
- Sivaprasad, S.; Adewoyin, T.; Bailey, T.A.; Dandekar, S.S.; Jenkins, S.; Webster, A.R.; Chong, N.V. Estimation of systemic complement C3 activity in age-related macular degeneration. Arch. Ophthalmol. 2007, 125, 515–519. [Google Scholar] [CrossRef]
- Scholl, H.P.; Charbel, I.P.; Walier, M.; Janzer, S.; Pollok-Kopp, B.; Borncke, F.; Fritsche, L.G.; Chong, N.V.; Fimmers, R.; Wienker, T.; et al. Systemic complement activation in age-related macular degeneration. PLoS One 2008, 3, e2593. [Google Scholar] [CrossRef]
- Hecker, L.A.; Edwards, A.O.; Ryu, E.; Tosakulwong, N.; Baratz, K.H.; Brown, W.L.; Charbel Issa, P.; Scholl, H.P.; Pollok-Kopp, B.; Schmid-Kubista, K.E.; et al. Genetic control of the alternative pathway of complement in humans and age-related macular degeneration. Hum. Mol. Genet. 2010, 19, 209–215. [Google Scholar] [CrossRef]
- Reynolds, R.; Hartnett, M.E.; Atkinson, J.P.; Giclas, P.C.; Rosner, B.; Seddon, J.M. Plasma complement components and activation fragments: Associations with age-related macular degeneration genotypes and phenotypes. Invest Ophthalmol. Vis. Sci. 2009, 50, 5818–5827. [Google Scholar] [CrossRef]
- Stanton, C.M.; Yates, J.R.; den Hollander, A.I.; Seddon, J.M.; Swaroop, A.; Stambolian, D.; Fauser, S.; Hoyng, C.; Yu, Y.; Atsuhiro, K.; et al. Complement factor D in age-related macular degeneration. Invest Ophthalmol. Vis. Sci. 2011, 52, 8828–8834. [Google Scholar] [CrossRef]
- Smailhodzic, D.; Klaver, C.C.; Klevering, B.J.; Boon, C.J.; Groenewoud, J.M.; Kirchhof, B.; Daha, M.R.; den Hollander, A.I.; Hoyng, C.B. Risk alleles in CFH and ARMS2 are independently associated with systemic complement activation in age-related macular degeneration. Ophthalmology 2012, 119, 339–346. [Google Scholar] [CrossRef]
- Silva, A.S.; Teixeira, A.G.; Bavia, L.; Lin, F.; Velletri, R.; Belfort, R., Jr.; Isaac, L. Plasma levels of complement proteins from the alternative pathway in patients with age-related macular degeneration are independent of Complement Factor H Tyr(4)(0)(2)His polymorphism. Mol. Vis. 2012, 18, 2288–2299. [Google Scholar]
- Ristau, T.; Paun, C.; Ersoy, L.; Hahn, M.; Lechanteur, Y.; Hoyng, C.; de Jong, E.K.; Daha, M.R.; Kirchhof, B.; den Hollander, A.I.; et al. Impact of the common genetic associations of age-related macular degeneration upon systemic complement component C3d levels. PLoS One 2014, 9, e93459. [Google Scholar] [CrossRef] [Green Version]
- Machalinska, A.; Dziedziejko, V.; Mozolewska-Piotrowska, K.; Karczewicz, D.; Wiszniewska, B.; Machalinski, B. Elevated plasma levels of C3a complement compound in the exudative form of age-related macular degeneration. Ophthalmic Res. 2009, 42, 54–59. [Google Scholar] [CrossRef]
- Gibson, J.; Hakobyan, S.; Cree, A.J.; Collins, A.; Harris, C.L.; Ennis, S.; Morgan, B.P.; Lotery, A.J. Variation in complement component C1 inhibitor in age-related macular degeneration. Immunobiology 2012, 217, 251–255. [Google Scholar] [CrossRef]
- Khandhadia, S.; Cipriani, V.; Yates, J.R.; Lotery, A.J. Age-related macular degeneration and the complement system. Immunobiology 2012, 217, 127–146. [Google Scholar] [CrossRef]
- Dunkelberger, J.R.; Song, W.C. Complement and its role in innate and adaptive immune responses. Cell Res. 2010, 20, 34–50. [Google Scholar] [CrossRef]
- Walport, M.J. Complement. First of two parts. N. Engl. J. Med. 2001, 344, 1058–1066. [Google Scholar]
- Walport, M.J. Complement. Second of two parts. N. Engl. J. Med. 2001, 344, 1140–1144. [Google Scholar] [CrossRef]
- Manderson, A.P.; Botto, M.; Walport, M.J. The role of complement in the development of systemic lupus erythematosus. Annu. Rev. Immunol. 2004, 22, 431–456. [Google Scholar] [CrossRef]
- Kavanagh, D.; Goodship, T.H.; Richards, A. Atypical hemolytic uremic syndrome. Semin. Nephrol. 2013, 33, 508–530. [Google Scholar] [CrossRef]
- Barbour, T.D.; Pickering, M.C.; Terence, C.H. Dense deposit disease and C3 glomerulopathy. Semin. Nephrol. 2013, 33, 493–507. [Google Scholar] [CrossRef]
- Gallenga, C.E.; Parmeggiani, F.; Costagliola, C.; Sebastiani, A.; Gallenga, P.E. Inflammaging: Should this term be suitable for age related macular degeneration too? Inflamm. Res. 2014, 63, 105–107. [Google Scholar]
- Xu, H.; Chen, M.; Forrester, J.V. Para-inflammation in the aging retina. Prog. Retin. Eye Res. 2009, 28, 348–368. [Google Scholar] [CrossRef]
- Morgan, B.P.; Gasque, P. Extrahepatic complement biosynthesis: Where, when and why? Clin. Exp. Immunol. 1997, 107, 1–7. [Google Scholar] [CrossRef]
- Luo, C.; Chen, M.; Xu, H. Complement gene expression and regulation in mouse retina and retinal pigment epithelium/choroid. Mol. Vis. 2011, 17, 1588–1597. [Google Scholar]
- Laufer, J.; Katz, Y.; Passwell, J.H. Extrahepatic synthesis of complement proteins in inflammation. Mol. Immunol. 2001, 38, 221–229. [Google Scholar] [CrossRef]
- Yanamadala, V.; Friedlander, R.M. Complement in neuroprotection and neurodegeneration. Trends Mol. Med. 2010, 16, 69–76. [Google Scholar] [CrossRef]
- Yu, M.; Zou, W.; Peachey, N.S.; McIntyre, T.M.; Liu, J. A novel role of complement in retinal degeneration. Invest Ophthalmol. Vis. Sci. 2012, 53, 7684–7692. [Google Scholar] [CrossRef]
- Hoh, K.J.; Lenassi, E.; Malik, T.H.; Pickering, M.C.; Jeffery, G. Complement component C3 plays a critical role in protecting the aging retina in a murine model of age-related macular degeneration. Am. J. Pathol. 2013, 183, 480–492. [Google Scholar] [CrossRef]
- Seth, A.; Cui, J.; To, E.; Kwee, M.; Matsubara, J. Complement-associated deposits in the human retina. Invest Ophthalmol. Vis. Sci. 2008, 49, 743–750. [Google Scholar]
- Mandal, M.N.; Ayyagari, R. Complement factor H: Spatial and temporal expression and localization in the eye. Invest Ophthalmol. Vis. Sci. 2006, 47, 4091–4097. [Google Scholar] [CrossRef]
- Chen, H.; Liu, B.; Lukas, T.J.; Neufeld, A.H. The aged retinal pigment epithelium/choroid: A potential substratum for the pathogenesis of age-related macular degeneration. PLoS One 2008, 3, e2339. [Google Scholar] [CrossRef]
- Chen, M.; Muckersie, E.; Forrester, J.V.; Xu, H. Immune activation in retinal aging: A gene expression study. Invest Ophthalmol. Vis. Sci. 2010, 51, 5888–5896. [Google Scholar] [CrossRef]
- Faber, C.; Williams, J.; Juel, H.B.; Greenwood, J.; Nissen, M.H.; Moss, S.E. Complement factor H deficiency results in decreased neuroretinal expression of Cd59a in aged mice. Invest Ophthalmol. Vis. Sci. 2012, 53, 6324–6330. [Google Scholar] [CrossRef]
- Ma, W.; Cojocaru, R.; Gotoh, N.; Gieser, L.; Villasmil, R.; Cogliati, T.; Swaroop, A.; Wong, W.T. Gene expression changes in aging retinal microglia: Relationship to microglial support functions and regulation of activation. Neurobiol. Aging 2013, 34, 2310–2321. [Google Scholar] [CrossRef]
- Rutar, M.; Valter, K.; Natoli, R.; Provis, J.M. Synthesis and propagation of complement C3 by microglia/monocytes in the aging retina. PLoS One 2014, 9, e93343. [Google Scholar] [CrossRef]
- Lohr, H.R.; Kuntchithapautham, K.; Sharma, A.K.; Rohrer, B. Multiple, parallel cellular suicide mechanisms participate in photoreceptor cell death. Exp. Eye Res. 2006, 83, 380–389. [Google Scholar] [CrossRef]
- Rohrer, B.; Guo, Y.; Kunchithapautham, K.; Gilkeson, G.S. Eliminating complement factor D reduces photoreceptor susceptibility to light-induced damage. Invest Ophthalmol. Vis. Sci. 2007, 48, 5282–5289. [Google Scholar] [CrossRef]
- Rohrer, B.; Long, Q.; Coughlin, B.; Wilson, R.B.; Huang, Y.; Qiao, F.; Tang, P.H.; Kunchithapautham, K.; Gilkeson, G.S.; Tomlinson, S. A targeted inhibitor of the alternative complement pathway reduces angiogenesis in a mouse model of age-related macular degeneration. Invest Ophthalmol. Vis. Sci. 2009, 50, 3056–3064. [Google Scholar] [CrossRef]
- Lyzogubov, V.V.; Tytarenko, R.G.; Jha, P.; Liu, J.; Bora, N.S.; Bora, P.S. Role of ocular complement factor H in a murine model of choroidal neovascularization. Am. J. Pathol. 2010, 177, 1870–1880. [Google Scholar] [CrossRef]
- Bora, N.S.; Kaliappan, S.; Jha, P.; Xu, Q.; Sivasankar, B.; Harris, C.L.; Morgan, B.P.; Bora, P.S. CD59, a complement regulatory protein, controls choroidal neovascularization in a mouse model of wet-type age-related macular degeneration. J. Immunol. 2007, 178, 1783–1790. [Google Scholar] [CrossRef]
- Baudouin, C.; Peyman, G.A.; Fredj-Reygrobellet, D.; Gordon, W.C.; Lapalus, P.; Gastaud, P.; Bazan, N.G. Immunohistological study of subretinal membranes in age-related macular degeneration. Jpn. J. Ophthalmol. 1992, 36, 443–451. [Google Scholar]
- Lommatzsch, A.; Hermans, P.; Weber, B.; Pauleikhoff, D. Complement factor H variant Y402H and basal laminar deposits in exudative age-related macular degeneration. Graefes Arch. Clin. Exp. Ophthalmol. 2007, 245, 1713–1716. [Google Scholar] [CrossRef]
- Lommatzsch, A.; Hermans, P.; Muller, K.D.; Bornfeld, N.; Bird, A.C.; Pauleikhoff, D. Are low inflammatory reactions involved in exudative age-related macular degeneration? Morphological and immunhistochemical analysis of AMD associated with basal deposits. Graefes Arch. Clin. Exp. Ophthalmol. 2008, 246, 803–810. [Google Scholar] [CrossRef]
- Yuan, X.; Gu, X.; Crabb, J.S.; Yue, X.; Shadrach, K.; Hollyfield, J.G.; Crabb, J.W. Quantitative proteomics: Comparison of the macular Bruch membrane/choroid complex from age-related macular degeneration and normal eyes. Mol. Cell Proteomics 2010, 9, 1031–1046. [Google Scholar] [CrossRef]
- Loyet, K.M.; Deforge, L.E.; Katschke, K.J., Jr.; Diehl, L.; Graham, R.R.; Pao, L.; Sturgeon, L.; Lewin-Koh, S.C.; Hollyfield, J.G.; van Lookeren Campagne, M. Activation of the alternative complement pathway in vitreous is controlled by genetics in age-related macular degeneration. Invest Ophthalmol. Vis. Sci. 2012, 53, 6628–6637. [Google Scholar] [CrossRef]
- Mullins, R.F.; Dewald, A.D.; Streb, L.M.; Wang, K.; Kuehn, M.H.; Stone, E.M. Elevated membrane attack complex in human choroid with high risk complement factor H genotypes. Exp. Eye Res. 2011, 93, 565–567. [Google Scholar] [CrossRef]
- Mullins, R.F.; Faidley, E.A.; Daggett, H.T.; Jomary, C.; Lotery, A.J.; Stone, E.M. Localization of complement 1 inhibitor (C1INH/SERPING1) in human eyes with age-related macular degeneration. Exp. Eye Res. 2009, 89, 767–773. [Google Scholar] [CrossRef]
- Vogt, S.D.; Curcio, C.A.; Wang, L.; Li, C.M.; McGwin, G., Jr.; Medeiros, N.E.; Philp, N.J.; Kimble, J.A.; Read, R.W. Retinal pigment epithelial expression of complement regulator CD46 is altered early in the course of geographic atrophy. Exp. Eye Res. 2011, 93, 413–423. [Google Scholar] [CrossRef]
- Ebrahimi, K.B.; Fijalkowski, N.; Cano, M.; Handa, J.T. Decreased membrane complement regulators in the retinal pigmented epithelium contributes to age-related macular degeneration. J. Pathol. 2013, 229, 729–742. [Google Scholar] [CrossRef]
- Weismann, D.; Hartvigsen, K.; Lauer, N.; Bennett, K.L.; Scholl, H.P.; Charbel, I.P.; Cano, M.; Brandstätter, H.; Tsimikas, S.; Skerka, C.; et al. Complement factor H binds malondialdehyde epitopes and protects from oxidative stress. Nature 2011, 478, 76–81. [Google Scholar] [CrossRef]
- Johnson, P.T.; Betts, K.E.; Radeke, M.J.; Hageman, G.S.; Anderson, D.H.; Johnson, L.V. Individuals homozygous for the age-related macular degeneration risk-conferring variant of complement factor H have elevated levels of CRP in the choroid. Proc. Natl. Acad. Sci. USA 2006, 103, 17456–17461. [Google Scholar] [CrossRef]
- Shaw, P.X.; Zhang, L.; Zhang, M.; Du, H.; Zhao, L.; Lee, C.; Grob, S.; Lim, S.L.; Hughes, G.; Lee, J.; et al. Complement factor H genotypes impact risk of age-related macular degeneration by interaction with oxidized phospholipids. Proc. Natl. Acad. Sci. USA 2012, 109, 13757–13762. [Google Scholar] [CrossRef]
- Perkins, S.J.; Nan, R.; Li, K.; Khan, S.; Miller, A. Complement factor H-ligand interactions: Self-association, multivalency and dissociation constants. Immunobiology 2012, 217, 281–297. [Google Scholar] [CrossRef]
- Pangburn, M.K. Host recognition and target differentiation by factor H, a regulator of the alternative pathway of complement. Immunopharmacology 2000, 49, 149–157. [Google Scholar] [CrossRef]
- Zipfel, P.F.; Skerka, C.; Hellwage, J.; Jokiranta, S.T.; Meri, S.; Brade, V.; Kraiczy, P.; Noris, M.; Remuzzi, G. Factor H family proteins: On complement, microbes and human diseases. Biochem. Soc. Trans. 2002, 30, 971–978. [Google Scholar] [CrossRef]
- Montes, T.; Tortajada, A.; Morgan, B.P.; Rodriguez de, C.S.; Harris, C.L. Functional basis of protection against age-related macular degeneration conferred by a common polymorphism in complement factor B. Proc. Natl. Acad. Sci. USA 2009, 106, 4366–4371. [Google Scholar] [CrossRef]
- Tortajada, A.; Montes, T.; Martinez-Barricarte, R.; Morgan, B.P.; Harris, C.L.; de Cordoba, S.R. The disease-protective complement factor H allotypic variant Ile62 shows increased binding affinity for C3b and enhanced cofactor activity. Hum. Mol. Genet. 2009, 18, 3452–3461. [Google Scholar] [CrossRef]
- Heurich, M.; Martinez-Barricarte, R.; Francis, N.J.; Roberts, D.L.; Rodriguez de, C.S.; Morgan, B.P.; Harris, C.L. Common polymorphisms in C3, factor B, and factor H collaborate to determine systemic complement activity and disease risk. Proc. Natl. Acad. Sci. USA 2011, 108, 8761–8766. [Google Scholar] [CrossRef] [Green Version]
- Giannakis, E.; Jokiranta, T.S.; Male, D.A.; Ranganathan, S.; Ormsby, R.J.; Fischetti, V.A.; Mold, C.; Gordon, D.L. A common site within factor H SCR 7 responsible for binding heparin, C-reactive protein and streptococcal M protein. Eur. J. Immunol. 2003, 33, 962–969. [Google Scholar] [CrossRef]
- Clark, S.J.; Perveen, R.; Hakobyan, S.; Morgan, B.P.; Sim, R.B.; Bishop, P.N.; Day, A.J. Impaired binding of the age-related macular degeneration-associated complement factor H 402H allotype to Bruch’s membrane in human retina. J. Biol. Chem. 2010, 285, 30192–30202. [Google Scholar] [CrossRef]
- Kelly, U.; Yu, L.; Kumar, P.; Ding, J.D.; Jiang, H.; Hageman, G.S.; Arshavsky, V.Y.; Frank, M.M.; Hauser, M.A.; Rickman, C.B. Heparan sulfate, including that in Bruch’s membrane, inhibits the complement alternative pathway: Implications for age-related macular degeneration. J. Immunol. 2010, 185, 5486–5494. [Google Scholar] [CrossRef]
- Skerka, C.; Lauer, N.; Weinberger, A.A.; Keilhauer, C.N.; Suhnel, J.; Smith, R.; Schlotzer-Schrehardt, U.; Fritsche, L.; Heinen, S.; Hartmann, A.; et al. Defective complement control of factor H (Y402H) and FHL-1 in age-related macular degeneration. Mol. Immunol. 2007, 44, 3398–3406. [Google Scholar] [CrossRef]
- Lauer, N.; Mihlan, M.; Hartmann, A.; Schlotzer-Schrehardt, U.; Keilhauer, C.; Scholl, H.P.; Charbel Issa, P.; Holz, F.; Weber, B.H.; Skerka, C.; et al. Complement regulation at necrotic cell lesions is impaired by the age-related macular degeneration-associated factor-H His402 risk variant. J. Immunol. 2011, 187, 4374–4383. [Google Scholar] [CrossRef]
- Ormsby, R.J.; Ranganathan, S.; Tong, J.C.; Griggs, K.M.; Dimasi, D.P.; Hewitt, A.W.; Burdon, K.P.; Craig, J.E.; Hoh, J.; Gordon, D.L. Functional and structural implications of the complement factor H Y402H polymorphism associated with age-related macular degeneration. Invest Ophthalmol. Vis. Sci. 2008, 49, 1763–1770. [Google Scholar] [CrossRef]
- Laine, M.; Jarva, H.; Seitsonen, S.; Haapasalo, K.; Lehtinen, M.J.; Lindeman, N.; Anderson, D.H.; Johnson, P.T.; Jarvela, I.; Jokiranta, T.S.; et al. Y402H polymorphism of complement factor H affects binding affinity to C-reactive protein. J. Immunol. 2007, 178, 3831–3836. [Google Scholar] [CrossRef]
- Herbert, A.P.; Deakin, J.A.; Schmidt, C.Q.; Blaum, B.S.; Egan, C.; Ferreira, V.P.; Pangburn, M.K.; Lyon, M.; Uhrin, D.; Barlow, P.N. Structure shows that a glycosaminoglycan and protein recognition site in factor H is perturbed by age-related macular degeneration-linked single nucleotide polymorphism. J. Biol. Chem. 2007, 282, 18960–18968. [Google Scholar] [CrossRef]
- Sjoberg, A.P.; Trouw, L.A.; Clark, S.J.; Sjolander, J.; Heinegard, D.; Sim, R.B.; Day, A.J.; Blom, A.M. The factor H variant associated with age-related macular degeneration (His-384) and the non-disease-associated form bind differentially to C-reactive protein, fibromodulin, DNA, and necrotic cells. J. Biol. Chem. 2007, 282, 10894–10900. [Google Scholar] [CrossRef]
- Yu, J.; Wiita, P.; Kawaguchi, R.; Honda, J.; Jorgensen, A.; Zhang, K.; Fischetti, V.A.; Sun, H. Biochemical analysis of a common human polymorphism associated with age-related macular degeneration. Biochemistry 2007, 46, 8451–8461. [Google Scholar] [CrossRef]
- Okemefuna, A.I.; Nan, R.; Miller, A.; Gor, J.; Perkins, S.J. Complement factor H binds at two independent sites to C-reactive protein in acute phase concentrations. J. Biol. Chem. 2010, 285, 1053–1065. [Google Scholar] [CrossRef]
- Black, S.; Kushner, I.; Samols, D. C-reactive Protein. J. Biol. Chem. 2004, 279, 48487–48490. [Google Scholar] [CrossRef]
- Yeh, E.T. CRP as a mediator of disease. Circulation 2004, 109. [Google Scholar] [CrossRef]
- Hong, T.; Tan, A.G.; Mitchell, P.; Wang, J.J. A review and meta-analysis of the association between C-reactive protein and age-related macular degeneration. Surv. Ophthalmol. 2011, 56, 184–194. [Google Scholar] [CrossRef]
- Seddon, J.M.; George, S.; Rosner, B.; Rifai, N. Progression of age-related macular degeneration: Prospective assessment of C-reactive protein, interleukin 6, and other cardiovascular biomarkers. Arch. Ophthalmol. 2005, 123, 774–782. [Google Scholar]
- Robman, L.; Baird, P.N.; Dimitrov, P.N.; Richardson, A.J.; Guymer, R.H. C-reactive protein levels and complement factor H polymorphism interaction in age-related macular degeneration and its progression. Ophthalmology 2010, 117, 1982–1988. [Google Scholar] [CrossRef]
- Bhutto, I.A.; Baba, T.; Merges, C.; Juriasinghani, V.; McLeod, D.S.; Lutty, G.A. C-reactive protein and complement factor H in aged human eyes and eyes with age-related macular degeneration. Br. J. Ophthalmol. 2011, 95, 1323–1330. [Google Scholar] [CrossRef]
- McLeod, D.S.; Grebe, R.; Bhutto, I.; Merges, C.; Baba, T.; Lutty, G.A. Relationship between RPE and choriocapillaris in age-related macular degeneration. Invest Ophthalmol. Vis. Sci. 2009, 50, 4982–4991. [Google Scholar] [CrossRef]
- Suzuki, M.; Kamei, M.; Itabe, H.; Yoneda, K.; Bando, H.; Kume, N.; Tano, Y. Oxidized phospholipids in the macula increase with age and in eyes with age-related macular degeneration. Mol. Vis. 2007, 13, 772–778. [Google Scholar]
- Wu, Z.; Lauer, T.W.; Sick, A.; Hackett, S.F.; Campochiaro, P.A. Oxidative stress modulates complement factor H expression in retinal pigmented epithelial cells by acetylation of FOXO3. J. Biol. Chem. 2007, 282, 22414–22425. [Google Scholar] [CrossRef]
- Lau, L.I.; Chiou, S.H.; Liu, C.J.; Yen, M.Y.; Wei, Y.H. The effect of photo-oxidative stress and inflammatory cytokine on complement factor H expression in retinal pigment epithelial cells. Invest Ophthalmol. Vis. Sci. 2011, 52, 6832–6841. [Google Scholar] [CrossRef]
- Ebrahimi, K.B.; Fijalkowski, N.; Cano, M.; Handa, J.T. Oxidized Low-Density-Lipoprotein-Induced Injury in Retinal Pigment Epithelium Alters Expression of the Membrane Complement Regulatory Factors CD46 and CD59 through Exosomal and Apoptotic Bleb Release. Adv. Exp. Med. Biol. 2014, 801, 259–265. [Google Scholar]
- Thurman, J.M.; Renner, B.; Kunchithapautham, K.; Ferreira, V.P.; Pangburn, M.K.; Ablonczy, Z.; Tomlinson, S.; Holers, V.M.; Rohrer, B. Oxidative stress renders retinal pigment epithelial cells susceptible to complement-mediated injury. J. Biol. Chem. 2009, 284, 16939–16947. [Google Scholar] [CrossRef]
- Bandyopadhyay, M.; Rohrer, B. Matrix metalloproteinase activity creates pro-angiogenic environment in primary human retinal pigment epithelial cells exposed to complement. Invest Ophthalmol. Vis. Sci. 2012, 53, 1953–1961. [Google Scholar] [CrossRef]
- Nozaki, M.; Raisler, B.J.; Sakurai, E.; Sarma, J.V.; Barnum, S.R.; Lambris, J.D.; Chen, Y.; Zhang, K.; Ambati, B.K.; Baffi, J.Z.; et al. Drusen complement components C3a and C5a promote choroidal neovascularization. Proc. Natl. Acad. Sci. USA 2006, 103, 2328–2333. [Google Scholar] [CrossRef]
- Bora, N.S.; Jha, P.; Lyzogubov, V.V.; Kaliappan, S.; Liu, J.; Tytarenko, R.G.; Fraser, D.A.; Morgan, B.P.; Bora, P.S. Recombinant membrane-targeted form of CD59 inhibits the growth of choroidal neovascular complex in mice. J. Biol. Chem. 2010, 285, 33826–33833. [Google Scholar] [CrossRef]
- Liu, J.; Jha, P.; Lyzogubov, V.V.; Tytarenko, R.G.; Bora, N.S.; Bora, P.S. Relationship between complement membrane attack complex, chemokine (C-C motif) ligand 2 (CCL2) and vascular endothelial growth factor in mouse model of laser-induced choroidal neovascularization. J. Biol. Chem. 2011, 286, 20991–21001. [Google Scholar] [CrossRef]
- Rohrer, B.; Coughlin, B.; Bandyopadhyay, M.; Holers, V.M. Systemic human CR2-targeted complement alternative pathway inhibitor ameliorates mouse laser-induced choroidal neovascularization. J. Ocul. Pharmacol. Ther. 2012, 28, 402–409. [Google Scholar] [CrossRef]
- Rohrer, B.; Coughlin, B.; Kunchithapautham, K.; Long, Q.; Tomlinson, S.; Takahashi, K.; Holers, V.M. The alternative pathway is required, but not alone sufficient, for retinal pathology in mouse laser-induced choroidal neovascularization. Mol. Immunol. 2011, 48, e1–e8. [Google Scholar] [CrossRef]
- Smith, W.; Assink, J.; Klein, R.; Mitchell, P.; Klaver, C.C.; Klein, B.E.; Hofman, A.; Jensen, S.; Wang, J.J.; de Jong, P.T. Risk factors for age-related macular degeneration: Pooled findings from three continents. Ophthalmology 2001, 108, 697–704. [Google Scholar] [CrossRef]
- Wang, A.L.; Lukas, T.J.; Yuan, M.; Du, N.; Handa, J.T.; Neufeld, A.H. Changes in retinal pigment epithelium related to cigarette smoke: Possible relevance to smoking as a risk factor for age-related macular degeneration. PLoS One 2009, 4, e5304. [Google Scholar]
- Woodell, A.; Coughlin, B.; Kunchithapautham, K.; Casey, S.; Williamson, T.; Ferrell, W.D.; Atkinson, C.; Jones, B.W.; Rohrer, B. Alternative complement pathway deficiency ameliorates chronic smoke-induced functional and morphological ocular injury. PLoS One 2013, 8, e67894. [Google Scholar] [CrossRef]
- Wang, L.; Kondo, N.; Cano, M.; Ebrahimi, K.; Yoshida, T.; Barnett, B.P.; Biswal, S.; Handa, J.T. Nrf2 signaling modulates cigarette smoke-induced complement activation in retinal pigmented epithelial cells. Free Radic. Biol. Med. 2014, 70, 155–166. [Google Scholar] [CrossRef]
- Kunchithapautham, K.; Atkinson, C.; Rohrer, B. Smoke-exposure causes endoplasmic reticulum stress and lipid accumulation in retinal pigment epithelium through oxidative stress and complement activation. J. Biol. Chem. 2014. [Google Scholar] [CrossRef]
- Zhou, J.; Jang, Y.P.; Kim, S.R.; Sparrow, J.R. Complement activation by photooxidation products of A2E, a lipofuscin constituent of the retinal pigment epithelium. Proc. Natl. Acad. Sci. USA 2006, 103, 16182–16187. [Google Scholar] [CrossRef]
- Zhou, J.; Kim, S.R.; Westlund, B.S.; Sparrow, J.R. Complement activation by bisretinoid constituents of RPE lipofuscin. Invest Ophthalmol. Vis. Sci. 2009, 50, 1392–1399. [Google Scholar] [CrossRef]
- Sparrow, J.R.; Ueda, K.; Zhou, J. Complement dysregulation in AMD: RPE-Bruch’s membrane-choroid. Mol. Aspects Med. 2012, 33, 436–445. [Google Scholar] [CrossRef]
- Bian, Q.; Gao, S.; Zhou, J.; Qin, J.; Taylor, A.; Johnson, E.J.; Tang, G.; Sparrow, J.R.; Gierhart, D.; Shang, F. Lutein and zeaxanthin supplementation reduces photooxidative damage and modulates the expression of inflammation-related genes in retinal pigment epithelial cells. Free Radic. Biol. Med. 2012, 53, 1298–1307. [Google Scholar] [CrossRef]
- Ma, W.; Coon, S.; Zhao, L.; Fariss, R.N.; Wong, W.T. A2E accumulation influences retinal microglial activation and complement regulation. Neurobiol. Aging 2013, 34, 943–960. [Google Scholar] [CrossRef]
- Chen, M.; Forrester, J.V.; Xu, H. Synthesis of complement factor H by retinal pigment epithelial cells is down-regulated by oxidized photoreceptor outer segments. Exp. Eye Res. 2007, 84, 635–645. [Google Scholar] [CrossRef]
- Radu, R.A.; Hu, J.; Jiang, Z.; Bok, D. Bisretinoid-mediated complement activation on retinal pigment epithelial cells is dependent on complement factor H haplotype. J. Biol. Chem. 2014, 289, 9113–9120. [Google Scholar] [CrossRef]
- Berchuck, J.E.; Yang, P.; Toimil, B.A.; Ma, Z.; Baciu, P.; Jaffe, G.J. All-trans-retinal sensitizes human RPE cells to alternative complement pathway-induced cell death. Invest Ophthalmol. Vis. Sci. 2013, 54, 2669–2677. [Google Scholar] [CrossRef]
- Hollyfield, J.G.; Bonilha, V.L.; Rayborn, M.E.; Yang, X.; Shadrach, K.G.; Lu, L.; Ufret, R.L.; Salomon, R.G.; Perez, V.L. Oxidative damage-induced inflammation initiates age-related macular degeneration. Nat. Med. 2008, 14, 194–198. [Google Scholar] [CrossRef]
- Isas, J.M.; Luibl, V.; Johnson, L.V.; Kayed, R.; Wetzel, R.; Glabe, C.G.; Langen, R.; Chen, J. Soluble and mature amyloid fibrils in drusen deposits. Invest Ophthalmol. Vis. Sci. 2010, 51, 1304–1310. [Google Scholar] [CrossRef]
- Johnson, L.V.; Leitner, W.P.; Rivest, A.J.; Staples, M.K.; Radeke, M.J.; Anderson, D.H. The Alzheimer’s A beta -peptide is deposited at sites of complement activation in pathologic deposits associated with aging and age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2002, 99, 11830–11835. [Google Scholar] [CrossRef]
- Wang, J.; Ohno-Matsui, K.; Yoshida, T.; Kojima, A.; Shimada, N.; Nakahama, K.; Safranova, O.; Iwata, N.; Saido, T.C.; Mochizuki, M.; et al. Altered function of factor I caused by amyloid beta: Implication for pathogenesis of age-related macular degeneration from Drusen. J. Immunol. 2008, 181, 712–720. [Google Scholar] [CrossRef]
- Wang, J.; Ohno-Matsui, K.; Yoshida, T.; Shimada, N.; Ichinose, S.; Sato, T.; Mochizuki, M.; Morita, I. Amyloid-beta up-regulates complement factor B in retinal pigment epithelial cells through cytokines released from recruited macrophages/microglia: Another mechanism of complement activation in age-related macular degeneration. J. Cell Physiol. 2009, 220, 119–128. [Google Scholar] [CrossRef]
- Luo, C.; Zhao, J.; Madden, A.; Chen, M.; Xu, H. Complement expression in retinal pigment epithelial cells is modulated by activated macrophages. Exp. Eye Res. 2013, 112, 93–101. [Google Scholar] [CrossRef]
- Catchpole, I.; Germaschewski, V.; Hoh, K.J.; Lundh von, L.P.; Ford, S.; Gough, G.; Adamson, P.; Overend, P.; Hilpert, J.; Lopez, F.J.; et al. Systemic administration of Abeta mAb reduces retinal deposition of Abeta and activated complement C3 in age-related macular degeneration mouse model. PLoS One 2013, 8, e65518. [Google Scholar] [CrossRef]
- Hakobyan, S.; Harris, C.L.; Tortajada, A.; Goicochea de, J.E.; Garcia-Layana, A.; Fernandez-Robredo, P.; Rodriguez de Cordoba, S.; Morgan, B.P. Measurement of factor H variants in plasma using variant-specific monoclonal antibodies: Application to assessing risk of age-related macular degeneration. Invest Ophthalmol. Vis. Sci. 2008, 49, 1983–1990. [Google Scholar] [CrossRef]
- Lipo, E.; Cashman, S.M.; Kumar-Singh, R. Aurintricarboxylic acid inhibits complement activation, membrane attack complex, and choroidal neovascularization in a model of macular degeneration. Invest Ophthalmol. Vis. Sci. 2013, 54, 7107–7114. [Google Scholar] [CrossRef]
- Birke, M.T.; Lipo, E.; Adhi, M.; Birke, K.; Kumar-Singh, R. AAV-mediated expression of human PRELP inhibits complement activation, choroidal neovascularization and deposition of membrane attack complex in mice. Gene Ther. 2014, 21, 507–513. [Google Scholar] [CrossRef]
- Cashman, S.M.; Ramo, K.; Kumar-Singh, R. A non membrane-targeted human soluble CD59 attenuates choroidal neovascularization in a model of age related macular degeneration. PLoS One 2011, 6, e19078. [Google Scholar] [CrossRef]
- Kim, S.J.; Kim, J.; Lee, J.; Cho, S.Y.; Kang, H.J.; Kim, K.Y.; Jin, D.K. Intravitreal human complement factor H in a rat model of laser-induced choroidal neovascularisation. Br. J. Ophthalmol. 2013, 97, 367–370. [Google Scholar] [CrossRef]
- Weber, B.H.; Charbel, I.P.; Pauly, D.; Herrmann, P.; Grassmann, F.; Holz, F.G. The role of the complement system in age-related macular degeneration. Dtsch. Arztebl. Int. 2014, 111, 133–138. [Google Scholar]
- Hetwick, C.C. MAHALO Finds New Biomarker for Dry Macular Degeneration, Medscape Medical News 21 November 2013. Available online: http://www.medscape.com/viewarticle/814822 (accessed on 1 August 2014).
- Hoffmann-La Roche. A Study Investigating the Safety and Efficacy of Lampalizumab Intravitreal Injections in Patients with Geographic Atrophy Secondary to Age-Related Macular Degeneration (CHROMA). Available online: https://clinicaltrials.gov/ct2/show/NCT02247479 (accessed on 19 October 2014).
- Hoffmann-La Roche. A Study Investigating the Safety and Efficacy of Lampalizumab Intravitreal Injections in Patients with Geographic Atrophy Secondary to Age-Related Macular Degeneration (SPECTRI). Available online: https://clinicaltrials.gov/ct2/show/NCT02247531 (accessed on 19 October 2014).
- Yehoshua, Z.; de Amorim Garcia Filho, C.A.; Nunes, R.P.; Gregori, G.; Penha, F.M.; Moshfeghi, A.A.; Zhang, K.; Sadda, S.; Feuer, W.; Rosenfeld, P.J. Systemic complement inhibition with eculizumab for geographic atrophy in age-related macular degeneration: The COMPLETE study. Ophthalmology 2014, 121, 693–701. [Google Scholar] [CrossRef]
- Moore, D.J.; Clover, G.M. The effect of age on the macromolecular permeability of human Bruch’s membrane. Invest. Ophthalmol. Vis. Sci. 2001, 42, 2970–2975. [Google Scholar]
- D’souza, Y.B.; Jones, C.J.; Short, C.D.; Roberts, I.S.; Bonshek, R.E. Oligosaccharide composition is similar in drusen and dense deposits in membranoproliferative glomerulonephritis type II. Kidney Int. 2009, 75, 824–827. [Google Scholar] [CrossRef]
- McAvoy, C.E.; Silvestri, G. Retinal changes associated with type 2 glomerulonephritis. Eye (Lond) 2005, 19, 985–989. [Google Scholar] [CrossRef]
- Bomback, A.S.; Smith, R.J.; Barile, G.R.; Zhang, Y.; Heher, E.C.; Herlitz, L.; Stokes, M.B.; Markowitz, G.S.; D’Agati, V.D.; Canetta, P.A.; et al. Eculizumab for dense deposit disease and C3 glomerulonephritis. Clin. J. Am. Soc. Nephrol. 2012, 7, 748–756. [Google Scholar] [CrossRef]
- Noris, M.; Caprioli, J.; Bresin, E.; Mossali, C.; Pianetti, G.; Gamba, S.; Daina, E.; Fenili, C.; Castelletti, F.; Sorosina, A.; et al. Relative role of genetic complement abnormalities in sporadic and familial aHUS and their impact on clinical phenotype. Clin. J. Am. Soc. Nephrol. 2010, 5, 1844–1859. [Google Scholar] [CrossRef]
- Geerdink, L.M.; Westra, D.; van Wijk, J.A.; Dorresteijn, E.M.; Lilien, M.R.; Davin, J.C.; Komhoff, M.; Van Hoeck, K.; van der Vlugt, A.; van den Heuvel, L.P.; et al. Atypical hemolytic uremic syndrome in children: Complement mutations and clinical characteristics. Pediatr. Nephrol. 2012, 27, 1283–1291. [Google Scholar] [CrossRef]
- Saland, J.M.; Ruggenenti, P.; Remuzzi, G. Liver-kidney transplantation to cure atypical hemolytic uremic syndrome. J. Am. Soc. Nephrol. 2009, 20, 940–949. [Google Scholar] [CrossRef]
- Brown, J.H.; Tellez, J.; Wilson, V.; Mackie, I.J.; Scully, M.; Tredger, M.M.; Moore, I.; McDougall, N.I.; Strain, L.; Marchbank, K.J.; et al. Postpartum aHUS secondary to a genetic abnormality in factor H acquired through liver transplantation. Am. J. Transplant. 2012, 12, 1632–1636. [Google Scholar] [CrossRef]
- Khandhadia, S.; Hakobyan, S.; Heng, L.Z.; Gibson, J.; Adams, D.H.; Alexander, G.J.; Gibson, J.M.; Martin, K.R.; Menon, G.; Nask, K.; et al. Age-related macular degeneration and modification of systemic complement factor H production through liver transplantation. Ophthalmology 2013, 120, 1612–1618. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Warwick, A.; Khandhadia, S.; Ennis, S.; Lotery, A. Age-Related Macular Degeneration: A Disease of Systemic or Local Complement Dysregulation? J. Clin. Med. 2014, 3, 1234-1257. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm3041234
Warwick A, Khandhadia S, Ennis S, Lotery A. Age-Related Macular Degeneration: A Disease of Systemic or Local Complement Dysregulation? Journal of Clinical Medicine. 2014; 3(4):1234-1257. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm3041234
Chicago/Turabian StyleWarwick, Alasdair, Samir Khandhadia, Sarah Ennis, and Andrew Lotery. 2014. "Age-Related Macular Degeneration: A Disease of Systemic or Local Complement Dysregulation?" Journal of Clinical Medicine 3, no. 4: 1234-1257. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm3041234