1. Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterized by progressive loss of cortical and spinal motor neurons, resulting in muscle denervation and paralysis, which lead to death within 3–5 years after diagnosis, normally due to respiratory failure [
1,
2]. ALS is the most common degenerative disorder affecting motor neurons in adults, with an incidence that ranges from 2 to 5 cases per 100,000 individuals worldwide. More than 90% of cases are sporadic (sALS), while 5 to 10% of cases have a familial origin (fALS) [
3]. Genetic mutations were found in a huge number of genes, but only few of them are epidemiologically relevant, including: SOD1, encoding for the copper/zinc superoxide dismutase 1, two RNA binding proteins (RBPs), trans-activation response element (TAR) DNA binding protein (TARDBP) and fused in sarcoma/translocated in liposarcoma (FUS/TLS, herein referred to as FUS), and C9ORF72, containing a pathological hexanucleotide repeat expansion [
4,
5].
The maintenance of genome stability and integrity is fundamental for cellular viability especially in neurons. Neurons are highly susceptible to DNA damage, both single-strand breaks (SSBs) and double-strand breaks (DSBs), because they are post-mitotic cells with a high metabolic rate, and they are also vulnerable to oxidative stress, which is one of the sources of DNA damage [
6,
7]. Furthermore, in post-mitotic neurons, because of their lack of self-renewal and replication, there are lesser choices to repair SSBs compared to proliferating cells. Thus, SSBs more probably could be transformed into more genotoxic DSBs [
6]. To date, genome damage and their inadequate repair have been linked to degenerating neurons in ALS patients; however, the underlying mechanisms remain unknown [
8].
Furthermore, some evidence suggests that DNA repair pathways and the DNA damage response (DDR) are impaired in numerous neurological disorders, including ALS [
9]. Two protein kinases, ataxia-telangiectasia-mutated (ATM) and ATM and Rad3-related (ATR), have a key role in the DDR pathways that respond to genotoxic stress. While ATM is essential for the DSB response, ATR is important in the proliferating cells surviving, due to its role in the response to replication stress. Despite differences in substrate specificity both ATM and ATR phosphorylate hundreds of protein targets at Ser/Thr-Gln motifs, regulating DNA repair, replication, transcription, cell cycle checkpoint signaling, and cell fate pathways, such as apoptosis or senescence [
10,
11,
12]. In neurodegenerative disease, persistent accumulation of DNA damage and defective repair mechanisms could lead to alteration of gene expression and neuroinflammation [
13]. Recent breakthroughs, that permit to unravel these pathways and their role in neurodegeneration, could open to new therapeutic approaches and targets, involving all the factors that play a key role in DNA repair.
Recent evidence suggests that also non-neural cells, such as peripheral blood mononuclear cells (PBMCs), could participate in triggering moton neurons degeneration, thus could be used in studying neurodegeneration [
14,
15]. Moreover, using PBMCs of ALS patients, we reported a discrepancy between low SOD1 protein concentration and high SOD1 mRNA expression level [
16,
17]. In 2013, Cereda and co-authors demonstrated that the “missing” protein has two fates: Accumulates in cytoplasmic aggregates or relocates in the nuclear fraction, hence two sub-groups of sporadic patients were identified [
15]. The presence of two sub-groups can explain the diverse severity of the phenotype of the pathology in patients. Unfortunately, the role of SOD1 impairment in DNA damage formation, i.e., DNA SSBs and DSBs has been controversially debated [
18,
19,
20], and no data exist on its role in DDR.
Thus, in this study we aimed to investigate DNA damage in PBMCs of ALS patients, evaluating differences between the two sub-groups of patients, and in human neuroblastoma cells SH-SY5Y treated with hydrogen peroxide, used as a cellular model of ALS disease [
17,
21]. In addition, we wanted to evaluate the expression of DDR’s proteins, including ATM and ATR, and eventually to explore a new pathway in which they and SOD1 are involved.
2. Materials and Methods
2.1. Chemicals and Reagents
All chemicals were purchased from Sigma-Aldrich (Saint Louis, MO, USA) and were of analytical grade or the highest grade available. All reagents for cell culture were purchased from PAA Laboratories (Toronto, ON, Canada). Lipofectamine Plus was purchased from Life Technologies (Carlsbad, CA, USA). Rabbit anti-SOD1 (FL-154) (sc-11407); rabbit anti-Sp1 (pep2) (sc-59); mouse anti-pThr (H2) (sc-5367); mouse anti-Chk2 (A-12) (sc-5278); protein A/G plus agarose immunoprecipitation reagent (sc-2003) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Rabbit anti-alpha tubulin (GTX 112141) and rabbit anti-GAPDH (GTX 100118) were purchased from GeneTex (Irvine, CA, USA). Rabbit anti-pSer (ab9332) was purchased from Abcam (Cambridge, UK). Mouse anti-SOD1 (6F5) (MA5-15520) was purchased from Thermo Fisher Scientific (Waltham, MA, USA). Donkey anti-rabbit (NA9340) and anti-mouse (NA931) secondary peroxidase-conjugated antibody and Amersham ECL Select Western Blotting Detection reagent (RPN 2235) were purchased from GE Healthcare (Chicago, IL, USA). Anti-mouse CF™488A (sab4600036) and anti-rabbit CF™594 antibodies (sab4600099) were purchased from Sigma-Aldrich. Primers were purchased from Sigma-Aldrich.
2.2. Cell Culture and Treatment
Human neuroblastoma cells SH-SY5Y were maintained in Dulbecco’s modified Eagle/F12 medium (Life Technologies), supplemented with 15% fetal bovine serum (Life Technologies), 2 mM l-glutamine, 100 U/mL penicillin, 10 mg/mL streptomycin, at 37 °C in an atmosphere with 5% of CO2 and 95% humidity. Cells were monitored every day and, at the 80–90% of confluence, they were exposed to phosphate buffer saline solution (1× PBS) (T0) or treated with oxidative agents, such as 1 mM H2O2 (Sigma-Aldrich) for 30 (T30) and 60 (T60) min. SH-SY5Y were washed with cold 1× PBS, collected using a cell scraper, and centrifuged to allow the formation of a cellular pellet, ready to undergo the fractionated protein extraction. The interaction between Chk2 and SOD1 interaction was further confirmed by adding AZD7762, an inhibitor of both Chk1 and Chk2, to SH-SY5Y cells. Briefly, cells were exposed to 5 nM of AZD7762 and 1 mM H2O2 for 30 and 60 min. Successively, cells were washed, scraped and centrifuged as previously described.
2.3. Patients’ Enrolment
Patients affected by ALS were enrolled at the IRCCS Mondino Foundation in Pavia. Experiments were done using PBMCs isolated from 40 sporadic ALS (sALS) patients (mean age: 59.9 ± 9.4). ALS diagnosis was made according to the revised El Escorial Criteria [
22]. SALS individuals harboring mutations in the SOD1, FUS/TLS, TARDBP, C9ORF72 and ANG genes were excluded from this study. Thirty-nine sex- and age-matched healthy volunteers, free from any pharmacological treatment (mean age: 54.7 ± 11), were recruited at the Transfusion Centre of the IRCCS Policlinico S. Matteo Foundation in Pavia. Healthy volunteers were all unrelated and the normal phenotype was confirmed by interviews based on personal health histories. The study design was examined by the IRBs of the enrolling Institutions. All individuals joined in the study signing the Consensus after reading Informative note. (see
Section 2.14).
2.4. Isolation of PBMCs from ALS Patients and Healthy Controls
PBMCs were immediately isolated from peripheral venous blood by Histopaque®-1077 (Sigma-Aldrich) following the manufacturer’s instructions. Cells viability was assessed by trypan blue exclusion test. Aliquots of PBMCs were collected from each subject and processed for the following experiments.
2.5. Subcellular Fractionation
Both for PBMCs and SH-SY5Y, the subcellular fractionation was performed according to the method of Schreiber and colleagues [
23], with some modifications. After cells have been washed with ice-cold 1× PBS, the cellular pellet was resuspended in ice-cold hypotonic lysis buffer (10 mM HEPES, pH 7.9, 10 mM KCl, 0.1 mM EDTA, 1 mM dithiothreitol, 0.5 mM phenylmethylsulfonyl fluoride, 1% of protease and phosphatase inhibitor cocktail). Cells were allowed to swell on ice for 25 min, after which 25 μL of 10% Nonidet NP-40 (Fluka, St. Gallen, Switzerland) was added. Samples were vortexed and centrifuged at the maximum speed. The supernatant, containing the cytoplasm proteins, was collected and stored at –80 °C until the use. The nuclear pellets were resuspended in ice-cold hypertonic nuclear extraction buffer (20 mM HEPES, pH 7.9, 0.4 M NaCl, 1 mM EDTA, 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, 1% of protease and phosphatase inhibitor cocktail), and incubated on ice for 20 min with agitation. The nuclear extracts were then centrifuged at the maximum speed for 5 min at 4 °C and the supernatant containing the nuclear proteins was collected and frozen at –80 °C.
2.6. Cell Transfection
To better distinguish cytoplasm and nuclear localization, we used chimeric fluorescent-tagged SOD1 proteins bearing either a nuclear export signal (NES) or a nuclear localization signal (NLS): YFP-NLS-wtSOD1 and YFP-NES-wtSOD1, respectively [
18]. The plasmids YFP-NLS-wtSOD1, YFP-NES-wtSOD1 were used for transient transfections of SH-SY5Y cells using Lipofectamine 2000 (Life Technologies) following the manufacturer’s instructions, as also described in Crippa et al. [
24]. About 1 × 10
5 cells/mL were transfected using 0.7 μg of YFP-NLS-SOD1s or YFP-NES-SOD1s plasmids in addition to 2 µL of Lipofectamine 2000 (amount for 1 well of a 12 wells/multiwell).
2.7. Western Blotting Analysis
A Western blotting analysis was performed by SDS–polyacrylamide gel electrophoresis (SDS-PAGE). Thirty μg of nuclear and cytoplasm proteins were loaded onto 12.5% SDS–PAGE gel (Bio-Rad Laboratories, Hercules, CA, USA). After electrophoresis, samples were transferred to nitrocellulose membrane (Trans-blot, Bio-Rad Laboratories) using a liquid transfer apparatus (Bio-Rad Laboratories). Nitrocellulose membranes were treated with a blocking solution (5% of non-fat dry milk in TBS-T buffer, 10 mM Tris-HCl, 100 mM NaCl, 0.1% Tween, pH 7.5) to block unspecific protein binding sites and incubated with primary antibody overnight at 4 °C.
Immunoreactivity was detected using donkey anti-rabbit or anti-mouse secondary peroxidase-conjugated antibody (GE Healthcare; dilution 1:8000) and bands were visualized using enhanced chemiluminescence detection kit (ECL Select, Ge Healthcare). Both primary and secondary antibodies were removed from the membrane by means of stripping solution (mercaptoethanol, 2% SDS, and 62.5 mM Tris/HCl, pH 6.7), and then processed as described above. Densitometric analysis of the bands was performed using the ImageJ software (version number 1.51,
http://rsb.info.nih.gov/ij/).
2.8. Immunocytochemistry
SH-SY5Y both at T0 and after treatment with 1 mM H
2O
2 (Sigma-Aldrich) for 30 (T30) and 60 (T60) min were harvested, washed with 1× PBS and suspended in RPMI-1640 medium (for PBMCs) and in DMEM-F12 medium (for SH-SY5Y). About 1 × 10
5 cells were placed on a poly-L-Lysine slide (Thermo Fisher Scientific) and incubated at 37 °C to allow cell attachment to the slide. Cells were rinsed with 1× PBS and then fixed using a solution of 4% paraformaldehyde in 1× PBS [
25]. Fixed cells were washed with 1× PBS and treated with a blocking solution (5% normal goat serum in 0.1% Tween-PBS) for 1 h to block unspecific protein binding sites, cells were then incubated ON at 4 °C with primary antibodies: Rabbit polyclonal anti-SOD1 (1:250 dilution), mouse monoclonal anti-SOD1 (1:250 dilution), mouse monoclonal anti-pThr (1:250 dilution) and rabbit polyclonal anti-pSer (1:250 dilution). Cells were washed with 1× PBS and incubated at room temperature for 1 h with secondary antibodies: CFTM 594 goat anti-mouse, (1:700 dilution) and CFTM 488A goat anti-rabbit (1:700 dilution) at room temperature for 1 h. Both primary and secondary antibodies were prepared in blocking buffer. Finally, samples were washed with 1× PBS, mounted with Prolong
® Gold antifade reagent with DAPI (Invitrogen, Carlsbad, CA, USA), dried, nail-polished and analyzed by confocal microscopy (Leica DM IRBE, Leica Microsystems Srl, Wetzlar, Germany).
2.9. Comet Assay
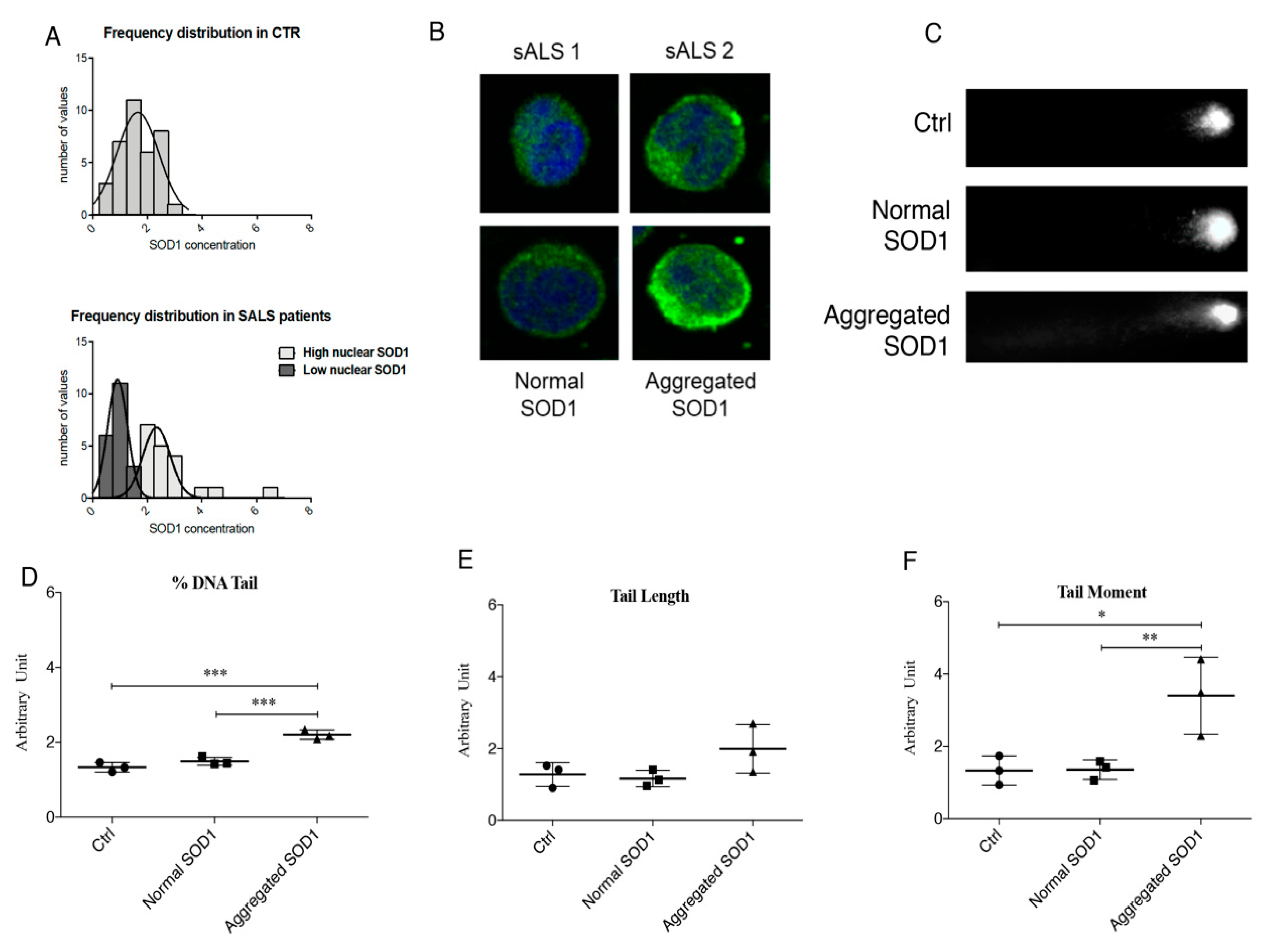
In order to study DNA damage, comet assay was performed in SH-SY5Y, non-transfected (NT) and transfected with NLS- and NES- SOD1, treated with 1 mM of H2O2 for 60 min, and in PBMCs of healthy controls and sALS.
Approximately 5 × 104 cells, both SH-SY5Y and PBMCs, were collected and centrifuged at 4000 rpm for 5 min, the supernatant was discarded, and the pellet was suspended in 0.75% low-melting-point agarose (Sigma-Aldrich). The cell suspension was placed onto microscope slides coated with a layer of 1% agarose in 1× PBS. Slides with SH-SY5Y were immersed in cold lysing buffer (2.5 M NaCl, 0.1 M EDTA, 10 mM Tris, 1% DMSO and 1% Triton X-114, pH 12.5–13) for 30 min at 4 °C; while slides with PBMCs were immersed in alkaline lysis buffer (2.5 M NaCl, 0.1 M EDTA, 10 mM Tris, pH 10) for 1 h at 4 °C. Then, all slides were put in a horizontal electrophoresis tank, filled with cold electrophoresis buffer (0.3 M NaOH, 1 mM EDTA, pH 13), equilibrate for 40 min before starting electrophoresis (run conditions: 300 mA, 25 V, 30 min at 4 °C). Slides were neutralized with 0.4 M Tris, pH 7.5 for 15 min at 4 °C, covered and stored in a humidity chamber. After the addition of the nuclear dye Hoechst (Sigma-Aldrich), individual cells or ‘Comets’ were analyzed using a fluorescence microscope (Axio Imager 2, Zeiss, Oberkochen, Germany).
2.10. Immunoprecipitation
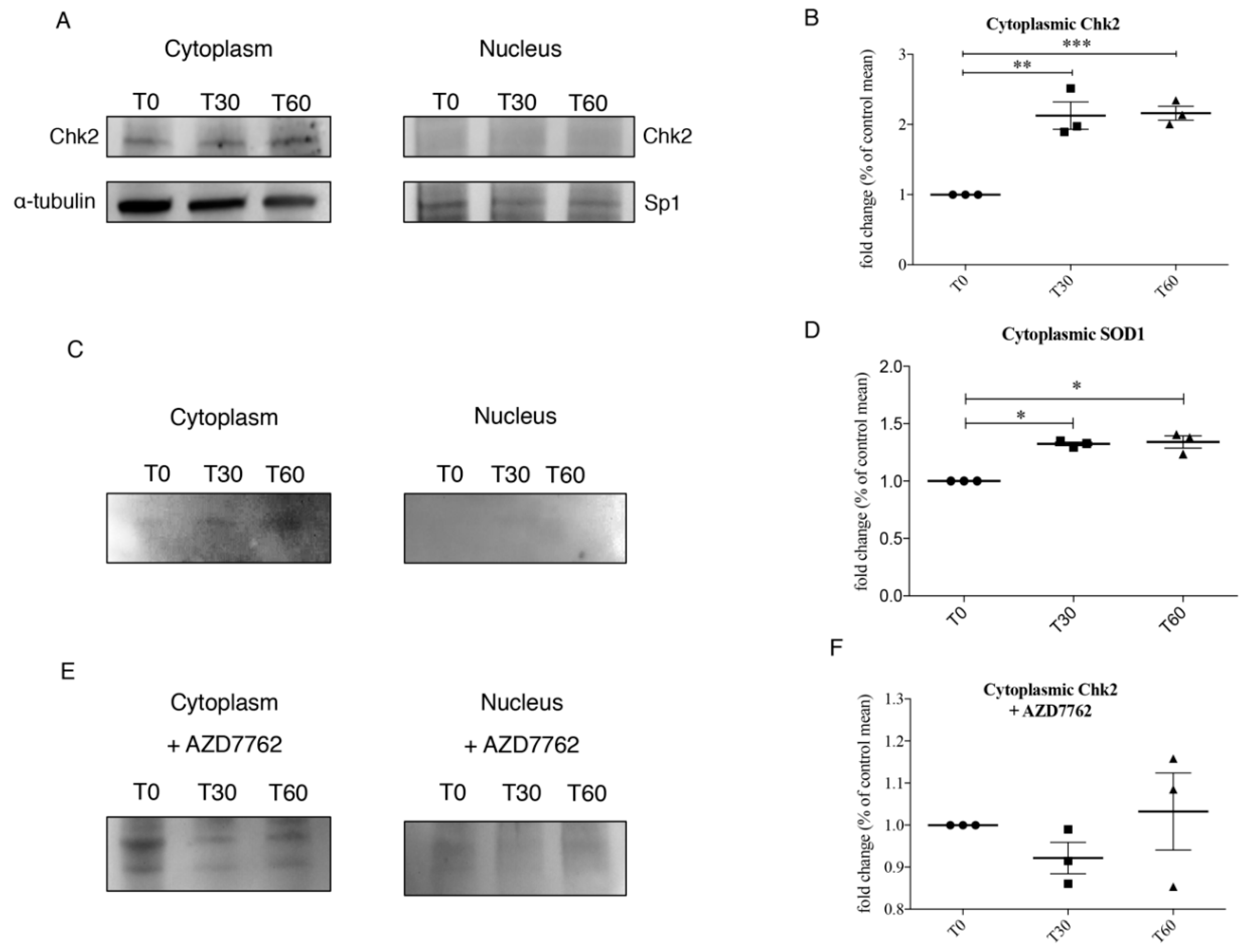
In order to evaluate the interaction between Chk2 and SOD1, we performed immunoprecipitation, that was performed according to Dell’Orco and co-authors [
21] with minor modifications. After a pre-clearing phase, immunoprecipitation was carried out overnight at 4 °C using 2 μg of rabbit polyclonal primary antibody anti-SOD1 and anti-Chk2 for 300 μg of proteins diluted both in cytoplasm ice-cold lysis buffer (10 mM HEPES, pH 7.9, 10 mM KCl, 0.1 mM EDTA, 1 mM dithiothreitol, 0.5 mM phenylmethylsulfonyl fluoride, 1% of protease and phosphatase inhibitor cocktail) and in ice-cold nuclear extraction buffer (20 mM HEPES, pH 7.9, 0.4 M NaCl, 1 mM EDTA, 1 mM dithiothreitol, 1 mM phenylmethylsulfonyl fluoride, 1% of protease and phosphatase inhibitor cocktail) in the presence of 30 μL of protein A/G plus agarose (Santa Cruz Biotechnology). Samples were finally subjected to Western blotting for: Anti-Phosphoserine antibody (dilution 2 μg/mL in 5% bovine serum albumin (BSA)); anti-Phosphothreonine antibody (dilution 1:200 in 5% BSA); anti ChK2 (A-12) antibody (dilution 1:200 in 5% BSA). The negative control was obtained in the same conditions, in the presence of an irrelevant antibody with the same isotype of the specific immunoprecipitating antibody.
2.11. Mass Spectrometry
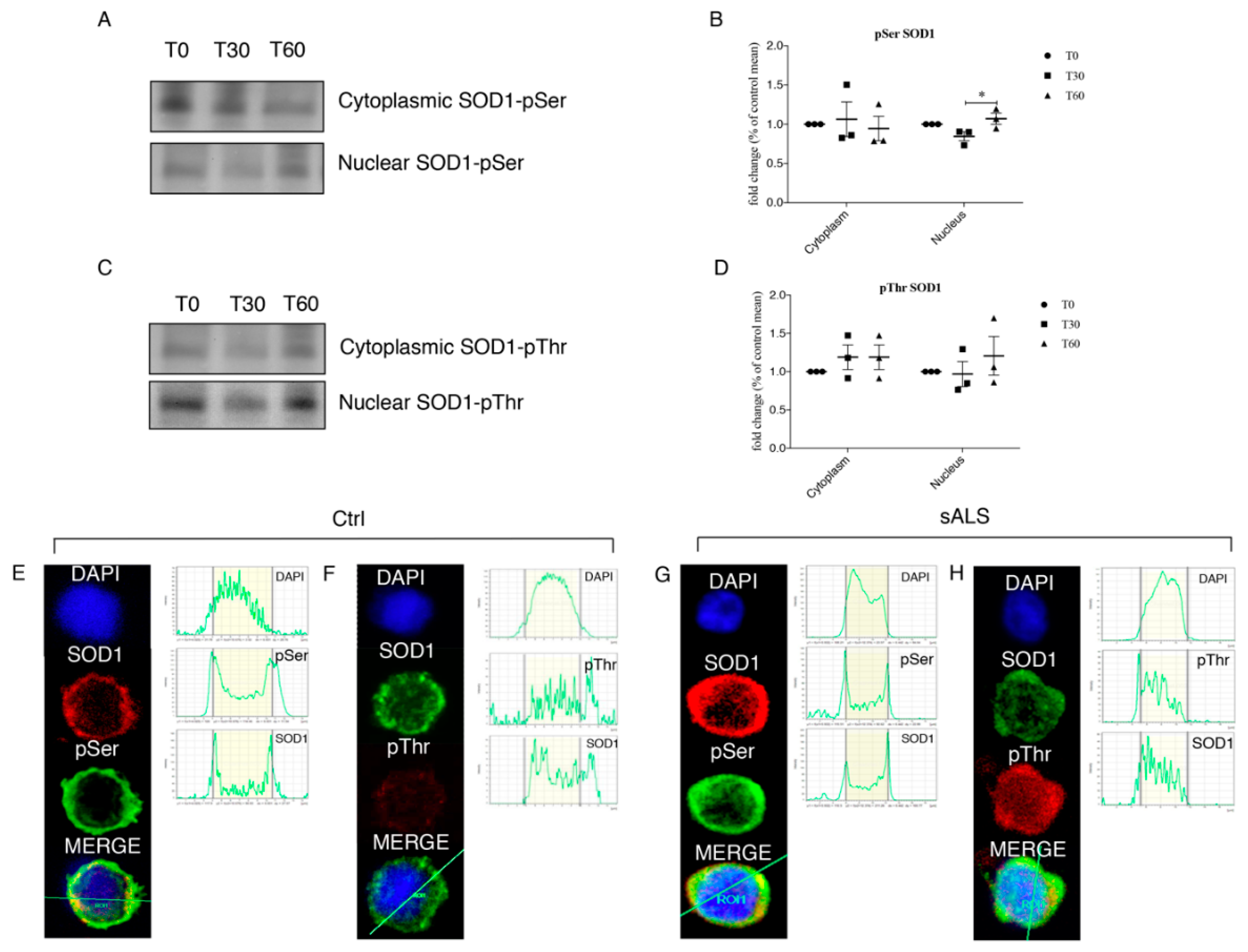
We analyzed the phosphorylation of SOD1 by means of mass spectrometry. To prepare mass spectrometry sample, 500 μg of both cytoplasm and nuclear proteins obtained from SH-SY5Y cells were immunoprecipitated by the Anti-FLAG M2 Magnetic Beads (Sigma-Aldrich) and eluted with the 3× FLAG peptide (Sigma-Aldrich). Samples were also treated with 50 U/µL of Phosphatase Alkaline (Sigma-Aldrich). Sample adequacy was verified by SDS-PAGE gel electrophoresis followed by silver staining of the gel and then processed for mass spectrometry. Upon immunoprecipitation, each sample was reduced with DTT, alkylated with 55 mM iodoacetamide at RT for 45 min and digested overnight with trypsin sequence grade, at 37 °C using a protease: protein ratio (1:20). The tryptic digest was desalted/concentrated on a ZipTipC18 (Millipore), and analyzed by mass spectrometry [
26]. Nano LC-electrospray ionization-MS/MS analysis was performed on a Dionex UltiMate 3000 HPLC System with a PicoFrit ProteoPrep C18 column (200 mm, internal diameter of 75 m) (New Objective. Gradient: 1% ACN in 0.1% formic acid for 10 min, 1–4% ACN in 0.1% formic acid for 6 min, 4–30% ACN in 0.1% formic acid for 147 min, and 30–50% ACN in 0.1% formic for 3 min at a flow rate of 0.3 L/min. The eluate was electrosprayed into an LTQ-Orbitrap Velos (Thermo Fisher Scientific) through a Proxeon nanoelectrospray ion source (Thermo Fisher Scientific). Data acquisition was controlled by Xcalibur 2.0 and Tune 2.4 software (Thermo Fisher Scientific). The LTQ-Orbitrap was operated in positive mode in data-dependent acquisition mode to automatically alternate between a full scan (m/z 350–2000) in the LTQ-Orbitrap (at resolution, 60,000 (CID) or 30,000 (HCD); AGC target, 1,000,000) and subsequent CID and HCD MS/MS in the linear ion trap of the 20 (CID) or 10 (HCD) most intense peaks from full scan (normalized collision energy of 35%, 10-ms activation). Data Base searching was performed using the Sequest search engine contained in the Proteome Discoverer 1.3 software (Thermo Fisher Scientific). The following parameters were used: 10 ppm for MS and 0.5 Da for MS/MS tolerance, carbamidomethylation of Cys as fixed modification; phosphorylation of Ser, Tyr, and Thr, as variable modifications, trypsin (two misses) as protease. To generate the list of phosphosites reported in
Table 1, we considered only the sites with the highest X Correlation value (Xcorr) in Sequest (1.5), the rank value of 1 and the best fragmentation pattern, selected manually after visual inspection of the MS/MS spectra. Three different tools for phosphorylation site prediction where applied: NetPhos 2.0 Server (DTU Health Tech, Lyngby, Denmark), Phosphosite plus (Cell Signaling Technology, Danvers, MA, USA) and Phosida (Max-Plank-Gesellschaft, München, Germany). Samples were analyzed by MS/MS before and after treatment with 50 U/µL of Phosphatase Alkaline to unambiguously verify the phosphosites listed in
Table 1.
2.12. Real Time-qPCR
SH-SY5Y cell total RNA was extracted with the Trizol® reagent (Invitrogen) using the manufacturer’s specifications and quantified by spectrophotometric analysis. One μg of RNA was reverse transcribed using an iScript cDNA synthesis kit (Bio-Rad Laboratories) according to the manufacturer’s recommendations. Primer and probe sequences: ATM Fw CAGGCGAAAAGAATCTGGGG, Rv GCACAAAGTAGGGTGGGAAAGC; ATR Fw TGAAAGGGCATTCCAAAGCG, Rv CAATAGATAACGGCAGTCCTGTCAC; CHEK1 Fw CAGGTCTTTCCTTATGGGATACCAG, Rv TGGGGTGCCAAGTAACTGACTATTC; CHEK2 Fw GCTATTGGTTCAGCAAGAGAGGC, Rv TCAGGCGTTTATTCCCCACC.
Real-time PCR was done on the LightCycler 480 II (Roche using iQ Sybr Green Supermix (Bio-Rad Laboratories). The qPCR incubations were run at 95 °C for 3 min, 95 °C for 15 s for 40 cycles, 60 °C for 45 s, 95 °C for 5 s, 65 °C for 1 min, then reach a peak of 97 °C and stay at 40 °C for 30 s. Averages of the Ct values from three replications for each sample with a standard deviation less than or the same as 0.15 was considered a valid result. Analysis of the ATM, ATR, CHEK1 and CHEK2 gene expression was made using GADPH (Fw CTTTTGCGTCGCCAG; Rv TTGATGGCAACAATATCCAC) to normalize. The amplification efficiencies for all genes were observed to be in the 90–110% range.
2.13. Statistical Analysis
All the experiments were performed at least three times. Values were expressed as means ± S.D. Statistical analysis was performed by One-Way Analysis of Variance (ANOVA) followed by Newman-Keuls Multiple Comparison as a post-hoc test (GraphPad Prism version 5, GraphPad Software, San Diego, CA, USA). Values were considered statistically significant when p values were <0.05.
2.14. Ethic Statement
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the Helsinki declaration and its later amendments or comparable ethical standards. The study design was examined by the IRBs of the enrolling Institutions (Protocol n°375/04–version 07/01/2004).
4. Discussion
Mutations in the gene encoding SOD1 were the first genetic cause of ALS to be identified [
29] and are implicated in ~20% of all fALS and in ~1% of sALS cases. In our previous works we provided evidence for the involvement of wild-type SOD1 in sporadic cases, since a reduced SOD1 expression profile in lysates from PBMCs was observed [
30]. On the other hand, abnormally high levels of SOD1 transcript were found in sporadic patients thus raising the question about the discordance between protein and mRNA expression levels [
16]. To explain this discrepancy, we hypothesize a re-localization of the ‘‘missing’’ SOD1 protein in other cellular compartments, such as the insoluble fraction. Accurate information on the exact subcellular localization of a protein is critical for understanding its physiological and pathophysiological function, in particular aggregated SOD1 could be non-functional, generating oxidative stress that leads to DNA damage [
30].
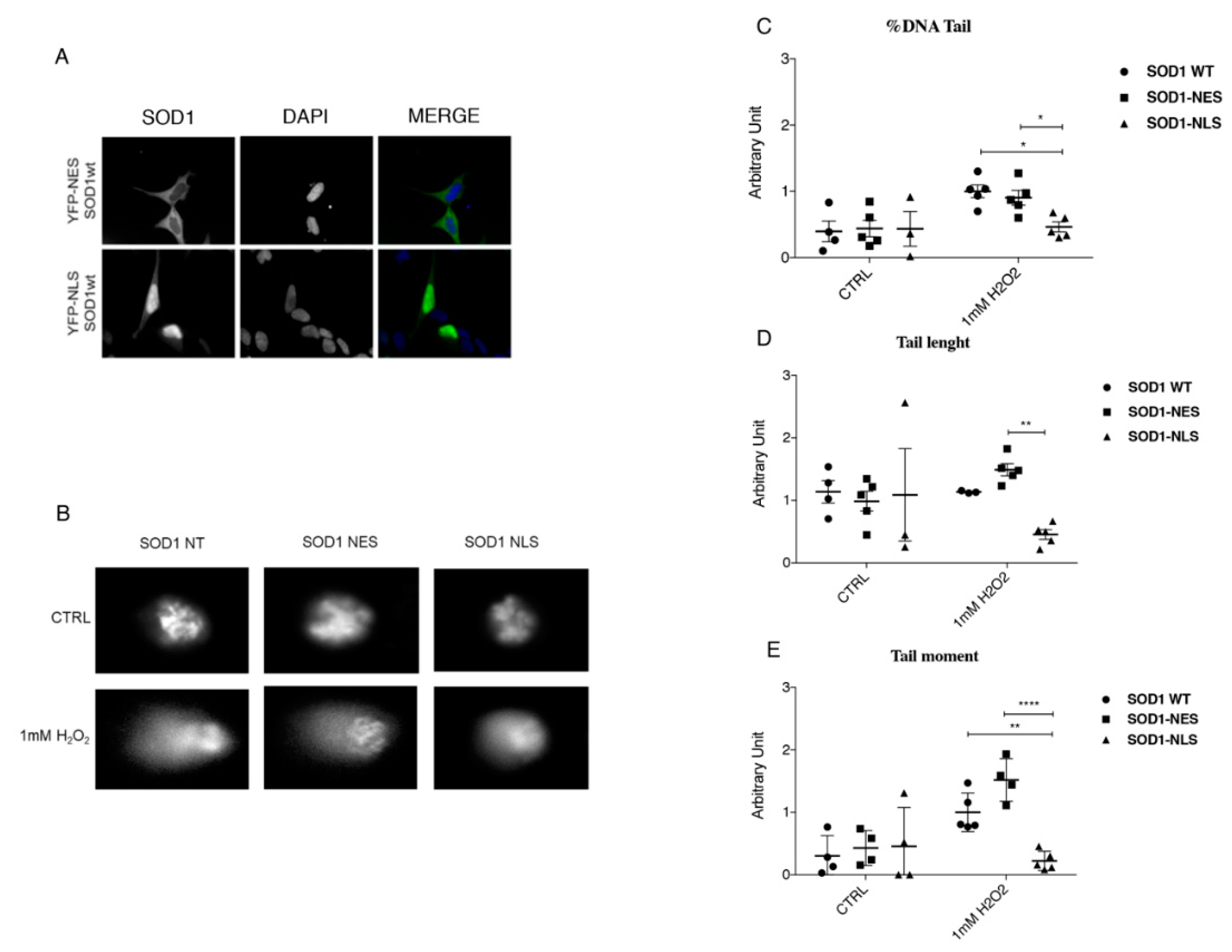
In PBMCs of sALS patients, we carried out a Comet assay and we clearly demonstrated the lack of DNA damage in the sub-group of sALS patients characterized by high levels of soluble nuclear SOD1, and an extensive DNA damage in sALS patients with high levels of insoluble/aggregate SOD1. This finding confirms our hypothesis that normal soluble SOD1 could have a potential protective role in DNA damage mediated by oxidative stress. On the contrary, aggregate SOD1 could be non-functional, leading to an increase of oxidative stress. Moreover, since SOD1 has a cytoplasmic peroxidase activity and it protects from oxidative stress, it could protect also DNA when it translocates to the nuclear compartment. Thus, we aimed to study such a pathway in our cellular model of neurotoxicity, SH-SY5Y treated with hydrogen peroxide.
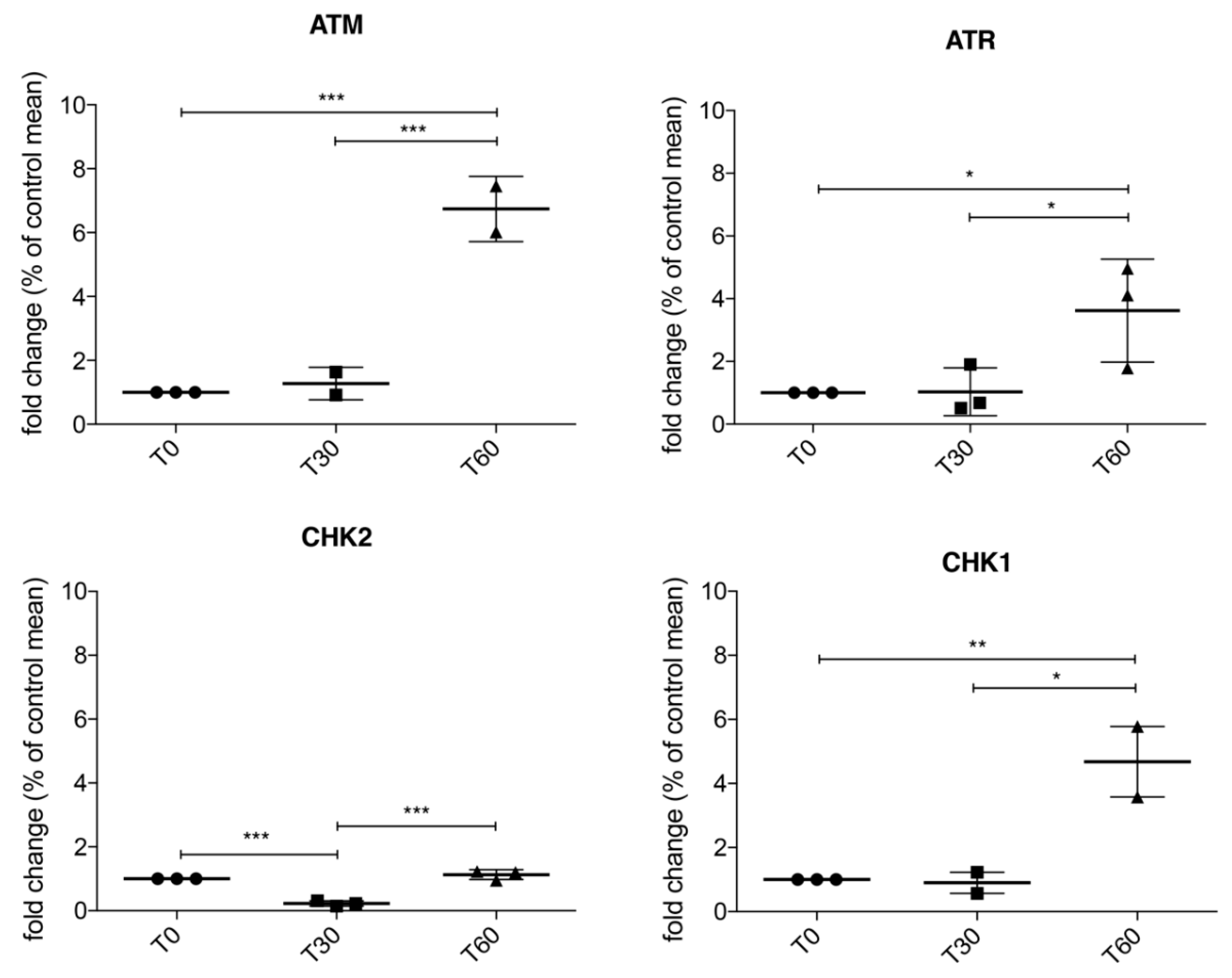
We sought to confirm in our cellular model activation of DDR pathways after oxidative stress, and we found that the ATM/Chk2 and ATR/Chk1 pathways are both activated after 60 min of 1 mM H
2O
2 treatment. In particular, according to data reported in the literature [
31] these two pathways are activated in a biphasic way, first ATM/Chk2 and then ATR/Chk1. Thus, we can postulate that, following H
2O
2-induced oxidative stress, different mechanisms crosstalk in order to coordinate DNA repair, cell cycle progression, transcription, apoptosis, and senescence.
Our observations lead us to suppose that the activation of DDR’s pathways could result in the phosphorylation of SOD1 thanks to their interaction. These data raised the hypothesis that oxidative stress lead to the activation of ATM/Chk2 and ATR/Chk1 pathways, in particular Chk2 could phosphorylate SOD1, as suggested by the work of Tsang and colleagues [
32]. Mass spectrometry data directed our attention to SOD1 phosphorylation at several Thr and Ser residues. SOD1 phosphorylation could lead to the translocation of the protein in the nuclear compartment, in order to protect DNA from oxidative damage.
Thus, we finally evaluated the protection of SOD1 in the nuclear compartment by using specific localization tag. We confirmed that a higher concentration of SOD1 in the nucleus have a protective role in the DNA damage mediated by oxidative stress. On the contrary, when SOD1 is forced into the cytoplasm, it cannot protect DNA from damage.
These data should open a new scenario into the development of new therapies of ALS, in particular targeting molecules involved in oxidative stress protection. As yet to reported in cancer research [
33], the nucleus could be a persuasive drug target, in particular by delivering nanoparticles directed in the proximity of the nucleus or against the nucleus itself. Targeted drugs that can reduce oxidative stress damage can have a pivotal role in delay the progression of the pathology.

 ,
, 






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}