Inflammation and Cell Death in Age-Related Macular Degeneration: An Immunopathological and Ultrastructural Model

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Innate Immunity in AMD
2.1. Macrophage Phenotypes and Polarization
2.2. Microglia
2.3. Complement Involvement
2.4. Pattern Recognition Receptors (PRRs)
2.5. NLRs: The NLRP3 Inflammasome

3. IL-17 in AMD
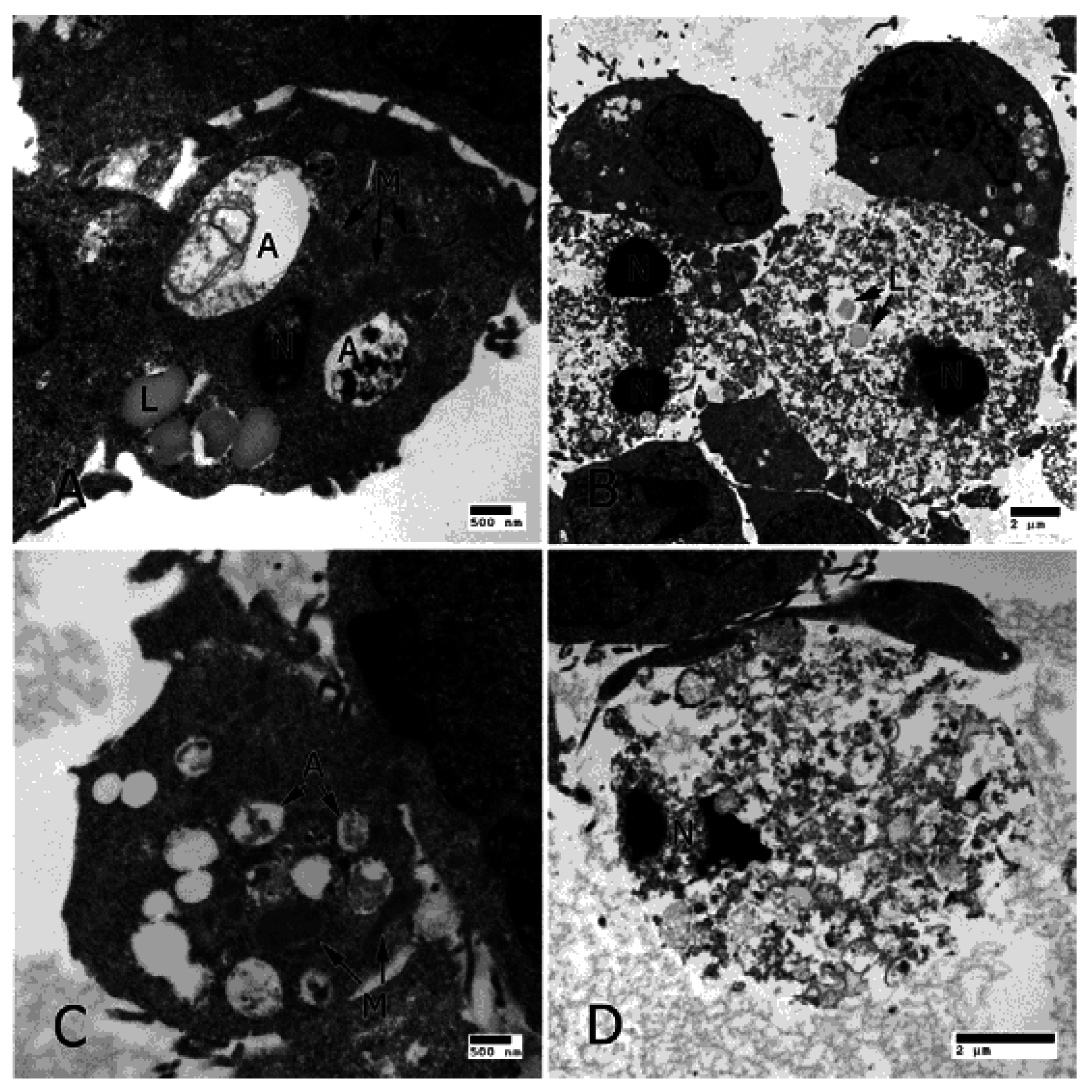
4. Apoptosis, Pyroptosis, Necroptosis and Autophagy in AMD


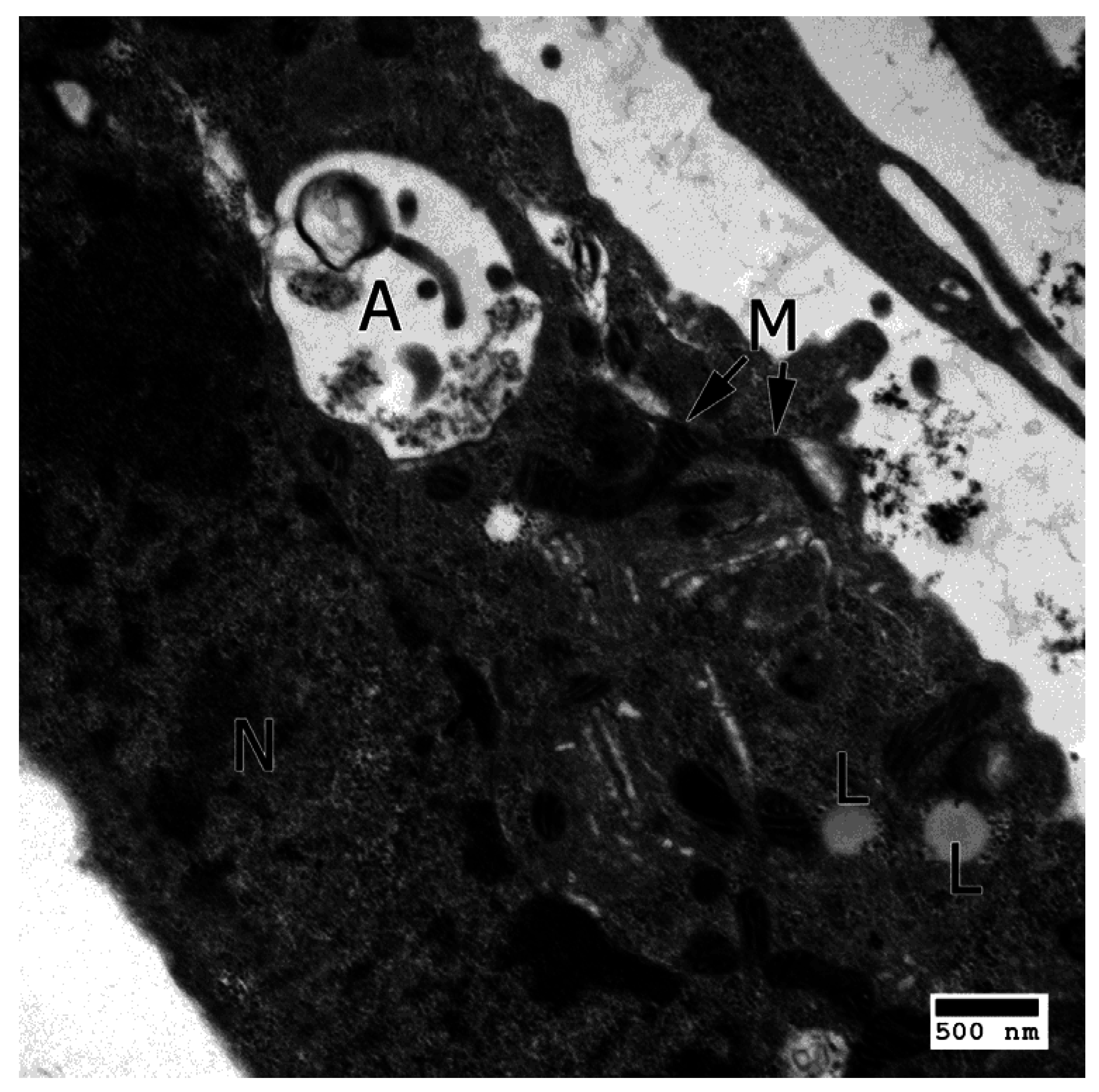
4.1. Apoptosis
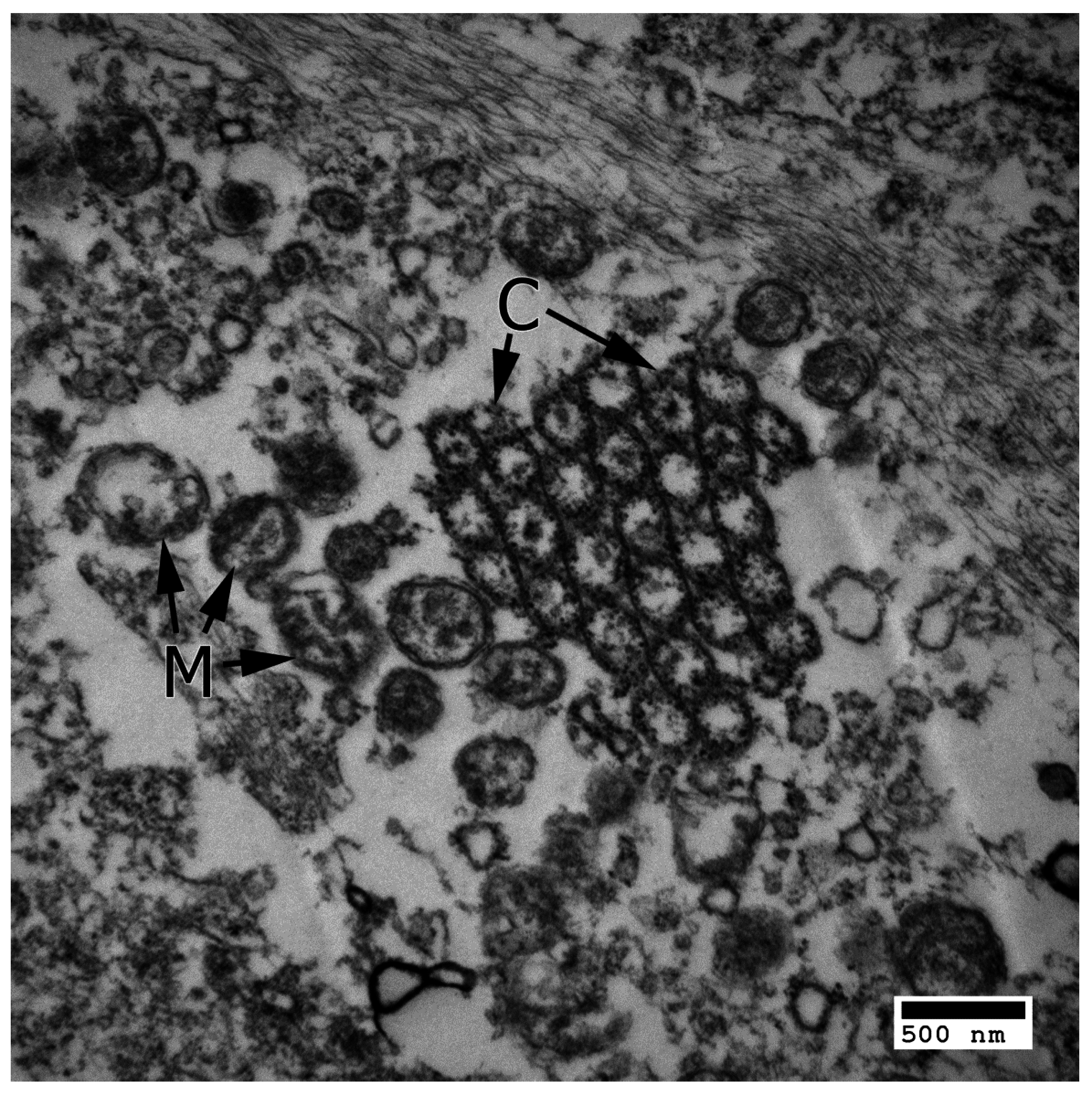
4.2. Pyroptosis
4.3. Necroptosis
4.4. Autophagy
5. Conclusions
Acknowledgment
Author Contributions
Conflict of Interest
References
- Congdon, N.; O’Colmain, B.; Klaver, C.C.W.; Klein, R.; Munoz, B.; Friedman, D.S.; Kempen, J.; Taylor, H.R.; Mitchell, P.; Hyman, L.; et al. Causes and prevalence of visual impairment among adults in the United States. Arch. Ophthalmol. Chic 2004, 122, 477–485. [Google Scholar] [CrossRef]
- Pascolini, D.; Mariotti, S.P. Global estimates of visual impairment: 2010. Br. J. Ophthalmol. 2012, 96, 614–618. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.; Chou, C.F.; Klein, B.E.; Zhang, X.; Meuer, S.M.; Saaddine, J.B. Prevalence of age-related macular degeneration in the US population. Arch. Ophthalmol. 2011, 129, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Griffith, T.S.; Brunner, T.; Fletcher, S.M.; Green, D.R.; Ferguson, T.A. Fas ligand-induced apoptosis as a mechanism of immune privilege. Science 1995, 270, 1189–1192. [Google Scholar] [CrossRef] [PubMed]
- Zamiri, P.; Sugita, S.; Streilein, J.W. Immunosuppressive properties of the pigmented epithelial cells and the subretinal space. Chem. Immunol. Allergy 2007, 92, 86–93. [Google Scholar] [PubMed]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Mosser, D.M. Macrophage activation by endogenous danger signals. J. Pathol. 2008, 214, 161–178. [Google Scholar] [CrossRef]
- Janeway, C. Immunobiology: The Immune System in Health and Disease, 8th ed.; Garland Science: New York, NY, USA, 2011; pp. 85–94. [Google Scholar]
- Erwig, L.P.; Henson, P.M. Immunological consequences of apoptotic cell phagocytosis. Am. J. Pathol. 2007, 171, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Sarks, S.H.; Arnold, J.J.; Killingsworth, M.C.; Sarks, J.P. Early drusen formation in the normal and aging eye and their relation to age related maculopathy: A clinicopathological study. Br. J. Ophthalmol. 1999, 83, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Penfold, P.L.; Killingsworth, M.C.; Sarks, S.H. Senile macular degeneration: The involvement of immunocompetent cells. Graefe’s Arch. Clin. Exp. Ophthalmol. 1985, 223, 69–76. [Google Scholar] [CrossRef]
- Dastgheib, K.; Green, W.R. Granulomatous reaction to bruch’s membrane in age-related macular degeneration. Arch. Ophthalmol. 1994, 112, 813–818. [Google Scholar] [CrossRef] [PubMed]
- Csaky, K.; Baffi, J.; Chan, C.C.; Byrnes, G.A. Clinicopathologic correlation of progressive fibrovascular proliferation associated with occult choroidal neovascularization in age-related macular degeneration. Arch. Ophthalmol. 2004, 122, 650–652. [Google Scholar] [CrossRef] [PubMed]
- Grossniklaus, H.E.; Ling, J.X.; Wallace, T.M.; Dithmar, S.; Lawson, D.H.; Cohen, C.; Elner, V.M.; Elner, S.G.; Sternberg, P., Jr. Macrophage and retinal pigment epithelium expression of angiogenic cytokines in choroidal neovascularization. Mol. Vis. 2002, 8, 119–126. [Google Scholar] [PubMed]
- Holtkamp, G.M.; de Vos, A.F.; Peek, R.; Kijlsta, A. Analysis of the secretion pattern of monocyte chemotactic protein-1 (MCP-1) and transforming growth factor-β2 (TGF-β2) by human retinal pigment epithelial cells. Clin. Exp. Immunol. 1999, 118, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Sica, A.; Locati, M. Macrophage polarization comes of age. Immunity 2005, 23, 344–346. [Google Scholar] [CrossRef] [PubMed]
- Badylak, S.F.; Valentin, J.E.; Ravindra, A.K.; McCabe, G.P.; Stewart-Akers, A.M. Macrophage phenotype as a determinant of biologic scaffold remodeling. Tissue Eng. Part A 2008, 14, 1835–1842. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Shen, D.; Patel, M.M.; Tuo, J.; Johnson, T.M.; Olsen, T.W.; Chan, C.C. Macrophage polarization in the maculae of age-related macular degeneration: A pilot study. Pathol. Int. 2011, 61, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Guilloty, F.; Saeed, A.M.; Echegaray, J.J.; Duffort, S.; Ballmick, A.; Tan, Y.; Betancourt, M.; Viteri, E.; Ramkhellawan, G.C.; Ewald, E.; et al. Infiltration of proinflammatory M1 macrophages into the outer retina precedes damage in a mouse model of age-related macular degeneration. Int. J. Inflamm. 2013, 2013. [Google Scholar] [CrossRef]
- Ebrahem, Q.; Renganathan, K.; Sears, J.; Vasanji, A.; Gu, X.; Lu, L.; Salomon, R.G.; Crabb, J.W.; Anand-Apte, B. Carboxyethylpyrrole oxidative protein modifications stimulate neovascularization: Implications for age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2006, 103, 13480–13484. [Google Scholar] [CrossRef] [PubMed]
- Hollyfield, J.G.; Bonilha, V.L.; Rayborn, M.E.; Yang, X.; Shadrach, K.G.; Lu, L.; Ufret, R.L.; Salomon, R.G.; Perez, V.L. Oxidative damage-induced inflammation initiates age-related macular degeneration. Nat. Med. 2008, 14, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Hollyfield, J.G.; Perez, V.L.; Salomon, R.G. A hapten generated from an oxidation fragment of docosahexaenoic acid is sufficient to initiate age-related macular degeneration. Mol. Neurobiol. 2010, 41, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Chen, M.; Mayer, E.J.; Forrester, J.V.; Dick, A.D. Turnover of resident retinal microglia in the normal adult mouse. Glia 2007, 55, 1189–1198. [Google Scholar] [CrossRef] [PubMed]
- Streit, W.J. Microglia as neuroprotective, immunocompetent cells of the CNS. Glia 2002, 40, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Hanisch, U.K.; Kettenmann, H. Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 2007, 10, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Rivest, S. Regulation of innate immune responses in the brain. Nat. Rev. Immunol. 2009, 9, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Combadiere, C.; Feumi, C.; Raoul, W.; Keller, N.; Rodero, M.; Pezard, A.; Lavalette, S.; Houssier, M.; Jonet, L.; Picard, E.; et al. Cx3cr1-dependent subretinal microglia cell accumulation is associated with cardinal features of age-related macular degeneration. J. Clin. Investig. 2007, 117, 2920–2928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuo, J.; Smith, B.C.; Bojanowski, C.M.; Meleth, A.D.; Gery, I.; Csaky, K.G.; Chew, E.Y.; Chan, C.C. The involvement of sequence variation and expression of Cx3cr1 in the pathogenesis of age-related macular degeneration. FASEB J. 2004, 18, 1297–1299. [Google Scholar] [PubMed]
- Lee, J.E.; Liang, K.J.; Fariss, R.N.; Wong, W.T. Ex vivo dynamic imaging of retinal microglia using time-lapse confocal microscopy. Investig. Ophthalmol. Vis. Sci. 2008, 49, 4169–4176. [Google Scholar] [CrossRef]
- Gupta, N.; Brown, K.E.; Milam, A.H. Activated microglia in human retinitis pigmentosa, late-onset retinal degeneration, and age-related macular degeneration. Exp. Eye Res. 2003, 76, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Buschini, E.; Piras, A.; Nuzzi, R.; Vercelli, A. Age related macular degeneration and drusen: Neuroinflammation in the Retina. Prog. Neurobiol. 2011, 95, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Hartung, H.P.; Hadding, U. Complement components in relation to macrophage function. Agents Actions 1983, 13, 415–428. [Google Scholar] [CrossRef] [PubMed]
- Markiewski, M.M.; Lambris, J.D. The role of complement in inflammatory diseases from behind the scenes into the spotlight. Am. J. Pathol. 2007, 171, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Tuo, J.; Grob, S.; Zhang, K.; Chan, C.C. Genetics of immunological and inflammatory components in age-related macular degeneration. Ocul. Immunol. Inflamm. 2012, 20, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.J.; Zeiss, C.; Chew, E.Y.; Tsai, J.Y.; Sackler, R.S.; Haynes, C.; Henning, A.K.; SanGiovanni, J.P.; Mane, S.M.; Mayne, S.T.; et al. Complement factor H polymorphism in age-related macular degeneration. Science 2005, 308, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Haines, J.L.; Hauser, M.A.; Schmidt, S.; Scott, W.K.; Olson, L.M.; Gallins, P.; Spencer, K.L.; Kwan, S.Y.; Noureddine, M.; Gilbert, J.R.; et al. Complement factor h variant increases the risk of age-related macular degeneration. Science 2005, 308, 419–421. [Google Scholar] [CrossRef] [PubMed]
- Gold, B.; Merriam, J.E.; Zernant, J.; Hancox, L.S.; Taiber, A.J.; Gehrs, K.; Cramer, K.; Neel, J.; Bergeron, J.; Barile, G.R.; et al. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat. Genet. 2006, 38, 458–462. [Google Scholar] [CrossRef] [PubMed]
- Yates, J.R.; Sepp, T.; Matharu, B.K.; Khan, J.C.; Thurlby, D.A.; Shahid, H.; Clayton, D.G.; Hayward, C.; Morgan, J.; Wright, A.F.; et al. Complement C3 variant and the risk of age-related macular degeneration. N. Engl. J. Med. 2007, 357, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Chen, L.J.; Tam, P.O.; Shi, Y.; Lai, T.Y.; Liu, D.T.; Chiang, S.W.; Yang, M.; Yang, Z.; Pang, C.P. Associations of the C2-CFB-RDBP-SKIV2l locus with age-related macular degeneration and polypoidal choroidal vasculopathy. Ophthalmology 2013, 120, 837–843. [Google Scholar] [CrossRef] [PubMed]
- Zareparsi, S.; Buraczynska, M.; Branham, K.E.; Shah, S.; Eng, D.; Li, M.; Pawar, H.; Yashar, B.M.; Moroi, S.E.; Lichter, P.R.; et al. Toll-like receptor 4 variant D299G is associated with susceptibility to age-related macular degeneration. Human Mol. Genet. 2005, 14, 1449–1455. [Google Scholar] [CrossRef]
- Yang, Z.; Stratton, C.; Francis, P.J.; Kleinman, M.E.; Tan, P.L.; Gibbs, D.; Tong, Z.; Chen, H.; Constantine, R.; Yang, X.; et al. Toll-like receptor 3 and geographic atrophy in age-related macular degeneration. N. Engl. J. Med. 2008, 359, 1456–1463. [Google Scholar] [CrossRef] [PubMed]
- Edwards, A.O.; Chen, D.; Fridley, B.L.; James, K.M.; Wu, Y.; Abecasis, G.; Swaroop, A.; Othman, M.; Branham, K.; Iyengar, S.K.; et al. Toll-like receptor polymorphisms and age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2008, 49, 1652–1659. [Google Scholar] [CrossRef]
- Cho, Y.; Wang, J.J.; Chew, E.Y.; Ferris, F.L.; Mitchell, P.; Chan, C.C.; Tuo, J. Toll-like receptor polymorphisms and age-related macular degeneration: Replication in three case-control samples. Investig. Ophthalmol. Vis. Sci. 2009, 50, 5614–5618. [Google Scholar] [CrossRef]
- Bok, D. Evidence for an inflammatory process in age-related macular degeneration gains new support. Proc. Natl. Acad. Sci. USA 2005, 102, 7053–7054. [Google Scholar] [CrossRef] [PubMed]
- Radu, R.A.; Hu, J.; Jiang, Z.; Bok, D. Bisretinoid-mediated complement activation on retinal pigment epithelial cells is dependent on complement factor H haplotype. J. Biol. Chem. 2014, 289, 9113–9120. [Google Scholar] [CrossRef] [PubMed]
- Laudisi, F.; Spreafico, R.; Evrard, M.; Hughes, T.R.; Mandriani, B.; Kandasamy, M.; Morgan, B.P.; Sivasankar, B.; Mortellaro, A. Cutting edge: The NLRP3 inflammasome links complement-mediated inflammation and IL-1β release. J. Immunol. 2013, 191, 1006–1010. [Google Scholar] [CrossRef] [PubMed]
- Morgan, B.P.; Campbell, A.K. The recovery of human polymorphonuclear leucocytes from sublytic complement attack is mediated by changes in intracellular free calcium. Biochem. J. 1985, 231, 205–208. [Google Scholar] [PubMed]
- Morgan, B.P.; Luzio, J.P.; Campbell, A.K. Intracellular Ca2+ and cell injury: A paradoxical role of Ca2+ in complement membrane attack. Cell Calcium 1986, 7, 399–411. [Google Scholar] [CrossRef] [PubMed]
- Triantafilou, K.; Hughes, T.R.; Triantafilou, M.; Morgan, B.P. The complement membrane attack complex triggers intracellular Ca2+ fluxes leading to NLRP3 inflammasome activation. J. Cell Sci. 2013, 126, 2903–2913. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proil-β. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Tarallo, V.; Hirano, Y.; Gelfand, B.D.; Dridi, S.; Kerur, N.; Kim, Y.; Cho, W.G.; Kaneko, H.; Fowler, B.J.; Bogdanovich, S.; et al. Dicer1 loss and Alu RNA induce age-related macular degeneration via the NLRP3 inflammasome and MyD88. Cell 2012, 149, 847–859. [Google Scholar] [CrossRef] [PubMed]
- Tseng, W.A.; Thein, T.; Kinnunen, K.; Lashkari, K.; Gregory, M.S.; D’Amore, P.A.; Ksander, B.R. NLRP3 inflammasome activation in retinal pigment epithelial cells by lysosomal destabilization: Implications for age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2013, 54, 110–120. [Google Scholar] [CrossRef]
- Chan, C.-C.; Shen, D.; Wang, Y.; Chu, X.K.; Abu-Asab, M.S.; Tuo, J. Inflammasomes in human eyes with amd and mouse retinas with focal retinal degeneration. In ARVO; ARVO: Seattle, WA, USA, 2013. [Google Scholar]
- Wang, Y.; Abu-Asab, M.; Shen, D.; Chu, X.; Ogilvy, A.; Tuo, J.; Chan, C.-C. NLRP3 inflammasome activation in human retinal pigment epithelium under inflammation and oxidative stress. In ARVO Abstr#149; ARVO: Seattle, WA, USA, 2013. [Google Scholar]
- Cao, S.; Ko, A.; Partanen, M.; Pakzad-Vaezi, K.; Merkur, A.B.; Albiani, D.A.; Kirker, A.W.; Wang, A.; Cui, J.Z.; Forooghian, F.; et al. Relationship between systemic cytokines and complement factor H Y402H polymorphism in patients with dry age-related macular degeneration. Am. J. Ophthalmol. 2013, 156, 1176–1183. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Meer, S.G.; Miyagi, M.; Rayborn, M.E.; Hollyfield, J.G.; Crabb, J.W.; Salomon, R.G. Carboxyethylpyrrole protein adducts and autoantibodies, biomarkers for age-related macular degeneration. J. Biol. Chem. 2003, 278, 42027–42035. [Google Scholar] [CrossRef] [PubMed]
- Doyle, S.L.; Campbell, M.; Ozaki, E.; Salomon, R.G.; Mori, A.; Kenna, P.F.; Farrar, G.J.; Kiang, A.S.; Humphries, M.M.; Lavelle, E.C.; et al. NLRP3 has a protective role in age-related macular degeneration through the induction of IL-18 by drusen components. Nat. Med. 2012, 18, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, H.; Dridi, S.; Tarallo, V.; Gelfand, B.D.; Fowler, B.J.; Cho, W.G.; Kleinman, M.E.; Ponicsan, S.L.; Hauswirth, W.W.; Chiodo, V.A.; et al. Dicer1 deficit induces Alu RNA toxicity in age-related macular degeneration. Nature 2011, 471, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Kerur, N.; Hirano, Y.; Tarallo, V.; Fowler, B.J.; Bastos-Carvalho, A.; Yasuma, T.; Yasuma, R.; Kim, Y.; Hinton, D.R.; Kirschning, C.J.; et al. TLR-independent and P2X7-dependent signaling mediate Alu Rna-induced NLRP3 inflammasome activation in geographic atrophy. Investig. Ophthalmol. Vis. Sci. 2013, 54, 7395–7401. [Google Scholar] [CrossRef]
- Lalor, S.J.; Dungan, L.S.; Sutton, C.E.; Basdeo, S.A.; Fletcher, J.M.; Mills, K.H. Caspase-1-processed cytokines IL-1β and IL-18 promote IL-17 production by γδ and CD4 T cells that mediate autoimmunity. J. Immunol. 2011, 186, 5738–5748. [Google Scholar] [CrossRef] [PubMed]
- Sutton, C.E.; Lalor, S.J.; Sweeney, C.M.; Brereton, C.F.; Lavelle, E.C.; Mills, K.H. Interleukin-1 and IL-23 induce innate IL-17 production from γδ T cells, amplifying TH17 responses and autoimmunity. Immunity 2009, 31, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Mills, K.H.; Dungan, L.S.; Jones, S.A.; Harris, J. The role of inflammasome-derived IL-1 in driving IL-17 responses. J. Leukocyte Biol. 2013, 93, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Wei, L.; Meyerle, C.; Tuo, J.; Sen, H.N.; Li, Z.; Chakrabarty, S.; Agron, E.; Chan, C.C.; Klein, M.L.; et al. Complement component C5A promotes expression of IL-22 and IL-17 from human T cells and its implication in age-related macular degeneration. J. Transl. Med. 2011, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Nassar, K.; Grisanti, S.; Elfar, E.; Luke, J.; Luke, M.; Grisanti, S. Serum cytokines as biomarkers for age-related macular degeneration. Graefe’s Arch. Clin. Exp. Ophthalmol. 2014. [Google Scholar] [CrossRef]
- Ardeljan, D.; Wang, Y.; Park, S.; Shen, D.; Chu, X.K.; Yu, C.R.; Abu-Asab, M.; Tuo, J.; Eberhart, C.G.; Olsen, T.W.; et al. Interleukin-17 retinotoxicity is prevented by gene transfer of a soluble interleukin-17 receptor acting as a cytokine blocker: Implications for age-related macular degeneration. PLoS One 2014, 9, e95900. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Liu, B.; Tuo, J.; Shen, D.; Chen, P.; Li, Z.; Liu, X.; Ni, J.; Dagur, P.; Sen, H.N.; et al. Hypomethylation of the IL17RC promoter associates with age-related macular degeneration. Cell Rep. 2012, 2, 1151–1158. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liu, Y.; Lu, S.; Cai, X. Genetic variants of interleukin 17A are functionally associated with increased risk of age-related macular degeneration. Inflammation 2014. [Google Scholar] [CrossRef]
- Ogilvy, A.J.; Shen, D.; Wang, Y.; Chan, C.C.; Abu-Asab, M.S. Implications of DNA leakage in eyes of mutant mice. Ultrastruct. Pathol. 2014, 38, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galluzzi, L.; Brenner, C. Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 2007, 87, 99–163. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Salvesen, G.S.; Riedl, S.J. Caspase mechanisms. Adv. Exp. Med. Biol. 2008, 615, 13–23. [Google Scholar] [PubMed]
- Kroemer, G.; Galluzzi, L.; Vandenabeele, P.; Abrams, J.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; el-Deiry, W.S.; Golstein, P.; Green, D.R.; et al. Classification of cell death: Recommendations of the nomenclature committee on cell death 2009. Cell Death Differ. 2009, 16, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Fadok, V.A.; Voelker, D.R.; Campbell, P.A.; Cohen, J.J.; Bratton, D.L.; Henson, P.M. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J. Immunol. 1992, 148, 2207–2216. [Google Scholar] [PubMed]
- Li, M.O.; Sarkisian, M.R.; Mehal, W.Z.; Rakic, P.; Flavell, R.A. Phosphatidylserine receptor is required for clearance of apoptotic cells. Science 2003, 302, 1560–1563. [Google Scholar] [CrossRef] [PubMed]
- Henson, P.M.; Bratton, D.L. Antiinflammatory effects of apoptotic cells. J. Clin. Investig. 2013, 123, 2773–2774. [Google Scholar] [CrossRef] [PubMed]
- Chiarugi, A. “Simple but not simpler”: Toward a unified picture of energy requirements in cell death. FASEB J. 2005, 19, 1783–1788. [Google Scholar] [CrossRef] [PubMed]
- Cookson, B.T.; Brennan, M.A. Pro-inflammatory programmed cell death. Trends Microbiol. 2001, 9, 113–114. [Google Scholar] [CrossRef] [PubMed]
- Fink, S.L.; Cookson, B.T. Apoptosis, pyroptosis, and necrosis: Mechanistic description of dead and dying eukaryotic cells. Infect. Immun. 2005, 73, 1907–1916. [Google Scholar] [CrossRef] [PubMed]
- Fink, S.L.; Cookson, B.T. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell. Microbiol. 2006, 8, 1812–1825. [Google Scholar] [CrossRef] [PubMed]
- Degterev, A.; Hitomi, J.; Germscheid, M.; Ch’en, I.L.; Korkina, O.; Teng, X.; Abbott, D.; Cuny, G.D.; Yuan, C.; Wagner, G.; et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 2008, 4, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; McQuade, T.; Siemer, A.B.; Napetschnig, J.; Moriwaki, K.; Hsiao, Y.S.; Damko, E.; Moquin, D.; Walz, T.; McDermott, A.; et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 2012, 150, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Vercammen, D.; Beyaert, R.; Denecker, G.; Goossens, V.; Van Loo, G.; Declercq, W.; Grooten, J.; Fiers, W.; Vandenabeele, P. Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J. Exp. Med. 1998, 187, 1477–1485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005, 1, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Kaczmarek, A.; Vandenabeele, P.; Krysko, D.V. Necroptosis: The release of damage-associated molecular patterns and its physiological relevance. Immunity 2013, 38, 209–223. [Google Scholar] [CrossRef] [PubMed]
- Cook, K.L.; Soto-Pantoja, D.R.; Abu-Asab, M.; Clarke, P.A.; Roberts, D.D.; Clarke, R. Mitochondria directly donate their membrane to form autophagosomes during a novel mechanism of parkin-associated mitophagy. Cell Biosci. 2014, 4, 16. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Shen, D.; Popp, N.; Tuo, J.; Abu-Asab, M.; Xie, T.; Chan, C.C. The involvement of IL-17rc pathway in the inflammatory stimuli of the multipotent retinal stem cells. In ARVO Abstr#3984; ARVO: Orlando, FL, USA, 2014. [Google Scholar]
- Nakamura, T.; Ito, T.; Igarashi, H.; Uchida, M.; Hijioka, M.; Oono, T.; Fujimori, N.; Niina, Y.; Suzuki, K.; Jensen, R.T.; et al. Cytosolic double-stranded DNA as a damage-associated molecular pattern induces the inflammatory response in rat pancreatic stellate cells: A plausible mechanism for tissue injury-associated pancreatitis. Int. J. Inflamm. 2012, 2012. [Google Scholar] [CrossRef]
- Ardeljan, D.; Chan, C.C. Aging is not a disease: Distinguishing age-related macular degeneration from aging. Prog. Retin. Eye Res. 2013, 37, 68–89. [Google Scholar] [CrossRef] [PubMed]
- Dungan, L.S.; Mills, K.H. Caspase-1-processed IL-1 family cytokines play a vital role in driving innate IL-17. Cytokine 2011, 56, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, M.F.; Kopf, M. On the role of the innate immunity in autoimmune disease. J. Exp. Med. 2001, 193, F47–F50. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Tschopp, J. Inflammatory caspases: Linking an intracellular innate immune system to autoinflammatory diseases. Cell 2004, 117, 561–574. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Rhebergen, A.M.; Eisenbarth, S.C. Licensing adaptive immunity by NOD-like receptors. Front. Immunol. 2013, 4. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ardeljan, C.P.; Ardeljan, D.; Abu-Asab, M.; Chan, C.-C. Inflammation and Cell Death in Age-Related Macular Degeneration: An Immunopathological and Ultrastructural Model. J. Clin. Med. 2014, 3, 1542-1560. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm3041542
Ardeljan CP, Ardeljan D, Abu-Asab M, Chan C-C. Inflammation and Cell Death in Age-Related Macular Degeneration: An Immunopathological and Ultrastructural Model. Journal of Clinical Medicine. 2014; 3(4):1542-1560. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm3041542
Chicago/Turabian StyleArdeljan, Christopher P., Daniel Ardeljan, Mones Abu-Asab, and Chi-Chao Chan. 2014. "Inflammation and Cell Death in Age-Related Macular Degeneration: An Immunopathological and Ultrastructural Model" Journal of Clinical Medicine 3, no. 4: 1542-1560. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm3041542