Can Metabolic Pathways Be Therapeutic Targets in Rheumatoid Arthritis?


1
Department of Pharmacology, UCSD School of Medicine, La Jolla, CA 92093-0663, USA
2
Department of Medicine, UCSD School of Medicine, La Jolla, CA 92093-0663, USA
*
Author to whom correspondence should be addressed.
J. Clin. Med. 2019, 8(5), 753; https://0-doi-org.brum.beds.ac.uk/10.3390/jcm8050753
Submission received: 1 April 2019
/
Revised: 5 May 2019
/
Accepted: 20 May 2019
/
Published: 27 May 2019
(This article belongs to the Section Endocrinology & Metabolism)
Abstract
:The metabolic rewiring of tumor cells and immune cells has been viewed as a promising source of novel drug targets. Many of the molecular pathways implicated in rheumatoid arthritis (RA) directly modify synovium metabolism and transform the resident cells, such as the fibroblast-like synoviocytes (FLS), and the synovial tissue macrophages (STM), toward an overproduction of enzymes, which degrade cartilage and bone, and cytokines, which promote immune cell infiltration. Recent studies have shown metabolic changes in stromal and immune cells from RA patients. Metabolic disruption in the synovium provide the opportunity to use in vivo metabolism-based imaging techniques for patient stratification and to monitor treatment response. In addition, these metabolic changes may be therapeutically targetable. Thus, resetting metabolism of the synovial membrane offers additional opportunities for disease modulation and restoration of homeostasis in RA. In fact, rheumatologists already use the antimetabolite methotrexate, a chemotherapy agent, for the treatment of patients with inflammatory arthritis. Metabolic targets that do not compromise systemic homeostasis or corresponding metabolic functions in normal cells could increase the drug armamentarium in rheumatic diseases for combination therapy independent of systemic immunosuppression. This article summarizes what is known about metabolism in synovial tissue cells and highlights chemotherapies that target metabolism as potential future therapeutic strategies for RA.
1. Introduction
Several recent reviews have highlighted metabolic changes in immunometabolism, stromal metabolism, and systemic metabolism in rheumatoid arthritis (RA) [1,2,3,4,5]. Qualitative changes to cellular metabolism are indeed essential to support physiological and pathological responses seen in the RA synovium. The phenotypic transformation of fibroblast-like synoviocytes (FLS) from quiescent cells to aggressive, metabolically active cells, the activation of synovial tissue macrophages (STM), and the recruitment of immune cells to the synovial tissue, all require an increased bioenergetic and biosynthetic demand. This is associated with changes in metabolism and energy production networks to support and enable rapid proliferation, migration, invasion, and proinflammatory mediator production in the hypoxic and nutrient deprived microenvironment that develops in the RA joint. We will focus in this review on clinical options for better stratification of patients through prognostic metabolomic analysis and on whether or not some of the therapeutic options explored in cancer could potentially increase the drug armamentarium in rheumatic diseases.
2. Joint Metabolism and Diagnostic Imaging
The use of metabolomic profiles to find novel biomarkers to help diagnose or stratify RA patients has been described [6]. Analysis of metabolites using one-dimensional nuclear magnetic resonance (1D NMR) spectroscopy or mass spectrometry coupled to gas or liquid phase separation techniques, have shown unique metabolic and lipid profiles in the plasma of RA patients and pre-symptomatic subjects compared with healthy donors [6]. For instance, acyl-carnitines, lysophosphatidylcholines (LPCs), and metabolites from tryptophan metabolism, were found to be enriched in plasma from pre-symptomatic patients [7]. This approach has highlighted urinary [8], serum, and synovial fluid metabolite signatures that distinguish RA from psoriatic arthritis and other diseases [6,9,10,11,12,13]. It has also described urine and plasma metabolic profiles that predict patient responses to biological therapies including etanercept, rituximab, and tocilizumab [14,15,16,17], highlighting the power of metabolomics in stratifying patients and directing RA treatment.
However, metabolomic profiles in serum, plasma, or urine do not necessarily correlate with joint metabolism. Other approaches are needed to identify synovial metabolic changes. Structural imaging techniques including radiography, ultrasound, and MRI, though very useful, fail to provide information on the underlying biochemical processes. Thus, non-invasive bioimaging techniques are of increasing interest to improve clinical diagnostics or to monitor arthritic disease. The ideal synovial biomarker probe would be a non-invasive probe able to identify cellular or molecular markers, which could help to discriminate at baseline between responders and non-responders to treatment, possibly leading to a more efficient and personalized treatment. Also, the analysis of serial synovial images would be particularly advantageous to detect changes in the synovial membrane so it can be used to determine the early effects of treatment. Thus, patient stratification based on pathological metabolic pathways prior to therapeutic intervention could be exploited in order to identify biomarker predictors of clinical outcomes and responses to therapy [18,19]. Noninvasive metabolic imaging modalities that include positron emission tomography (PET) and magnetic resonance spectroscopy (MRS) could help in patient stratification. Stable isotope resolved metabolomics studies of the synovium using liquid chromatography and gas chromatography mass spectrometry (LC-MS and GC-MS) can be utilized to complement noninvasive imaging techniques (Figure 1).
2.1. Positron Emission Tomography
PET imaging works by detection of gamma rays from positron emitting radionuclides that have been injected into the patient. The most commonly used radionuclide is 18F but there is a wide range of radionuclides available—the more commonly used isotopes include 18F, 11C, and 15O.
2.1.1. 18F-FDG PET/CT (Positron Emission Tomography with 2-deoxy-2-(fluorine-18)fluoro-D-glucose Integrated with Computed Tomography)
18F-FDG works by entering the cell through glucose transporters where it is rapidly phosphorylated by hexokinase into 18F-FDG-6-phosphate where it can no longer be metabolized. The high consumption of glucose by advanced tumors made PET imaging with 18F-FDG an ideal probe to detect glycolytic tumors. However, the use of FDG to visualize tumor metabolic activity can also identify non-tumor cells that also have an increased metabolic activity at the inflammatory sites. In arthritis, synovial FLS and STM were shown to contribute to a high level of FDG-PET accumulation in the RA pannus [20]. Recent work has shown that the number of PET-positive joints in 28 and 68 joints was significantly correlated with the swollen and tender joint counts in RA patients [21]. 18F-FDG PET activity within days or weeks of initiating therapy correlates significantly with clinical endpoints. Thus, quantitative FDG-PET/CT-based assessment of inflammatory activity present in the joints of RA patients might be a promising approach for the whole-body assessment of RA disease activity and treatment response [22,23,24,25,26]. Additionally, it can also be used to detect high-risk disease complications at an early stage [27,28], such as atlanto-axial joint involvement, or co-morbidities including aortic inflammation [28]. A major drawback of PET imaging with 18F-FDG is that some normal cells in the brain, heart, and brown adipose tissue also have high metabolic rates and utilize above-average amounts of glucose, which often leads to the generation of false positive results.
2.1.2. 11C-Choline PET/CT
Choline is a vitamin-like essential nutrient that is phosphorylated by choline kinase (ChoK), the enzyme that catalyzes the first step in the de novo synthesis of the phosphatidylcholine pathway [29]. Choline transporters CTL1 and CTL2 are expressed in the majority of the cells within the joint, in particular FLS and STM [30,31]. 11C-choline PET scanning, which is already in clinical use for identifying prostate cancer metastasis, showed increased choline uptake in inflammatory arthritis [32] and elevated levels of choline is present in RA FLS and synovium [33,34,35]. Although the use of choline radiotracers for diagnosis or stratification needs further examination, some studies have shown that there is a substantial difference in the synovial choline levels in RA patients compared to that of osteoarthritis (OA) patients, in particular in female RA patients [34].
2.1.3. 18F-(2S,4R)4-fluoroglutamine Glutamine-, 11C-acetate-, 11C-methonine-PET/CT, O-(2-18Fluoroethyl)-L-tyrosine (FET), L-3-(18F)-Fluoro-α-methyl tyrosine (FAMT), and 11C-DASA23
Tumors or inflamed tissues do not solely rely on glucose. In recent years, the use of amino acid PET tracers is gaining attention for the diagnosis and evaluation of disease progression in several types of tumors, such as low-grade gliomas, and lung and breast cancer [36,37]. Some studies suggest that amino acid tracers are more sensitive in differentiating cancer cells from inflammatory cells, in part due to the upregulation of amino acid transport systems in cancer cells [38]. Transfer of amino acids across the plasma membrane is observed during metabolic stress. Several amino acid transporters have also been involved in the pathogenesis of other diseases including metabolic diseases such as obesity and diabetes [39]. Therefore, radiolabeling additional metabolites such as acetate, methionine, and glutamine with either 18F or 11C provide opportunities to perform broad profiling of tissue metabolism. For instance, it is believed that some tumors cannot be imaged with 18F-FDG as they do not derive their energy through glycolysis and instead probably use the glutaminolysis pathway as an alternate source of energy. Preliminary results suggest that 18F-(2S,4R)4-fluoroglutamine PET may be a new tool for probing in vivo metabolism of glutamine in cancer patients and for guiding glutamine targeted therapeutics [40,41]. 11C-acetate is converted to acetyl-CoA and used in mitochondria in the TCA cycle or incorporated into cell membranes. 11C-methionine and tyrosine tracers are used as a marker of amino acid uptake and protein synthesis primarily in cancer where uptake of the radiotracer correlates with tumor grade. Finally, Gambhir and colleagues reported the generation of a PET imaging probe specific for PKM2 using a class of N, N-diarylsulfonamides (DASA) known to promote PKM2 tetramer formation [42]. Although it was suggested that glutamine metabolism and glutaminase might be involved in the pathogenesis of RA fibroblasts [43], further studies are needed to determine whether or not these PET modalities could be useful for RA patient stratification.
2.2. Magnetic Resonance Spectroscopy Imaging (MRSI)
Magnetic Resonance (MR) imaging is most frequently used to determine anatomical registration of the tissues. Single-voxel MR spectroscopy (MRS) and multi-voxel MR spectroscopic imaging (MRSI) constitute another technique that enables detailed detection of cellular metabolic activity. The chemical composition forms a well-defined region of interest (ROI) or volume of interest in any organ of the human body that can be characterized using radiofrequency signals generated by nuclear spins of magnetic resonance active nuclei including 1H, 31P and 13C [44,45,46]. Most commonly, MRS has been used to evaluate endogenous 1H signals from choline-containing molecules, especially in the brain. 31P gathers information on the energy status of the tissue through the observation of various phosphate metabolites. Finally, 13C-labeled substrates provide dynamic metabolic flux information. However, one of the limitations of 1H MRS imaging is low sensitivity. Although several methods have allowed for an increase in the signal to noise ratio, this technology can mostly detect a few abundant metabolites in the imaged tissue, namely choline, glutamate, glutamine, lactate, aspartate, and phospholipid metabolites. Of interest, a few reports detected cerebral magnetic resonance spectroscopy changes in rheumatic diseases. Compared with healthy controls, patients with fibromyalgia had significantly higher levels of glutamate + glutamine and higher glutamate + glutamine/creatine (Glx/Cr) ratios in the posterior gyrus among other metabolic changes [47,48]. In another study, patients with active RA had a significantly higher ratio of choline to creatine and a significantly lower ratio of N-acetylaspartate to choline than did patients with inactive RA [49]. However, no studies have determined MRS changes in the arthritis synovium. As with other modalities of non-invasive imaging, monitoring response to therapy is one of the most promising aspects of MRS imaging.
2.3. Stable Isotope Resolved Metabolomics Studies
The noninvasive metabolism imaging methods discussed above can be complemented with mass spectrometry analysis of the inflamed synovial tissue. Little is known about metabolic or lipidomic profiling of the synovial tissue [34,35] although the increasing interest in synovial biopsies to obtain inflamed synovial tissue from joints [50] could improve understanding of the metabolic events in these diseases. It should be noted that although identification and quantification of endogenous and exogenous metabolic biomarkers can provide a metabolic snapshot of the status of a living organism, it cannot provide an unambiguous picture of the metabolic flux between different cellular compartments. For instance, an increase in the concentration of a metabolite can be associated with either the upregulation of the enzyme responsible for the metabolite synthesis, or the downregulation of the one consuming it. Several heavy isotopes including deuterium (2H), carbon (13C), nitrogen (15N), and oxygen (18O) have been used to aid in capturing the direction of a metabolic perturbation via the interpretation of stable isotope patterns and these datasets are nowadays increasingly being used in different mathematical modeling approaches, such as metabolic flux analysis. Stable isotope resolved metabolomics studies utilizes liquid chromatography mass spectrometry (LC-MS) or gas chromatography mass spectrometry (GC-MS) as a direct means to measure the distribution of labeled metabolites in the tissue and could potentially inform understanding of metabolic events in the RA synovium [51,52].
3. Metabolic Pathways as Therapeutic Targets in Rheumatoid Arthritis
Recent findings demonstrate the additional and consequent alterations in cellular signaling pathways and in the tumor microenvironment, including changes in the metabolism of glucose, lipids, and amino acids [53,54]. Therefore, in addition to ATP synthesis, metabolic changes appear to be a means of supplying cancer cells with the precursors of proteins, lipids, amino acids, and nucleic acids for building their cellular structure and maintaining their upregulated proliferation.
Due to the importance of metabolic alterations in the development and progression of cancer, several agents targeting cancer metabolism have been developed and evaluated under preclinical and clinical studies [55,56,57,58,59,60,61]. Some metabolism-targeting agents, such as mTOR inhibitors (rapamycin -sirolimus-, everolimus, and temsirolius) and metformin (an AMPK activator and mitochondrial Complex I inhibitor) are now approved for clinical use. Strategies targeting different metabolic alterations for anticancer therapy that could potentially be used in RA are detailed in the following sections and summarized in Figure 2, Table 1 and Table 2. In fact, rheumatologists already use the antimetabolites methotrexate (MTX) and leflunomide for the treatment of patients with inflammatory arthritis. Teriflunomide, the active metabolite of leflunomide, achieves its effects by inhibiting the mitochondrial enzyme dihydroorotate dehydrogenase (an enzyme involved in de novo pyrimidine synthesis). Methotrexate, developed as a folic acid analogue, inhibits purine and pyrimidine synthesis, although recent studies have indicated that other mechanisms such as adenosine accumulation can contribute to its effect in RA.
3.1. Glycolysis
The shift from oxidative phosphorylation to glycolytic ATP production is a common feature of activated and reactive cells such as cancer cells, fibroblasts, and macrophages [1,62,63]. As mentioned above, glucose uptake has been used to monitor tumor growth and to identify metabolically active sites such as RA joints or other inflammatory sites. Glycolysis is a multistep process that mobilizes glucose to produce pyruvate with a net yield of two adenosine triphosphate molecules. Although glycolysis is significantly less efficient producing ATP than the highly efficient mitochondria that generates 30-36 ATP molecules from a single glucose molecule, it is the preferential source of ATP under hypoxic conditions [64]. Inflammatory sites, such as inflamed joints, are overcrowded environments with scarce oxygen supply [65,66]. Synovial fluid is enriched in hypoxia-inducible factor 1 alpha (HIF1α), which contributes to RA pathogenesis, increases angiogenesis, inflammation, apoptosis, oxidative damage, and cartilage erosion [66]. Importantly, HIF1α is also a crucial regulator of glycolysis. In patients, synovial glycolytic marker expression positively correlated with reduced oxygen and macroscopic and microscopic changes in the joint, linking the glycolytic switch with hypoxia and inflammation [67]. The effect of HIF1α on glycolysis contributes to the pathogenic capacity of the majority of cells in the RA joint, including the production of proinflammatory cytokines such as IL-1β by macrophages [68], FLS survival [69], and FLS migration and invasion [66]. In addition, by inducing glycolysis, HIF1α serves as a metabolic checkpoint that supports Th17 development in detriment of Treg cells differentiation [70]. Yet, a recent report demonstrated a key function of HIF1α in driving the IL-10 expression in B cells [71].
Among the HIF1α−transcriptionally regulated genes, glucose transporter 1 (GLUT1) and lactate dehydrogenase (LDH) are upregulated in RA [1,60,72,73]. In the synovial lining, uptake of glucose is provided through Glut1 overexpression, which is accompanied by an increase in the glycolytic signature in the stromal compartment [60]. Glucose availability and uptake is crucial for the proliferative and invasive capacities of FLS [60]. The therapeutic use of GLUT1 blockers has been proposed to attenuate cancer cell proliferation [55], although none of the small molecules designed to block or reduce GLUT1 activity have met the standards to move forward to human studies. In contrast, a pilot study and Phase 1 study using an antisense oligonucleotide inhibitor of HIF1α has been tested in adults with advanced solid tumors, although the safety and efficacy reducing glycolysis is still unknown [74].
Another key glycolysis regulator downstream of HIF1α is the rate-limiting enzyme Hexokinase II (HK2), which is predominantly expressed in FLS within the RA joint [75]. Overexpression of HK2 in FLS provides a migratory and invasive advantage that is abolished when HK2 is ablated, and has attenuated the severity of bone and cartilage damage in a mouse model of inflammatory arthritis [75]. Importantly, ablation of glycolytic genes or treatment with 3-bromopyruvate, which antagonizes hexokinase II, significantly reduced the severity of mouse arthritis [60,76,77,78]. Methotrexate is a first-line therapeutic option for many RA patients. Interestingly, methotrexate treatment significantly reduced HK2 expression and glucose/fructose carriers (SLC2A5, a member of the solute carrier family 2) in human FLS, suggesting that FLS glycolytic activity can be modulated by methotrexate [79]. Although HK2 specific inhibitors are not available, steps downstream of HK2 can be inhibited by the use of 2-deoxyglucose (2-DG), which is a derivative of glucose that can be phosphorylated by HK2 but cannot be mobilized through the succeeding steps of glycolysis. This results in the accumulation of phosphorylated 2-DG causing product inhibition of HK2. Mouse studies have shown that 2-DG reduces cancer cell proliferation and the severity of the spontaneous murine models of arthritis [76]. These preclinical data have precipitated the examination of 2-DG in phase I/II trials for treatment of advanced cancer. Although the efficacy of 2-DG treatment in cancer progression is still unknown, only mild adverse effects have been observed, which include nausea and glucopenia, encouraging evaluation of this agent for treatment of inflammatory diseases such as RA, in which glycolysis is hyperactive.
Another important rate-limiting enzyme in glycolysis is pyruvate kinase, PKM2, which catalyzes phosphoenolpyruvic acid and ADP to pyruvate and ATP. PKM2 is overexpressed in many cancers and has a crucial role in glycolytic shift in immune cells [80]. PKM2 not only generates pyruvate, but also has multiple binding partners, such as HIF1α and Oct-4, which control inflammation and stem cell maintenance [81]. Monocytes/macrophages from patients with cardiovascular diseases, a common comorbidity of RA, have an increased glucose uptake and glycolytic flux, which causes mitochondrial stress and ROS production [82]. As a result, PKM2 dimerizes and translocates to the nucleus to activate the STAT3 transcriptional program that controls cytokine production [82]. Interestingly, inhibition of JAK/STAT3 signaling with Tofacinib, a drug approved for severe RA and active psoriasis, induces oxidative phosphorylation and maximal respiratory capacity of FLS while shutting down key glycolytic enzymes including HK2 and LDHA [83]. Currently, PKM2 inhibitor TLN-232/CAP-232 is evaluated in Phase II trials for refractory and metastatic renal cell carcinoma (NCT00422786) and recurrent metastatic melanoma (NCT00735332). It is reasonable to think that PKM2 inhibition may also attenuate the progression of the pathogenesis not only of RA but also of OA, as PKM2 overexpression controls glycolysis and extracellular matrix dynamic in chondrocytes [84]. Another critical enzyme in the glycolytic breakdown of glucose is the bifunctional 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase (PFKFB) enzymes. In particular, PFKFB3 activity is defective in CD4+ T cells in RA patients which results in energy deprivation that prone cells to undergo apoptosis [85]. PFKFB3 inhibition reduced glucose uptake and utilization which resulted in decreased lactate production [86]. These inhibition of the glycolytic flux by small molecule inhibitors of PFKFB3 significantly reduces FLS migration and invasion, and the production of inflammatory mediators [67,86]. Conversely, fructose 1,6-bisphosphate (FBP), a high-energy intermediate of glycolysis, attenuated experimental arthritis by activating the anti-inflammatory adenosinergic pathway [87].
Active and sustained glycolytic activity also leads to lactate overproduction and release to the extracellular milieu. In tumors, lactate contributes to cell-to-cell communication and has an immunosuppressive effect on T cells [88]. Blockage of subtype-specific lactate transporters on T cells results in their release from the inflammatory site during peritonitis, supporting the role of lactate in T cell entrapment and function [89]. In RA patients, acidosis of synovial fluid occurs but it varies significantly between individuals. In obese mice, lactate dependent activation of HIF1α induces proinflammatory cytokine production [90]. Four members of the SLC16A family (SLC16A1, SLC16A3, SLC16A7 and SLC16A8), and two sodium-coupled lactate cotransporters (SLC5A12 and SLC5A8) are monocarboxylate transporters (MCTs) involved in lactate homeostasis [91,92]. Inhibition of MCTs or LDH activity by several non-specific inhibitors have been evaluated in preclinical cancer models; a specific inhibitor for human evaluation in patients remains to be discovered [93,94]. Lactate levels, or more precisely, the conversion of pyruvate into lactate, can be monitored by hyperpolarized MRS using 13C-pyruvate to assess tumor response [95]. As mentioned above, T cells in RA patients show deficient glycolytic flux, which results in low intracellular pyruvate levels and ATP scarcity. Metabolically challenged T cells initiate fatty acid synthesis and the formation of lipid droplets, which induces podosome scaffolding protein TKS5 overexpression, and results in hypermotility and T cell infiltration of synovial tissue [96].
Pyruvate molecules generated during glycolysis is converted to acetyl CoA to fuel the Krebs cycle. Emerging evidences show that Krebs cycle intermediates classically associated with metabolic functions also possess signaling functions as inflammatory mediators. Metabolic profiling has revealed itaconic acid as a potential marker of RA. Importantly, this increased levels of itaconic acid can be attenuated by treatment with infliximab, a biologic drug targeting tumor necrosis factor (TNF) [97]. In macrophages, itaconate acts as an anti-inflammatory factor that connect metabolism with oxidative and electrophilic stress responses and immune responses limiting HIF1α and cytokines production [98]. Succinate is another Krebs cycle intermediate that is abundant in RA synovial fluids. Synovial succinate correlates with enhanced release of IL-1β by macrophages in a mechanism that involves the overexpression of succinate receptor SUCNR1/GPR91 [99]. In addition, SUCNR1/GPR91 functions as a chemotactic signal for dendritic cells recruitment into lymph nodes which leads to Th17 cells expansion and the development of experimental antigen-induced arthritis [100]. Consistently, Sucnr1 ablation prevented articular hyperplasia, neutrophils infiltration, Th17 expansion, and the number of cytokines in the joint [100].
3.2. Glutaminolysis
Glutamine is another important carbon source that provides energy for respiration and serves as a precursor for the synthesis of nucleotides and proteins. Elevated glycolytic activity results in conversion of pyruvate to lactate rather than being incorporated into the tricarboxylic acid (TCA) cycle. To compensate, cancer cells rely on increased glutaminolysis [101]. Consistent with their metabolic similarities with cancer cells, reliance on glutamine is also a feature of FLS [43]. Inhibition or genetic ablation of glutaminase 1 (GLS1), the enzyme that converts glutamine to glutamate, inhibits RA-FLS proliferation and ameliorate the severity of experimental autoimmune arthritis [43]. Glutamate, which is converted to alpha ketoglutarate (a-KG) and channeled to the TCA cycle, is increased in the synovial fluid of RA patients and correlates with increased inflammation and IL-6 production by FLS [102]. In addition to RA, global and targeted metabolomic studies have shown that there is an enrichment of glutamine in the synovial fluid of OA patients compared to the synovial fluid from control individuals [103]. The use of radiotracers to visualize glutamine flux and metabolism in human tumors is under evaluation. 18F-(2S,4R)-4-fluoroglutamine has effectively depicted predominantly aggressive tumors of those which carried mutations related to glutamine metabolism [104]. Whether this technique can be used as an alternative to 18F-FDG or be used for RA diagnosis is still unknown. However, modulation of glutamine utilization under pathological conditions has been evaluated in cancer preclinical models. In particular, the blockage of glutamine transporter SLC1A5 has shown potent antitumor activity; however, its use in humans has been ruled out due to its effect on healthy cells [105]. Several small molecules that target GLS also have shown therapeutic potential for cancer patients [106,107]. The GLS1 inhibitor, CB839, which has proved to reduce cancer cells viability and proliferation, is currently being evaluated in clinical trials (NCT02071862, NCT02071888, NCT03428217).
3.3. Choline Metabolism
Increased phospholipid synthesis was classically associated with enhanced proliferation of cells. However, in recent years, several studies have revealed that phospholipid synthesis has more biological functions. In particular, the uptake, mobilization, and phosphorylation of choline by ChoK is critical for the de novo synthesis of the phosphatidylcholine pathway, also known as the Kennedy pathway. This pathway is elevated in activated cells and play important roles in inflammation [32,108,109,110]. RA FLS exhibit the so-called ‘GPC-to-PCho switch’ that is observed in cancer cells. Its activation is characterized by increased levels of phosphocholine (PCho) and total choline-containing metabolites along with a decrease in glycerophosphocholine (GPC)/PCho ratio, which indicates activation of this pathway [109]. FLS activators, including the proinflammatory cytokines TNF and IL-1β, and growth factors such as PDGF, induced ChoK accumulation [109]. Blocking ChoK activity by using a small molecule inhibitor limits the proliferative and migratory capacity of FLS by interfering with metalloproteases activity and Akt activation. In vivo, ChoK inhibition attenuated joint inflammation and destruction [109]. Of interest, a recent paper identified the choline transporters in RA FLS [31]. Their results suggested that CTL1 (high-affinity) and CTL2 (low-affinity), which were highly expressed in RA FLS, were critical for choline transport. They also showed that the choline uptake was significantly increased compared with that in OA FLS, suggesting an increase of this metabolism in RA FLS [31].
Activated macrophages also have a special avidity for choline, and choline transporters were also described in RA STM [30]. Inflammatory macrophages exhibit an enhanced uptake of choline that is rapidly phosphorylated by ChoK and mobilized through the Kennedy pathway to supply the phospholipids required for maintaining proper membrane fluidity and composition, facilitating cytokine production and release [111,112]. We have recently showed that when choline is limited, or after ChoK inhibition, the mitochondrial lipid profile is disrupted resulting in a reduction of ATP synthase activity and intracellular ATP, which subsequently activates the energy sensor and anti-inflammatory molecule AMPK [111]. AMPK activation then facilitates mitophagy and decreased NLRP3 inflammasome and IL-1β and IL-18 production [111].
Although the role of choline metabolism and choline kinase activity is well known in cancer cells, and many choline kinase inhibitors have shown antitumor properties, only TCD-717 has been evaluated in phase I clinical studies for the treatment of solid advanced tumors (NCT01215864). Importantly, the use of choline as a tracer, in addition to glucose, for cancer diagnosis and its potential use as a predictive factor of therapy response is currently on going in several trials. Although the safety of altering choline metabolism in humans still needs to be determined, further evaluation of choline metabolism and ChoK inhibition might open new approaches for controlling the progression of inflammatory diseases such as RA, OA, and gout.
3.4. Metabolic Regulators of Mitochondrial Function and Biogenesis (AMPK, mTOR, PGC1a)
Shift towards a glycolytic phenotype implies a reduction in mitochondrial function, which is also accompanied by impaired mitochondrial biogenesis. Reduced cellular ATP triggers the activation of the energy sensor AMPK, which then phosphorylates multiple downstream targets and turns off biosynthetic pathways that consume ATP, including the synthesis of protein, fatty acids, and lipids, while facilitating glucose uptake and fatty acid oxidation to promote ATP production [113]. AMPK also is known for triggering autophagy/mitophagy through phosphorylation of ULK1 and MFF, a process that saves energy by reutilizing waste or unused materials [114,115]. AMPK is essential for RA T cells. AMPK activation is dependent on myristoylation, and in RA it has been shown that T cells aberrant N-myristoyltranferase prevented AMPK activation. This induced a mTORC1 overactivation that facilitated the differentiation of Th1 into Th17 cells [116]. Furthermore, pharmacological activation of AMPK using a specific AMPK agonist reduced the expression and release of IL-6 [117]. Importantly, methotrexate is able to induce the activation of AMPK, which correlates with reduced inflammatory response in macrophages stimulated with LPS and TNF [118]. Metformin and its analog phenformin are glucose-lowering drugs used for diabetes mellitus patients. Although their exact mechanism of action is not clearly understood, their well-known effect on AMPK activity may also contribute to the beneficial secondary effects of these drugs, as reducing inflammatory markers, improving lipid metabolism, and attenuating experimental autoimmune arthritis [119,120,121,122,123,124]. Its potential role as immune modulator indicate that treatment with metformin may be of special interest in inflammatory and autoimmune diseases [125]. In particular, several clinical trials are evaluating the role of metformin in RA and co-morbidities (NCT02246257, NCT03686657), and in psoriatic arthritis (NCT02188654).
AMPK activation is known to reduce mTOR signaling and activate autophagy. The mTOR signaling pathway, which is a key regulator of protein synthesis, is unusually active in many cancers and autoinflammatory and autoimmune diseases. mTOR signaling is also crucial for monocyte differentiation from myeloid progenitors [126]. Inhibition of mTOR can be achieved by rapamycin, also known as sirolimus, or its analogs, so-called rapalogs, that are FDA approved to treat advanced cancer [127]. Currently, the use of rapalogs is under evaluation in several clinical trials to determine their possible use in autoimmune diseases. The small molecule temsirolimus was evaluated in active RA patients, concomitant with methotrexate (NCT00076206), and rapamycin was also assessed by pharmacodynamic studies on hyperuricemia in gout patients (NCT02959918). In addition, a recently completed trial evaluating the effect of sirolimus in pediatric autoimmune diseases, including systemic lupus erythematosus (SLE), inflammatory bowel disease, and RA showed that sirolimus led to a complete and long-lasting response in the majority of pediatric patients, suggesting that sirolimus should be considered in the treatment of children with SLE [128].
Another important regulator of multiple cellular processes that is downregulated in RA is SIRT1. SIRT1 has an important role in suppressing the activation of NF-κB-dependent inflammatory responses including COX-2 and iNOS production, and promotes the activation of antioxidant transcriptional program [129]. Both SIRT1 and SIRT6 are essential for maintaining cartilage homeostasis and the use of SIRT1 activating compounds has been proposed as therapeutic approach in OA [130]. In this sense, resveratrol, a polyphenol found in wines, is extensively studied as an SIRT activator, and exhibit potent antioxidant, anti-inflammatory, and anticancer activities [131]. Recently, resveratrol was reported to suppress the severity of inflammatory arthritis in mice [132,133]. A randomized controlled clinical trial of 100 RA patients (68 female, 32 male) showed that the clinical markers and the disease activity score assessment was lowered by resveratrol [134]. More importantly, serum biochemical markers, such as C-reactive protein, erythrocyte sedimentation rate, undercarboxylated osteocalcin, matrix metalloproteinase-3, TNF, and IL-6 were also significantly decreased in resveratrol-treated patients [134]. In another study, treatment with resveratrol as an adjuvant with meloxicam improved pain and joint function [135]. The evaluation of resveratrol in knee OA is also currently under evaluation (NCT02905799).
3.5. Amino Acid Uptake
Increased amino acid uptake is often found in certain types of tumors to fuel metabolism and protein synthesis [37]. Besides glutamine, other amino acids, including serine, have been shown to be important for initiating proper inflammatory responses. In macrophages, serine metabolism to produce glutathione is required for transcriptional regulation of IL-1β [136]. In proinflammatory macrophages, influx of the branch-chain amino acid leucine also contributes to cytokine production via mTORC1-induced glycolytic reprogramming [137]. Consistently, branched-chain aminotransferase 1 (BCAT1) controls oxygen consumption and glycolysis in macrophages. Its inhibition by ERG240 limits infiltration of inflammatory macrophages and therefore reduces the severity of experimental inflammatory arthritis [138]. In addition, RA synovium is enriched in L-type amino acid transporter gene LAT1 [139]. Expression of SLC7A5/LAT1 was found to be significantly elevated in monocytes derived from patients with RA [137]. RA FLS also overexpress LAT1 and has an increased uptake of leucine after IL-17 stimulation, which potentiates the FLS migratory capacity that was eliminated by blocking LAT1 [139]. Thus, tracing and targeting the uptake of serine and leucine may be useful to determine and decrease the inflammatory status in RA joints.
Another important amino acid for macrophage function is arginine. Arginine is the substrate of two enzymes, arginase to produce ornithine and urea, and nitric oxide synthase (NOS) to generate citrulline and nitric oxide (NO). Macrophages couple the arginine metabolism with polarization and functional phenotype [140,141]. Inflammatory macrophages, classically called M1, preferentially overexpress NOS and use arginine for NO production, which is a key effector of microbicidal activity [141]. In contrast, M2 macrophages preferentially express arginase to generate ornithine, which is the precursor of polyamines that contributes to proliferation and restoration of tissue homeostasis [141]. Of interest, arginase protein and activity are elevated in serum from RA patients [142]. Inhibition of arginase by different approaches has been clinically assessed in human cancers.
In the past few years, the enzyme indoleamine 2,3-dioxygenase (IDO), involved in the metabolism of the essential amino acid tryptophan, has gained attention. IDO is overexpressed in cancer and mediates immune tolerance. T cells exhibit sensitivity to tryptophan deprivation and to kynurenines, the products of tryptophan degradation [143,144]. IDO inhibitors were well tolerated in phase I studies [145,146]. As IDO inhibitors boosted “immunogenic” chemotherapy or immune checkpoint drugs [147], current clinical trials focus on evaluating the effects of combining IDO inhibitors with taxotere, sipuleucel-T (dendritic cell vaccine), and anti-PD1 (NCT03047928, NCT01219348, NCT01982487). Although increased tryptophan degradation measured as kynurenines/tryptophan ratio is elevated in the blood of RA patients [148], the performance of IDO inhibitors in arthritis is under debate as some works showed that IDO inhibition exacerbated disease severity in mouse models of arthritis [149,150].
3.6. Fatty Acid Synthesis
Fatty acid metabolism is a dynamic process of anabolic and catabolic reactions to maintain energy homeostasis. Synthesis of fatty acids is essential for building up metabolic intermediates to store energy, maintain cell membrane structures, and participate in intracellular and intercellular communication. When energy is needed, the cell can break down the fatty acids through β-oxidation. Balance between synthesis and degradation is impaired in many diseases, which leads to lipid accumulation. Fatty acid synthase, FASN, a key enzyme in the de novo synthesis of lipids, is found to be overexpressed in many cancers [151,152,153]. In macrophages, FASN is essential for maintaining optimal membrane composition [154]. Deletion of FASN in macrophages impairs the retention of cholesterol in the plasma membrane and alters Rho GTPase-dependent cell adhesion and migration [154]. Inhibition of FASN is under evaluation in several clinical trials in cancer patients (NCT03179904, NCT02980029, NCT02595372), after promising preclinical studies in animal models [155].
Fatty acids elongation determines chain length of saturated, monosaturated, and polyunsaturated fatty acids in cellular lipids. In some cancers, the enzymes that ultimately control the elongation of the fatty acids, the fatty acid elongases (ELOVL1-7), are dysregulated and can be used as predictive factors [156,157,158]. The role of fatty acid synthesis and elongation in the behavioral changes of RA FLS is yet to be clarified. FLS have a potent migratory and invasive capability, which presumably will require an active lipid remodeling. In addition, many of the inflammatory molecules that are present in the synovium stimulate fatty acid synthesis. Evaluation of this biosynthetic pathway, not only in FLS but also in other cells that play a role in the arthritic joint, such as macrophages, may offer novel targets to attenuate joint damage.
4. Conclusions
In the last decade, the use of new technologies to conduct metabolomic and lipidomic studies in body fluids have provided new insights into immune-mediated inflammatory diseases such as RA. It now become clearer that alterations in the lipid profile and the hyperactivation of metabolic pathways are hallmarks of RA, and that they can be potential biomarkers and therapeutic targets. Findings in human synovium or peripheral blood and preclinical studies in mouse models of inflammatory arthritis strongly suggest that agents that interfere with lipid metabolism and certain steps of glycolysis or other energy-related pathways can be therapeutic in RA (Table 3). Yet, we must emphasize that we are far from understanding the pathways that discern normal from pathogenic metabolic phenotypes of synovial cells. The widespread use of imaging techniques or mass spectrometry analysis in larger and more heterogeneous cohorts can provide signatures of metabolic disruption under pathogenic conditions. Additionally, further efforts are needed to fine tune metabolism by designing or improving current medications, or by using prodrugs that can only be activated when the target is hyperactive, to ensure the successful use of metabolic drugs with minimal off-targets. Whether or not chemotherapies that modulate the metabolism truly present an option to increase the drug armamentarium in rheumatic diseases remains to be determined.
Author Contributions
E.S.L.: A.C. and M.G. reviewed literature and wrote the review.
Funding
M.G. is supported by the National Institutes of Health under Award Numbers R01AR073324, and by Rheumatology Research Foundation.
Conflicts of Interest
The authors report no conflict of interest.
References
- Bustamante, M.F.; Garcia-Carbonell, R.; Whisenant, K.D.; Guma, M. Fibroblast-like synoviocyte metabolism in the pathogenesis of rheumatoid arthritis. Arthritis Res. Ther. 2017, 19, 110. [Google Scholar] [CrossRef]
- Falconer, J.; Murphy, A.N.; Young, S.; Clark, A.R.; Tiziani, S.; Guma, M.; Buckley, C.D. Synovial cell metabolism and chronic inflammation in rheumatoid arthritis. Arthritis Rheumatol. 2018, 70, 984–999. [Google Scholar] [CrossRef] [PubMed]
- Fearon, U.; Hanlon, M.M.; Wade, S.M.; Fletcher, J.M. Altered metabolic pathways regulate synovial inflammation in rheumatoid arthritis. Clin. Exp. Immunol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Fearon, U.; Canavan, M.; Biniecka, M.; Veale, D.J. Hypoxia, mitochondrial dysfunction and synovial invasiveness in rheumatoid arthritis. Nat. Rev. Rheumatol. 2016, 12, 385–397. [Google Scholar] [CrossRef]
- Weyand, C.M.; Goronzy, J.J. Immunometabolism in early and late stages of rheumatoid arthritis. Nat. Rev. Rheumatol. 2017, 13, 291–301. [Google Scholar] [CrossRef]
- Guma, M.; Tiziani, S.; Firestein, G.S. Metabolomics in rheumatic diseases: Desperately seeking biomarkers. Nat. Rev. Rheumatol. 2016, 12, 269–281. [Google Scholar] [CrossRef]
- Surowiec, I.; Arlestig, L.; Rantapaa-Dahlqvist, S.; Trygg, J. Metabolite and Lipid Profiling of Biobank Plasma Samples Collected Prior to Onset of Rheumatoid Arthritis. PLoS ONE 2016, 11, e0164196. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.; Julià, A.; Vinaixa, M.; Domènech, E.; Fernández-Nebro, A.; Cañete, J.D.; Ferrandiz, C.; Tornero, J.; Gisbert, J.P.; Nos, P.; et al. Urine metabolome profiling of immune-mediated inflammatory diseases. BMC Med. 2016, 14, 133. [Google Scholar] [CrossRef]
- Madsen, R.K.; Lundstedt, T.; Gabrielsson, J.; Sennbro, C.J.; Alenius, G.M.; Moritz, T.; Rantapaa-Dahlqvist, S.; Trygg, J. Diagnostic properties of metabolic perturbations in rheumatoid arthritis. Arthritis Res. Ther. 2011, 13, R19. [Google Scholar] [CrossRef] [PubMed]
- Hugle, T.; Kovacs, H.; Heijnen, I.A.; Daikeler, T.; Baisch, U.; Hicks, J.M.; Valderrabano, V. Synovial fluid metabolomics in different forms of arthritis assessed by nuclear magnetic resonance spectroscopy. Clin. Exp. Rheumatol. 2012, 30, 240–245. [Google Scholar] [PubMed]
- Kim, S.; Hwang, J.; Xuan, J.; Jung, Y.H.; Cha, H.-S.; Kim, K.H. Global Metabolite Profiling of Synovial Fluid for the Specific Diagnosis of Rheumatoid Arthritis from Other Inflammatory Arthritis. PLoS ONE 2014, 9, e97501. [Google Scholar] [CrossRef]
- Dubey, D.; Kumar, S.; Chaurasia, S.; Guleria, A.; Ahmed, S.; Singh, R.; Kumari, R.; Modi, D.R.; Misra, R.; Kumar, D. NMR-Based Serum Metabolomics Revealed Distinctive Metabolic Patterns in Reactive Arthritis Compared with Rheumatoid Arthritis. J. Proteome Res. 2018, 18, 130–146. [Google Scholar] [CrossRef]
- Anderson, J.R.; Chokesuwattanaskul, S.; Phelan, M.M.; Welting, T.J.M.; Lian, L.Y.; Peffers, M.J.; Wright, H.L. 1H NMR Metabolomics Identifies Underlying Inflammatory Pathology in Osteoarthritis and Rheumatoid Arthritis Synovial Joints. J. Proteome Res. 2018, 17, 3780–3790. [Google Scholar] [CrossRef]
- Cuppen, B.V.J.; Fu, J.; van Wietmarschen, H.A.; Harms, A.C.; Koval, S.; Marijnissen, A.C.; peeters, J.J.; Bijlsma, J.W.; Tekstra, J.; van Laar, J.M.; et al. Exploring the Inflammatory Metabolomic Profile to Predict Response to TNF-α Inhibitors in Rheumatoid Arthritis. PLoS ONE 2016, 11, e0163087. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, S.R.; Kavanaugh, A.; Lodi, A.; Wang, B.; Boyle, D.; Tiziani, S.; Guma, M. Metabolomic profiling predicts outcome of rituximab therapy in rheumatoid arthritis. RMD Open 2016, 2, e000289. [Google Scholar] [CrossRef] [PubMed]
- Tatar, Z.; Migne, C.; Petera, M.; Gaudin, P.; Lequerre, T.; Marotte, H.; Tebib, J.; Pujos Guillot, E.; Soubrier, M. Variations in the metabolome in response to disease activity of rheumatoid arthritis. BMC Musculoskelet. Disord. 2016, 17, 353. [Google Scholar] [CrossRef]
- Teitsma, X.M.; Yang, W.; Jacobs, J.W.G.; Petho-Schramm, A.; Borm, M.E.A.; Harms, A.C.; Hankemeier, T.; van Laar, J.M.; Bijlsma, J.W.J.; Lafeber, F.P.J.G. Baseline metabolic profiles of early rheumatoid arthritis patients achieving sustained drug-free remission after initiating treat-to-target tocilizumab, methotrexate, or the combination: Insights from systems biology. Arthritis Res. Ther. 2018, 20, 230. [Google Scholar] [CrossRef]
- Astorri, E.; Nerviani, A.; Bombardieri, M.; Pitzalis, C. Towards a stratified targeted approach with biologic treatments in rheumatoid arthritis: Role of synovial pathobiology. Curr. Pharm. Des. 2015, 21, 2216–2224. [Google Scholar] [CrossRef]
- Pitzalis, C.; Kelly, S.; Humby, F. New learnings on the pathophysiology of RA from synovial biopsies. Curr. Opin. Rheumatol. 2013, 25, 334–344. [Google Scholar] [CrossRef]
- Matsui, T.; Nakata, N.; Nagai, S.; Nakatani, A.; Takahashi, M.; Momose, T.; Ohtomo, K.; Koyasu, S. Inflammatory cytokines and hypoxia contribute to 18F-FDG uptake by cells involved in pannus formation in rheumatoid arthritis. J. Nucl. Med. 2009, 50, 920–926. [Google Scholar] [CrossRef]
- Lee, S.J.; Jeong, J.H.; Lee, C.H.; Ahn, B.C.; Eun, J.S.; Kim, N.R.; Kang, J.W.; Nam, E.J.; Kang, Y.M. Development and validation of an 18F-FDG PET/CT-based tool for the evaluation of joint counts and disease activity in patients with rheumatoid arthritis. Arthritis Rheumatol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.S.; Shejul, Y.; Asopa, R.; Basu, S. Quantitative Metabolic Volumetric Product on 18Fluorine-2fluoro-2-deoxy-d-glucose-positron Emission Tomography/Computed Tomography in Assessing Treatment Response to Disease-modifying Antirheumatic Drugs in Rheumatoid Arthritis: Multiparametric Analysis Integrating American College of Rheumatology/European League Against Rheumatism Criteria. World J. Nucl. Med. 2017, 16, 293–302. [Google Scholar] [PubMed]
- Okamura, K.; Yonemoto, Y.; Okura, C.; Higuchi, T.; Tsushima, Y.; Takagishi, K. Evaluation of tocilizumab therapy in patients with rheumatoid arthritis based on FDG-PET/CT. BMC Musculoskelet. Disord. 2014, 15, 393. [Google Scholar] [CrossRef]
- Vijayant, V.; Sarma, M.; Aurangabadkar, H.; Bichile, L.; Basu, S. Potential of 18F-FDG-PET as a valuable adjunct to clinical and response assessment in rheumatoid arthritis and seronegative spondyloarthropathies. World J. Radiol. 2012, 4, 462–468. [Google Scholar] [CrossRef]
- Roivainen, A.; Hautaniemi, S.; Mottonen, T.; Nuutila, P.; Oikonen, V.; Parkkola, R.; Pricop, L.; Ress, R.; Seneca, N.; Seppanen, M.; et al. Correlation of 18F-FDG PET/CT assessments with disease activity and markers of inflammation in patients with early rheumatoid arthritis following the initiation of combination therapy with triple oral antirheumatic drugs. Eur. J. Nucl. Med. Mol. Imaging 2013, 40, 403–410. [Google Scholar] [CrossRef]
- Fosse, P.; Kaiser, M.J.; Namur, G.; de Seny, D.; Malaise, M.G.; Hustinx, R. 18F- FDG PET/CT joint assessment of early therapeutic response in rheumatoid arthritis patients treated with rituximab. Eur. J. Nucl. Med. Mol. Imaging 2018, 2, 6. [Google Scholar] [CrossRef]
- Kubota, K.; Ito, K.; Morooka, M.; Minamimoto, R.; Miyata, Y.; Yamashita, H.; Takahashi, Y.; Minori, A. FDG PET for rheumatoid arthritis: Basic considerations and whole-body PET/CT. Ann. N. Y. Acad. Sci. 2011, 1228, 29–38. [Google Scholar] [CrossRef]
- Kubota, K.; Ito, K.; Morooka, M.; Mitsumoto, T.; Kurihara, K.; Yamashita, H.; Takahashi, Y.; Minori, A. Whole-body FDG-PET/CT on rheumatoid arthritis of large joints. Ann. Nucl. Med. 2009, 23, 783–791. [Google Scholar] [CrossRef]
- Aoyama, C.; Liao, H.; Ishidate, K. Structure and function of choline kinase isoforms in mammalian cells. Prog. Lipid Res. 2004, 43, 266–281. [Google Scholar] [CrossRef]
- Beckmann, J.; Schubert, J.; Morhenn, H.G.; Grau, V.; Schnettler, R.; Lips, K.S. Expression of choline and acetylcholine transporters in synovial tissue and cartilage of patients with rheumatoid arthritis and osteoarthritis. Cell Tissue Res. 2015, 359, 465–477. [Google Scholar] [CrossRef]
- Seki, M.; Kawai, Y.; Ishii, C.; Yamanaka, T.; Odawara, M.; Inazu, M. Functional analysis of choline transporters in rheumatoid arthritis synovial fibroblasts. Mod. Rheumatol. 2017, 27, 995–1003. [Google Scholar] [CrossRef]
- Roivainen, A.; Parkkola, R.; Yli-Kerttula, T.; Lehikoinen, P.; Viljanen, T.; Mottonen, T.; Nuutila, P.; Minn, H. Use of positron emission tomography with methyl-11C-choline and 2-18F-fluoro-2-deoxy-D-glucose in comparison with magnetic resonance imaging for the assessment of inflammatory proliferation of synovium. Arthritis Rheum. 2003, 48, 3077–3084. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.K.; Kim, S.; Hwang, J.; Kim, J.; Kim, K.H.; Cha, H.S. GC/TOF-MS-based metabolomic profiling in cultured fibroblast-like synoviocytes from rheumatoid arthritis. Jt. Bone Spine 2016, 83, 707–713. [Google Scholar] [CrossRef]
- Volchenkov, R.; Dung Cao, M.; Elgstoen, K.B.; Goll, G.L.; Eikvar, K.; Bjorneboe, O.; Bathen, T.F.; Holen, H.L.; Kvien, T.K.; Skalhegg, B.S. Metabolic profiling of synovial tissue shows altered glucose and choline metabolism in rheumatoid arthritis samples. Scand. J. Rheumatol. 2017, 46, 160–161. [Google Scholar] [CrossRef] [PubMed]
- Hitchon, C.A.; El-Gabalawy, H.S.; Bezabeh, T. Characterization of synovial tissue from arthritis patients: A proton magnetic resonance spectroscopic investigation. Rheumatol. Int. 2009, 29, 1205–1211. [Google Scholar] [CrossRef] [PubMed]
- Naslund, O.; Smits, A.; Forander, P.; Laesser, M.; Bartek, J., Jr.; Gempt, J.; Liljegren, A.; Daxberg, E.L.; Jakola, A.S. Amino acid tracers in PET imaging of diffuse low-grade gliomas: A systematic review of preoperative applications. Acta Neurochir. 2018, 160, 1451–1460. [Google Scholar] [CrossRef]
- Sun, A.; Liu, X.; Tang, G. Carbon-11 and Fluorine-18 Labeled Amino Acid Tracers for Positron Emission Tomography Imaging of Tumors. Front. Chem. 2017, 5, 124. [Google Scholar] [CrossRef]
- Deng, H.; Tang, X.; Wang, H.; Tang, G.; Wen, F.; Shi, X.; Yi, C.; Wu, K.; Meng, Q. S-11C-methyl-L-cysteine: A new amino acid PET tracer for cancer imaging. J. Nucl. Med. 2011, 52, 287–293. [Google Scholar] [CrossRef]
- Kandasamy, P.; Gyimesi, G.; Kanai, Y.; Hediger, M.A. Amino acid transporters revisited: New views in health and disease. Trends Biochem. Sci. 2018, 43, 752–789. [Google Scholar] [CrossRef]
- Venneti, S.; Dunphy, M.P.; Zhang, H.; Pitter, K.L.; Zanzonico, P.; Campos, C.; Carlin, S.D.; La Rocca, G.; Lyashchenko, S.; Ploessl, K.; et al. Glutamine-based PET imaging facilitates enhanced metabolic evaluation of gliomas in vivo. Sci. Transl. Med. 2015, 7, 274ra217. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Ploessl, K.; Zhou, R.; Mankoff, D.; Kung, H.F. Metabolic Imaging of Glutamine in Cancer. J. Nucl. Med. 2017, 58, 533–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witney, T.H.; James, M.L.; Shen, B.; Chang, E.; Pohling, C.; Arksey, N.; Hoehne, A.; Shuhendler, A.; Park, J.H.; Bodapati, D.; et al. PET imaging of tumor glycolysis downstream of hexokinase through noninvasive measurement of pyruvate kinase M2. Sci. Transl. Med. 2015, 7, 310ra169. [Google Scholar] [CrossRef]
- Takahashi, S.; Saegusa, J.; Sendo, S.; Okano, T.; Akashi, K.; Irino, Y.; Morinobu, A. Glutaminase 1 plays a key role in the cell growth of fibroblast-like synoviocytes in rheumatoid arthritis. Arthritis Res. Ther. 2017, 19, 76. [Google Scholar] [CrossRef] [PubMed]
- Momcilovic, M.; Shackelford, D.B. Imaging Cancer Metabolism. Biomol. Ther. 2018, 26, 81–92. [Google Scholar] [CrossRef] [Green Version]
- Jagannathan, N.R.; Sharma, U. Breast Tissue Metabolism by Magnetic Resonance Spectroscopy. Metabolites 2017, 7, 25. [Google Scholar] [CrossRef]
- Kim, M.M.; Parolia, A.; Dunphy, M.P.; Venneti, S. Non-invasive metabolic imaging of brain tumours in the era of precision medicine. Nat. Rev. Clin. Oncol. 2016, 13, 725–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fayed, N.; Garcia-Campayo, J.; Magallon, R.; Andres-Bergareche, H.; Luciano, J.V.; Andres, E.; Beltran, J. Localized 1H-NMR spectroscopy in patients with fibromyalgia: A controlled study of changes in cerebral glutamate/glutamine, inositol, choline, and N-acetylaspartate. Arthritis Res. Ther. 2010, 12, R134. [Google Scholar] [CrossRef]
- Valdes, M.; Collado, A.; Bargallo, N.; Vazquez, M.; Rami, L.; Gomez, E.; Salamero, M. Increased glutamate/glutamine compounds in the brains of patients with fibromyalgia: A magnetic resonance spectroscopy study. Arthritis Rheum. 2010, 62, 1829–1836. [Google Scholar] [CrossRef]
- Emmer, B.J.; van der Bijl, A.E.; Huizinga, T.W.; Breedveld, F.C.; Steens, S.C.; Th Bosma, G.P.; van Buchem, M.A.; van der Grond, J. Brain involvement in rheumatoid arthritis: A magnetic resonance spectroscopy study. Arthritis Rheum. 2009, 60, 3190–3195. [Google Scholar] [CrossRef]
- Kelly, S.; Humby, F.; Filer, A.; Ng, N.; Di Cicco, M.; Hands, R.E.; Rocher, V.; Bombardieri, M.; D’Agostino, M.A.; McInnes, I.B.; et al. Ultrasound-guided synovial biopsy: A safe, well-tolerated and reliable technique for obtaining high-quality synovial tissue from both large and small joints in early arthritis patients. Ann. Rheum. Dis. 2015, 74, 611–617. [Google Scholar] [CrossRef]
- Fan, T.W.; Lane, A.N.; Higashi, R.M. Stable Isotope Resolved Metabolomics Studies in Ex Vivo TIssue Slices. Bio-Protocol 2016, 6, e1730. [Google Scholar] [CrossRef]
- Lane, A.N.; Tan, J.; Wang, Y.; Yan, J.; Higashi, R.M.; Fan, T.W. Probing the metabolic phenotype of breast cancer cells by multiple tracer stable isotope resolved metabolomics. Metab. Eng. 2017, 43, 125–136. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Kepp, O.; Vander Heiden, M.G.; Kroemer, G. Metabolic targets for cancer therapy. Nat. Rev. Drug Discov. 2013, 12, 829–846. [Google Scholar] [CrossRef]
- Cantor, J.R.; Sabatini, D.M. Cancer cell metabolism: One hallmark, many faces. Cancer Discov. 2012, 2, 881–898. [Google Scholar] [CrossRef]
- Liu, Y.; Cao, Y.; Zhang, W.; Bergmeier, S.; Qian, Y.; Akbar, H.; Colvin, R.; Ding, J.; Tong, L.; Wu, S.; et al. A small-molecule inhibitor of glucose transporter 1 downregulates glycolysis, induces cell-cycle arrest, and inhibits cancer cell growth in vitro and in vivo. Mol. Cancer Ther. 2012, 11, 1672–1682. [Google Scholar] [CrossRef]
- Huijts, C.M.; Lougheed, S.M.; Bodalal, Z.; van Herpen, C.M.; Hamberg, P.; Tascilar, M.; Haanen, J.B.; Verheul, H.M.; de Gruijl, T.D.; van der Vliet, H.J.; et al. The effect of everolimus and low-dose cyclophosphamide on immune cell subsets in patients with metastatic renal cell carcinoma: Results from a phase I clinical trial. Cancer Immunol. Immunother. 2019, 68, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Lansky, A.; Wijns, W.; Xu, B.; Kelbaek, H.; van Royen, N.; Zheng, M.; Morel, M.A.; Knaapen, P.; Slagboom, T.; Johnson, T.W.; et al. Targeted therapy with a localised abluminal groove, low-dose sirolimus-eluting, biodegradable polymer coronary stent (TARGET All Comers): A multicentre, open-label, randomised non-inferiority trial. Lancet 2018, 392, 1117–1126. [Google Scholar] [CrossRef]
- Petrera, M.; Paleari, L.; Clavarezza, M.; Puntoni, M.; Caviglia, S.; Briata, I.M.; Oppezzi, M.; Mislej, E.M.; Stabuc, B.; Gnant, M.; et al. The ASAMET trial: A randomized, phase II, double-blind, placebo-controlled, multicenter, 2 x 2 factorial biomarker study of tertiary prevention with low-dose aspirin and metformin in stage I-III colorectal cancer patients. BMC Cancer 2018, 18, 1210. [Google Scholar] [CrossRef]
- Oladghaffari, M.; Shabestani Monfared, A.; Farajollahi, A.; Baradaran, B.; Mohammadi, M.; Shanehbandi, D.; Asghari Jafar Abadi, M.; Pirayesh Islamian, J. MLN4924 and 2DG combined treatment enhances the efficiency of radiotherapy in breast cancer cells. Int. J. Radiat. Biol. 2017, 93, 590–599. [Google Scholar] [CrossRef]
- Garcia-Carbonell, R.; Divakaruni, A.S.; Lodi, A.; Vicente-Suarez, I.; Saha, A.; Cheroutre, H.; Boss, G.R.; Tiziani, S.; Murphy, A.N.; Guma, M. Critical Role of Glucose Metabolism in Rheumatoid Arthritis Fibroblast-like Synoviocytes. Arthritis Rheumatol. 2016, 68, 1614–1626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, S.; Pandey, S.K.; Kumar, A.; Kujur, P.K.; Singh, R.P.; Singh, S.M. Antitumor and chemosensitizing action of 3-bromopyruvate: Implication of deregulated metabolism. Chem. Biol. Interact. 2017, 270, 73–89. [Google Scholar] [CrossRef]
- Sun, L.; Suo, C.; Li, S.T.; Zhang, H.; Gao, P. Metabolic reprogramming for cancer cells and their microenvironment: Beyond the Warburg Effect. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 51–66. [Google Scholar] [CrossRef]
- Kelly, B.; O’Neill, L.A. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015, 25, 771–784. [Google Scholar] [CrossRef] [Green Version]
- Epstein, T.; Gatenby, R.A.; Brown, J.S. The Warburg effect as an adaptation of cancer cells to rapid fluctuations in energy demand. PLoS ONE 2017, 12, e0185085. [Google Scholar] [CrossRef]
- Quinonez-Flores, C.M.; Gonzalez-Chavez, S.A.; Pacheco-Tena, C. Hypoxia and its implications in rheumatoid arthritis. J. Biomed. Sci. 2016, 23, 62. [Google Scholar] [CrossRef]
- Hua, S.; Dias, T.H. Hypoxia-Inducible Factor (HIF) as a Target for Novel Therapies in Rheumatoid Arthritis. Front. Pharmacol. 2016, 7, 184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biniecka, M.; Canavan, M.; McGarry, T.; Gao, W.; McCormick, J.; Cregan, S.; Gallager, L.; Smith, T.; Phelan, J.J.; Ryan, J.; et al. Dysregulated bioenergetics: A key regulator of joint inflammation. Ann. Rheum. Dis. 2016, 75, 2192–2200. [Google Scholar] [CrossRef] [PubMed]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef]
- Del Rey, M.J.; Valin, A.; Usategui, A.; Garcia-Herrero, C.M.; Sanchez-Arago, M.; Cuezva, J.M.; Galindo, M.; Bravo, B.; Canete, J.D.; Blanco, F.J.; et al. Hif-1alpha Knockdown Reduces Glycolytic Metabolism and Induces Cell Death of Human Synovial Fibroblasts Under Normoxic Conditions. Sci. Rep. 2017, 7, 3644. [Google Scholar] [CrossRef]
- Shi, L.Z.; Wang, R.; Huang, G.; Vogel, P.; Neale, G.; Green, D.R.; Chi, H. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J. Exp. Med. 2011, 208, 1367–1376. [Google Scholar] [CrossRef]
- Meng, X.; Grotsch, B.; Luo, Y.; Knaup, K.X.; Wiesener, M.S.; Chen, X.X.; Jantsch, J.; Fillatreau, S.; Schett, G.; Bozec, A. Hypoxia-inducible factor-1alpha is a critical transcription factor for IL-10-producing B cells in autoimmune disease. Nat. Commun. 2018, 9, 251. [Google Scholar] [CrossRef] [PubMed]
- Hurter, K.; Spreng, D.; Rytz, U.; Schawalder, P.; Ott-Knusel, F.; Schmokel, H. Measurements of C-reactive protein in serum and lactate dehydrogenase in serum and synovial fluid of patients with osteoarthritis. Vet. J. 2005, 169, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.J.; Husson, Z.M.A.; Hu, D.E.; Callejo, G.; Brindle, K.M.; Smith, E.S.J. Increased hyperpolarized [1-13C] lactate production in a model of joint inflammation is not accompanied by tissue acidosis as assessed using hyperpolarized 13C C-labelled bicarbonate. NMR Biomed. 2018, 31, e3892. [Google Scholar] [CrossRef] [PubMed]
- Jeong, W.; Rapisarda, A.; Park, S.R.; Kinders, R.J.; Chen, A.; Melillo, G.; Turkbey, B.; Steinberg, S.M.; Choyke, P.; Doroshow, J.H.; et al. Pilot trial of EZN-2968, an antisense oligonucleotide inhibitor of hypoxia-inducible factor-1 alpha (HIF-1alpha), in patients with refractory solid tumors. Cancer Chemother. Pharmacol. 2014, 73, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Bustamante, M.F.; Oliveira, P.G.; Garcia-Carbonell, R.; Croft, A.P.; Smith, J.M.; Serrano, R.L.; Sanchez-Lopez, E.; Liu, X.; Kisseleva, T.; et al. Hexokinase 2 as a novel selective metabolic target for rheumatoid arthritis. Ann. Rheum. Dis. 2018, 77, 1636–1643. [Google Scholar] [CrossRef]
- Abboud, G.; Choi, S.C.; Kanda, N.; Zeumer-Spataro, L.; Roopenian, D.C.; Morel, L. Inhibition of Glycolysis Reduces Disease Severity in an Autoimmune Model of Rheumatoid Arthritis. Front. Immunol. 2018, 9, 1973. [Google Scholar] [CrossRef]
- Okano, T.; Saegusa, J.; Nishimura, K.; Takahashi, S.; Sendo, S.; Ueda, Y.; Morinobu, A. 3-bromopyruvate ameliorate autoimmune arthritis by modulating Th17/Treg cell differentiation and suppressing dendritic cell activation. Sci. Rep. 2017, 7, 42412. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Lu, Q.; Fan, H.; Zhang, X.; Ge, L.; Tian, R.; Wang, S.; Feng, T.; Pan, J.; Feng, J.; et al. Inhibition of hexokinases holds potential as treatment strategy for rheumatoid arthritis. Arthritis Res. Ther. 2019, 21, 87. [Google Scholar] [CrossRef]
- Shervington, L.; Darekar, A.; Shaikh, M.; Mathews, R.; Shervington, A. Identifying Reliable Diagnostic/Predictive Biomarkers for Rheumatoid Arthritis. Biomark. Insights 2018, 13, 1177271918801005. [Google Scholar] [CrossRef]
- Wiese, E.K.; Hitosugi, T. Tyrosine Kinase Signaling in Cancer Metabolism: PKM2 Paradox in the Warburg Effect. Front. Cell Dev. Biol. 2018, 6, 79. [Google Scholar] [CrossRef]
- Porporato, P.E.; Dhup, S.; Dadhich, R.K.; Copetti, T.; Sonveaux, P. Anticancer targets in the glycolytic metabolism of tumors: A comprehensive review. Front. Pharmacol. 2011, 2, 49. [Google Scholar] [CrossRef]
- Shirai, T.; Nazarewicz, R.R.; Wallis, B.B.; Yanes, R.E.; Watanabe, R.; Hilhorst, M.; Tian, I.; Harrison, D.G.; Giacomini, J.C.; Assimes, T.L.; et al. The glycolytic enzyme PKM2 bridges metabolic and inflammatory dysfunction in coronary artery disease. J. Exp. Med. 2016, 213, 337–354. [Google Scholar] [CrossRef] [PubMed]
- McGarry, T.; Orr, C.; Wade, S.; Biniecka, M.; Wade, S.; Gallagher, L.; Low, C.; Veale, D.J.; Fearon, U. JAK/STAT Blockade Alters Synovial Bioenergetics, Mitochondrial Function, and Proinflammatory Mediators in Rheumatoid Arthritis. Arthritis Rheumatol. 2018, 70, 1959–1970. [Google Scholar] [CrossRef]
- Yang, X.; Chen, W.; Zhao, X.; Chen, L.; Li, W.; Ran, J.; Wu, L. Pyruvate Kinase M2 Modulates the Glycolysis of Chondrocyte and Extracellular Matrix in Osteoarthritis. DNA Cell Biol. 2018, 37, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Fujii, H.; Mohan, S.V.; Goronzy, J.J.; Weyand, C.M. Phosphofructokinase deficiency impairs ATP generation, autophagy, and redox balance in rheumatoid arthritis T cells. J. Exp. Med. 2013, 210, 2119–2134. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.; Zeng, S.; Huang, M.; Qiu, Q.; Xiao, Y.; Shi, M.; Zhan, Z.; Liang, L.; Yang, X.; Xu, H. Inhibition of 6-phosphofructo-2-kinase suppresses fibroblast-like synoviocytes-mediated synovial inflammation and joint destruction in rheumatoid arthritis. Br. J. Pharmacol. 2017, 174, 893–908. [Google Scholar] [CrossRef] [Green Version]
- Veras, F.P.; Peres, R.S.; Saraiva, A.L.; Pinto, L.G.; Louzada-Junior, P.; Cunha, T.M.; Paschoal, J.A.; Cunha, F.Q.; Alves-Filho, J.C. Fructose 1,6-bisphosphate, a high-energy intermediate of glycolysis, attenuates experimental arthritis by activating anti-inflammatory adenosinergic pathway. Sci. Rep. 2015, 5, 15171. [Google Scholar] [CrossRef]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef]
- Haas, R.; Smith, J.; Rocher-Ros, V.; Nadkarni, S.; Montero-Melendez, T.; D’Acquisto, F.; Bland, E.J.; Bombardieri, M.; Pitzalis, C.; Perretti, M.; et al. Lactate Regulates Metabolic and Pro-inflammatory Circuits in Control of T Cell Migration and Effector Functions. PLoS Biol. 2015, 13, e1002202. [Google Scholar] [CrossRef]
- Miao, H.; Chen, L.; Hao, L.; Zhang, X.; Chen, Y.; Ruan, Z.; Liang, H. Stearic acid induces proinflammatory cytokine production partly through activation of lactate-HIF1alpha pathway in chondrocytes. Sci. Rep. 2015, 5, 13092. [Google Scholar] [CrossRef]
- Halestrap, A.P. The monocarboxylate transporter family--Structure and functional characterization. IUBMB Life 2012, 64, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, S.R.; Gopal, E.; Zhuang, L.; Itagaki, S.; Martin, P.M.; Fei, Y.J.; Ganapathy, V.; Prasad, P.D. Cloning and functional identification of slc5a12 as a sodium-coupled low-affinity transporter for monocarboxylates (SMCT2). Biochem. J. 2005, 392, 655–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doherty, J.R.; Cleveland, J.L. Targeting lactate metabolism for cancer therapeutics. J. Clin. Investig. 2013, 123, 3685–3692. [Google Scholar] [CrossRef]
- Pucino, V.; Cucchi, D.; Mauro, C. Lactate transporters as therapeutic targets in cancer and inflammatory diseases. Expert Opin. Ther. Targets 2018, 22, 735–743. [Google Scholar] [CrossRef] [PubMed]
- Miloushev, V.Z.; Granlund, K.L.; Boltyanskiy, R.; Lyashchenko, S.K.; DeAngelis, L.M.; Mellinghoff, I.K.; Brennan, C.W.; Tabar, V.; Yang, T.J.; Holodny, A.I.; et al. Metabolic Imaging of the Human Brain with Hyperpolarized 13C Pyruvate Demonstrates 13C Lactate Production in Brain Tumor Patients. Cancer Res. 2018, 78, 3755–3760. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Wen, Z.; Li, Y.; Matteson, E.L.; Hong, J.; Goronzy, J.J.; Weyand, C.M. Metabolic control of the scaffold protein TKS5 in tissue-invasive, proinflammatory T cells. Nat. Immunol. 2017, 18, 1025–1034. [Google Scholar] [CrossRef]
- Michopoulos, F.; Karagianni, N.; Whalley, N.M.; Firth, M.A.; Nikolaou, C.; Wilson, I.D.; Critchlow, S.E.; Kollias, G.; Theodoridis, G.A. Targeted Metabolic Profiling of the Tg197 Mouse Model Reveals Itaconic Acid as a Marker of Rheumatoid Arthritis. J. Proteome Res. 2016, 15, 4579–4590. [Google Scholar] [CrossRef]
- Lampropoulou, V.; Sergushichev, A.; Bambouskova, M.; Nair, S.; Vincent, E.E.; Loginicheva, E.; Cervantes-Barragan, L.; Ma, X.; Huang, S.C.; Griss, T.; et al. Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab. 2016, 24, 158–166. [Google Scholar] [CrossRef]
- Littlewood-Evans, A.; Sarret, S.; Apfel, V.; Loesle, P.; Dawson, J.; Zhang, J.; Muller, A.; Tigani, B.; Kneuer, R.; Patel, S.; et al. GPR91 senses extracellular succinate released from inflammatory macrophages and exacerbates rheumatoid arthritis. J. Exp. Med. 2016, 213, 1655–1662. [Google Scholar] [CrossRef] [PubMed]
- Saraiva, A.L.; Veras, F.P.; Peres, R.S.; Talbot, J.; de Lima, K.A.; Luiz, J.P.; Carballido, J.M.; Cunha, T.M.; Cunha, F.G.; Ryffel, B.; et al. Succinate receptor deficiency attenuates arthritis by reducing dendritic cell traffic and expansion of Th17 cells in the lymph nodes. FASEB J. 2018, 32, 6550–6558. [Google Scholar] [CrossRef]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Flood, S.L.; Duance, V.C.; Mason, D.J. The role of glutamate signalling in rheumatoid arthritis. Int. J. Exp. Pathol. 2008, 85, A19–A20. [Google Scholar] [CrossRef]
- Zheng, K.; Shen, N.; Chen, H.; Ni, S.; Zhang, T.; Hu, M.; Wang, J.; Sun, L.; Yang, X. Global and targeted metabolomics of synovial fluid discovers special osteoarthritis metabolites. J. Orthop. Res. 2017, 35, 1973–1981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunphy, M.P.S.; Harding, J.J.; Venneti, S.; Zhang, H.; Burnazi, E.M.; Bromberg, J.; Omuro, A.M.; Hsieh, J.J.; Mellinghoff, I.K.; Staton, K.; et al. In Vivo PET Assay of Tumor Glutamine Flux and Metabolism: In-Human Trial of 18F-(2S,4R)-4-Fluoroglutamine. Radiology 2018, 287, 667–675. [Google Scholar] [CrossRef]
- Scalise, M.; Pochini, L.; Console, L.; Losso, M.A.; Indiveri, C. The Human SLC1A5 (ASCT2) Amino Acid Transporter: From Function to Structure and Role in Cell Biology. Front. Cell Dev. Biol. 2018, 6, 96. [Google Scholar] [CrossRef] [PubMed]
- Nagana Gowda, G.A.; Barding, G.A., Jr.; Dai, J.; Gu, H.; Margineantu, D.H.; Hockenbery, D.M.; Raftery, D. A Metabolomics Study of BPTES Altered Metabolism in Human Breast Cancer Cell Lines. Front. Mol. Biosci. 2018, 5, 49. [Google Scholar] [CrossRef] [PubMed]
- Gross, M.I.; Demo, S.D.; Dennison, J.B.; Chen, L.; Chernov-Rogan, T.; Goyal, B.; Janes, J.R.; Laidig, G.J.; Lewis, E.R.; Li, J.; et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol. Cancer Ther. 2014, 13, 890–901. [Google Scholar] [CrossRef]
- Glunde, K.; Bhujwalla, Z.M.; Ronen, S.M. Choline metabolism in malignant transformation. Nat. Rev. Cancer 2011, 11, 835–848. [Google Scholar] [CrossRef] [Green Version]
- Guma, M.; Sanchez-Lopez, E.; Lodi, A.; Garcia-Carbonell, R.; Tiziani, S.; Karin, M.; Lacal, J.C.; Firestein, G.S. Choline kinase inhibition in rheumatoid arthritis. Ann. Rheum. Dis. 2015, 74, 1399–1407. [Google Scholar] [CrossRef]
- Hellberg, S.; Silvola, J.M.; Kiugel, M.; Liljenback, H.; Metsala, O.; Viljanen, T.; Metso, J.; Jauhiainen, M.; Saukko, P.; Nuutila, P.; et al. Type 2 diabetes enhances arterial uptake of choline in atherosclerotic mice: An imaging study with positron emission tomography tracer 18F-fluoromethylcholine. Cardiovasc. Diabetol. 2016, 15, 26. [Google Scholar] [CrossRef]
- Sanchez-Lopez, E.; Zhong, Z.; Stubelius, A.; Sweeney, S.R.; Booshehri, L.M.; Antonucci, L.; Liu-Bryan, R.; Lodi, A.; Terkeltaub, R.; Lacal, J.C.; et al. Choline uptake and metabolism modulate macrophage IL-1b and IL-18. Cell Metab. 2019, in press. [Google Scholar] [CrossRef]
- Snider, S.A.; Margison, K.D.; Ghorbani, P.; LeBlond, N.D.; O’Dwyer, C.; Nunes, J.R.; Nguyen, T.; Xu, H.; Bennett, S.A.L.; Fullerton, M.D. Choline transport links macrophage phospholipid metabolism and inflammation. J. Biol. Chem. 2018, 293, 11600–11611. [Google Scholar] [CrossRef] [Green Version]
- Liu-Bryan, R. Inflammation and intracellular metabolism: New targets in OA. Osteoarthr. Cartil. 2015, 23, 1835–1842. [Google Scholar] [CrossRef] [PubMed]
- Toyama, E.Q.; Herzig, S.; Courchet, J.; Lewis, T.L., Jr.; Loson, O.C.; Hellberg, K.; Young, N.P.; Chen, H.; Polleux, F.; Chan, D.C.; et al. Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 2016, 351, 275–281. [Google Scholar] [CrossRef]
- Zhao, M.; Klionsky, D.J. AMPK-dependent phosphorylation of ULK1 induces autophagy. Cell Metab. 2011, 13, 119–120. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.; Jin, K.; Shen, Y.; Yang, Z.; Li, Y.; Wu, B.; Tian, L.; Shoor, S.; Roche, N.E.; Goronzy, J.J.; et al. N-myristoyltransferase deficiency impairs activation of kinase AMPK and promotes synovial tissue inflammation. Nat. Immunol. 2019, 20, 313–325. [Google Scholar] [CrossRef]
- Guma, M.; Wang, Y.; Viollet, B.; Liu-Bryan, R. AMPK Activation by A-769662 Controls IL-6 Expression in Inflammatory Arthritis. PLoS ONE 2015, 10, e0140452. [Google Scholar] [CrossRef]
- Cudrici, C.D.; Pelletier, M.; Siegel, R. A potential target for methotrexate in macrophages: AMP-activated protein kinase. J. Immunol. 2017, 198, 11. [Google Scholar]
- Lu, Q.; Li, X.; Liu, J.; Sun, X.; Rousselle, T.; Ren, D.; Tong, N.; Li, J. AMPK is associated with the beneficial effects of antidiabetic agents on cardiovascular diseases. Biosci. Rep. 2019, 39, BSR20181995. [Google Scholar] [CrossRef]
- Lee, S.Y.; Moon, S.J.; Kim, E.K.; Seo, H.B.; Yang, E.J.; Son, H.J.; Kim, J.K.; Min, J.K.; Park, S.H.; Cho, M.L. Metformin Suppresses Systemic Autoimmunity in Roquin(san/san) Mice through Inhibiting B Cell Differentiation into Plasma Cells via Regulation of AMPK/mTOR/STAT3. J. Immunol. 2017, 198, 2661–2670. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Katerelos, M.; Gleich, K.; Galic, S.; Kemp, B.E.; Mount, P.F.; Power, D.A. Phosphorylation of Acetyl-CoA Carboxylase by AMPK Reduces Renal Fibrosis and Is Essential for the Anti-Fibrotic Effect of Metformin. J. Am. Soc. Nephrol. 2018, 29, 2326–2336. [Google Scholar] [CrossRef]
- Son, H.J.; Lee, J.; Lee, S.Y.; Kim, E.K.; Park, M.J.; Kim, K.W.; Park, S.H.; Cho, M.L. Metformin attenuates experimental autoimmune arthritis through reciprocal regulation of Th17/Treg balance and osteoclastogenesis. Mediat. Inflamm. 2014, 2014, 973986. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Zhou, H.F.; Hu, Y.; Pham, C.T. Suppression of experimental arthritis through AMP-activated protein kinase activation and autophagy modulation. J. Rheum. Dis. Treat. 2015, 1, 5. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.Y.; Kim, Y.K.; Yi, H.; Kim, J.; Jung, H.R.; Kim, I.J.; Cho, J.M.; Park, S.H.; Kim, H.Y.; Ju, J.H. Metformin downregulates Th17 cells differentiation and attenuates murine autoimmune arthritis. Int. Immunopharmacol. 2013, 16, 85–92. [Google Scholar] [CrossRef]
- Ursini, F.; Russo, E.; Pellino, G.; D’Angelo, S.; Chiaravalloti, A.; De Sarro, G.; Manfredini, R.; De Giorgio, R. Metformin and Autoimmunity: A “New Deal” of an Old Drug. Front. Immunol. 2018, 9, 1236. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.Y.; Sykes, D.B.; Ameri, S.; Kalaitzidis, D.; Charles, J.F.; Nelson-Maney, N.; Wei, K.; Cunin, P.; Morris, A.; Cardona, A.E.; et al. The metabolic regulator mTORC1 controls terminal myeloid differentiation. Sci. Immunol. 2017, 2, eaam6641. [Google Scholar] [PubMed]
- Meng, L.H.; Zheng, X.F. Toward rapamycin analog (rapalog)-based precision cancer therapy. Acta Pharmacol. Sin. 2015, 36, 1163–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bride, K.L.; Vincent, T.; Smith-Whitley, K.; Lambert, M.P.; Bleesing, J.J.; Seif, A.E.; Manno, C.S.; Casper, J.; Grupp, S.A.; Teachey, D.T. Sirolimus is effective in relapsed/refractory autoimmune cytopenias: Results of a prospective multi-institutional trial. Blood 2016, 127, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Mendes, K.L.; Lelis, D.F.; Santos, S.H.S. Nuclear sirtuins and inflammatory signaling pathways. Cytokine Growth Factor Rev. 2017, 38, 98–105. [Google Scholar] [CrossRef]
- Dvir-Ginzberg, M.; Mobasheri, A.; Kumar, A. Erratum to: The Role of Sirtuins in Cartilage Homeostasis and Osteoarthritis. Curr. Rheumatol. Rep. 2016, 18, 48. [Google Scholar] [CrossRef]
- Borra, M.T.; Smith, B.C.; Denu, J.M. Mechanism of human SIRT1 activation by resveratrol. J. Biol. Chem. 2005, 280, 17187–17195. [Google Scholar] [CrossRef] [PubMed]
- Correa, M.G.; Pires, P.R.; Ribeiro, F.V.; Pimentel, S.P.; Cirano, F.R.; Napimoga, M.H.; Casati, M.Z.; Casarin, R.C.V. Systemic treatment with resveratrol reduces the progression of experimental periodontitis and arthritis in rats. PLoS ONE 2018, 13, e0204414. [Google Scholar] [CrossRef]
- Riveiro-Naveira, R.R.; Valcarcel-Ares, M.N.; Almonte-Becerril, M.; Vaamonde-Garcia, C.; Loureiro, J.; Hermida-Carballo, L.; Lopez-Pelaez, E.; Blanco, F.J.; Lopez-Armada, M.J. Resveratrol lowers synovial hyperplasia, inflammatory markers and oxidative damage in an acute antigen-induced arthritis model. Rheumatology 2016, 55, 1889–1900. [Google Scholar] [CrossRef]
- Khojah, H.M.; Ahmed, S.; Abdel-Rahman, M.S.; Elhakeim, E.H. Resveratrol as an effective adjuvant therapy in the management of rheumatoid arthritis: A clinical study. Clin. Rheumatol. 2018, 37, 2035–2042. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.A.; Marouf, B.H.; Ali, Z.S.; Ahmmad, R.S. Efficacy and safety of co-administration of resveratrol with meloxicam in patients with knee osteoarthritis: A pilot interventional study. Clin. Interv. Aging 2018, 13, 1621–1630. [Google Scholar] [CrossRef]
- Rodriguez, A.E.; Ducker, G.S.; Billingham, L.K.; Martinez, C.A.; Mainolfi, N.; Suri, V.; Friedman, A.; Manfredi, M.G.; Weinberg, S.E.; Rabinowitz, J.D.; et al. Serine Metabolism Supports Macrophage IL-1beta Production. Cell Metab. 2019. [Google Scholar] [CrossRef]
- Yoon, B.R.; Oh, Y.J.; Kang, S.W.; Lee, E.B.; Lee, W.W. Role of SLC7A5 in Metabolic Reprogramming of Human Monocyte/Macrophage Immune Responses. Front. Immunol. 2018, 9, 53. [Google Scholar] [CrossRef] [Green Version]
- Papathanassiu, A.E.; Ko, J.H.; Imprialou, M.; Bagnati, M.; Srivastava, P.K.; Vu, H.A.; Cucchi, D.; McAdoo, S.P.; Ananieva, E.A.; Mauro, C.; et al. BCAT1 controls metabolic reprogramming in activated human macrophages and is associated with inflammatory diseases. Nat. Commun. 2017, 8, 16040. [Google Scholar] [CrossRef]
- Yu, Z.; Lin, W.; Rui, Z.; Jihong, P. Fibroblast-like synoviocyte migration is enhanced by IL-17-mediated overexpression of L-type amino acid transporter 1 (LAT1) via the mTOR/4E-BP1 pathway. Amino Acids 2018, 50, 331–340. [Google Scholar] [CrossRef] [PubMed]
- Mills, C.D. M1 and M2 Macrophages: Oracles of Health and Disease. Crit. Rev. Immunol. 2012, 32, 463–488. [Google Scholar] [CrossRef] [Green Version]
- Saha, S.; Shalova, I.N.; Biswas, S.K. Metabolic regulation of macrophage phenotype and function. Immunol. Rev. 2017, 280, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.W.; Chang, K.L.; Chen, C.J.; Liu, H.W. Arginase levels are increased in patients with rheumatoid arthritis. Kaohsiung J. Med. Sci. 2001, 17, 358–363. [Google Scholar] [PubMed]
- Sharma, M.D.; Hou, D.Y.; Liu, Y.; Koni, P.A.; Metz, R.; Chandler, P.; Mellor, A.L.; He, Y.; Munn, D.H. Indoleamine 2,3-dioxygenase controls conversion of Foxp3+ Tregs to TH17-like cells in tumor-draining lymph nodes. Blood 2009, 113, 6102–6111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frumento, G.; Rotondo, R.; Tonetti, M.; Damonte, G.; Benatti, U.; Ferrara, G.B. Tryptophan-derived catabolites are responsible for inhibition of T and natural killer cell proliferation induced by indoleamine 2,3-dioxygenase. J. Exp. Med. 2002, 196, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Beatty, G.L.; O’Dwyer, P.J.; Clark, J.; Shi, J.G.; Bowman, K.J.; Scherle, P.A.; Newton, R.C.; Schnaub, R.; Maleski, J.; Leopold, L.; et al. First-in-Human Phase I Study of the Oral Inhibitor of Indoleamine 2,3-Dioxygenase-1 Epacadostat (INCB024360) in Patients with Advanced Solid Malignancies. Clin. Cancer Res. 2017, 23, 3269–3276. [Google Scholar] [CrossRef]
- Kristeleit, R.; Davidenko, I.; Shirinkin, V.; El-Khouly, F.; Bondarenko, I.; Goodheart, M.J.; Gorbunova, V.; Penning, C.A.; Shi, J.G.; Liu, X.; et al. A randomised, open-label, phase 2 study of the IDO1 inhibitor epacadostat (INCB024360) versus tamoxifen as therapy for biochemically recurrent (CA-125 relapse)-only epithelial ovarian cancer, primary peritoneal carcinoma, or fallopian tube cancer. Gynecol. Oncol. 2017, 146, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Prendergast, G.C.; Malachowski, W.J.; Mondal, A.; Scherle, P.; Muller, A.J. Indoleamine 2,3-Dioxygenase and Its Therapeutic Inhibition in Cancer. Int. Rev. Cell Mol. Biol. 2018, 336, 175–203. [Google Scholar]
- Schroecksnadel, K.; Kaser, S.; Ledochowski, M.; Neurauter, G.; Mur, E.; Herold, M.; Fuchs, D. Increased degradation of tryptophan in blood of patients with rheumatoid arthritis. J. Rheumatol. 2003, 30, 1935–1939. [Google Scholar]
- Criado, G.; Simelyte, E.; Inglis, J.J.; Essex, D.; Williams, R.O. Indoleamine 2,3 dioxygenase-mediated tryptophan catabolism regulates accumulation of Th1/Th17 cells in the joint in collagen-induced arthritis. Arthritis Rheum. 2009, 60, 1342–1351. [Google Scholar] [CrossRef]
- Szanto, S.; Koreny, T.; Mikecz, K.; Glant, T.T.; Szekanecz, Z.; Varga, J. Inhibition of indoleamine 2,3-dioxygenase-mediated tryptophan catabolism accelerates collagen-induced arthritis in mice. Arthritis Res. Ther. 2007, 9, R50. [Google Scholar] [CrossRef]
- Menendez, J.A.; Lupu, R. Fatty acid synthase (FASN) as a therapeutic target in breast cancer. Expert Opin. Ther. Targets 2017, 21, 1001–1016. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Xi, Q.; Wu, G. Fatty acid synthase regulates invasion and metastasis of colorectal cancer via Wnt signaling pathway. Cancer Med. 2016, 5, 1599–1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bian, Y.; Yu, Y.; Wang, S.; Li, L. Up-regulation of fatty acid synthase induced by EGFR/ERK activation promotes tumor growth in pancreatic cancer. Biochem. Biophys. Res. Commun. 2015, 463, 612–617. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Song, H.; Yin, L.; Rizzo, M.G.; Sidhu, R.; Covey, D.F.; Ory, D.S.; Semenkovish, C.F. Fatty acid synthesis configures the plasma membrane for inflammation in diabetes. Nature 2016, 539, 294–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, S.F.; Infante, J.R. Molecular Pathways: Fatty Acid Synthase. Clin. Cancer Res. 2015, 21, 5434–5438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, Y.; Nishiumi, S.; Kono, S.; Takao, S.; Azuma, T.; Yoshida, M. Differences in elongation of very long chain fatty acids and fatty acid metabolism between triple-negative and hormone receptor-positive breast cancer. BMC Cancer 2017, 17, 589. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Makino, A.; Hullin-Matsuda, F.; Kobayashi, T.; Furihata, M.; Chung, S.; Ashida, S.; Miki, T.; Fujioka, T.; Shuin, T.; et al. Novel lipogenic enzyme ELOVL7 is involved in prostate cancer growth through saturated long-chain fatty acid metabolism. Cancer Res. 2009, 69, 8133–8140. [Google Scholar] [CrossRef]
- Su, Y.C.; Feng, Y.H.; Wu, H.T.; Huang, Y.S.; Tung, C.L.; Wu, P.; Chang, C.J.; Shiau, A.L.; Wu, C.L. Elovl6 is a negative clinical predictor for liver cancer and knockdown of Elovl6 reduces murine liver cancer progression. Sci. Rep. 2018, 8, 6586. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Positron emission tomography (PET) methods that provide information on the underlying biochemical processes. PET imaging not only can improve clinical diagnostics but also potentially predict treatment effects. 18F-FDG, 2-deoxy-2-(fluorine-18)fluoro-D-glucose, provides information on glycolysis and glucose uptake; 11C-DASA23, a class of N, N-diarylsulfonamides, is able to measure PKM2 uptake; 18F-Gln, 18F-(2S,4R)4-fluoroglutamine, allows for the monitoring of glutamine metabolism. 11C-Met, 11C-methionine; 18F-FET, O-(2-[18F]fluoroethyl)-L-tyrosine; 18F-FAMT, L-3-(18F)-Fluoro-α-methyl tyrosine; radiolabeled methionine and tyrosine can provide data on amino acid uptake and protein synthesis. Finally, 11C-acetate is converted to acetyl-CoA and used in mitochondria in TCA cycle or incorporated into cell membranes. MCT4, monocarboxylate transporter 4; GLUT1, glucose transporter 1; MCT1, monocarboxylate transporter 1; R-5-P, ribose-5-phosphate; PGD, phosphogluconate dehydrogenase; 6-PG, 6-phosphogluconate; G6PD, glucose-6-phosphate-dehydrogenase; HK, hexokinase; PFK1, phosphofructokinase 1; F2,6BP, fructose-2,6-bisphosphate; PFKFB3, 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3; dTMP, deoxythymidine monophosphate; dUMP, deoxyuridine monophosphate; TS, thymidylate synthase; THF, tetrahydrofolate; DHF, dihydrofolate; DHFR, dihydrofolate reductase; CK, choline kinase; PKM2, pyruvate kinase muscle isozyme M2; LDHα, lactate dehydrogenase A; CA, carbonic anhydrase; ACC, acetyl-CoA carboxylase; FAS, fatty acid synthase; PDK1, pyruvate dehydrogenase kinase 1; IDH, isocitrate dehydrogenase; α-KGDH, alpha-ketoglutarate dehydrogenase; GLS, glutaminase.
Figure 1.
Positron emission tomography (PET) methods that provide information on the underlying biochemical processes. PET imaging not only can improve clinical diagnostics but also potentially predict treatment effects. 18F-FDG, 2-deoxy-2-(fluorine-18)fluoro-D-glucose, provides information on glycolysis and glucose uptake; 11C-DASA23, a class of N, N-diarylsulfonamides, is able to measure PKM2 uptake; 18F-Gln, 18F-(2S,4R)4-fluoroglutamine, allows for the monitoring of glutamine metabolism. 11C-Met, 11C-methionine; 18F-FET, O-(2-[18F]fluoroethyl)-L-tyrosine; 18F-FAMT, L-3-(18F)-Fluoro-α-methyl tyrosine; radiolabeled methionine and tyrosine can provide data on amino acid uptake and protein synthesis. Finally, 11C-acetate is converted to acetyl-CoA and used in mitochondria in TCA cycle or incorporated into cell membranes. MCT4, monocarboxylate transporter 4; GLUT1, glucose transporter 1; MCT1, monocarboxylate transporter 1; R-5-P, ribose-5-phosphate; PGD, phosphogluconate dehydrogenase; 6-PG, 6-phosphogluconate; G6PD, glucose-6-phosphate-dehydrogenase; HK, hexokinase; PFK1, phosphofructokinase 1; F2,6BP, fructose-2,6-bisphosphate; PFKFB3, 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3; dTMP, deoxythymidine monophosphate; dUMP, deoxyuridine monophosphate; TS, thymidylate synthase; THF, tetrahydrofolate; DHF, dihydrofolate; DHFR, dihydrofolate reductase; CK, choline kinase; PKM2, pyruvate kinase muscle isozyme M2; LDHα, lactate dehydrogenase A; CA, carbonic anhydrase; ACC, acetyl-CoA carboxylase; FAS, fatty acid synthase; PDK1, pyruvate dehydrogenase kinase 1; IDH, isocitrate dehydrogenase; α-KGDH, alpha-ketoglutarate dehydrogenase; GLS, glutaminase.

Figure 2.
Anticancer agents targeting various metabolic pathways that are upregulated in activated cells. Since synovial tissue cells share many similar metabolic changes, these very same antimetabolites may also have potential uses in RA. MCT4, monocarboxylate transporter 4; GLUT1, glucose transporter 1; MCT1, monocarboxylate transporter 1; R-5-P, ribose-5-phosphate; PGD, phosphogluconate dehydrogenase; 6-PG, 6-phosphogluconate; G6PD, glucose-6-phosphate-dehydrogenase; HK, hexokinase; PFK1, phosphofructokinase 1; F2,6BP, fructose-2,6-bisphosphate; PFKFB3, 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3; dTMP, deoxythymidine monophosphate; dUMP, deoxyuridine monophosphate; TS, thymidylate synthase; THF, tetrahydrofolate; DHF, dihydrofolate; DHFR, dihydrofolate reductase; CK, choline kinase; PKM2, pyruvate kinase muscle isozyme M2; LDHα, lactate dehydrogenase A; CA, carbonic anhydrase; ACC, acetyl-CoA carboxylase; FAS, fatty acid synthase; PDK1, pyruvate dehydrogenase kinase 1; IDH, isocitrate dehydrogenase; α-KGDH, alpha-ketoglutarate dehydrogenase; GLS, glutaminase.
Figure 2.
Anticancer agents targeting various metabolic pathways that are upregulated in activated cells. Since synovial tissue cells share many similar metabolic changes, these very same antimetabolites may also have potential uses in RA. MCT4, monocarboxylate transporter 4; GLUT1, glucose transporter 1; MCT1, monocarboxylate transporter 1; R-5-P, ribose-5-phosphate; PGD, phosphogluconate dehydrogenase; 6-PG, 6-phosphogluconate; G6PD, glucose-6-phosphate-dehydrogenase; HK, hexokinase; PFK1, phosphofructokinase 1; F2,6BP, fructose-2,6-bisphosphate; PFKFB3, 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3; dTMP, deoxythymidine monophosphate; dUMP, deoxyuridine monophosphate; TS, thymidylate synthase; THF, tetrahydrofolate; DHF, dihydrofolate; DHFR, dihydrofolate reductase; CK, choline kinase; PKM2, pyruvate kinase muscle isozyme M2; LDHα, lactate dehydrogenase A; CA, carbonic anhydrase; ACC, acetyl-CoA carboxylase; FAS, fatty acid synthase; PDK1, pyruvate dehydrogenase kinase 1; IDH, isocitrate dehydrogenase; α-KGDH, alpha-ketoglutarate dehydrogenase; GLS, glutaminase.


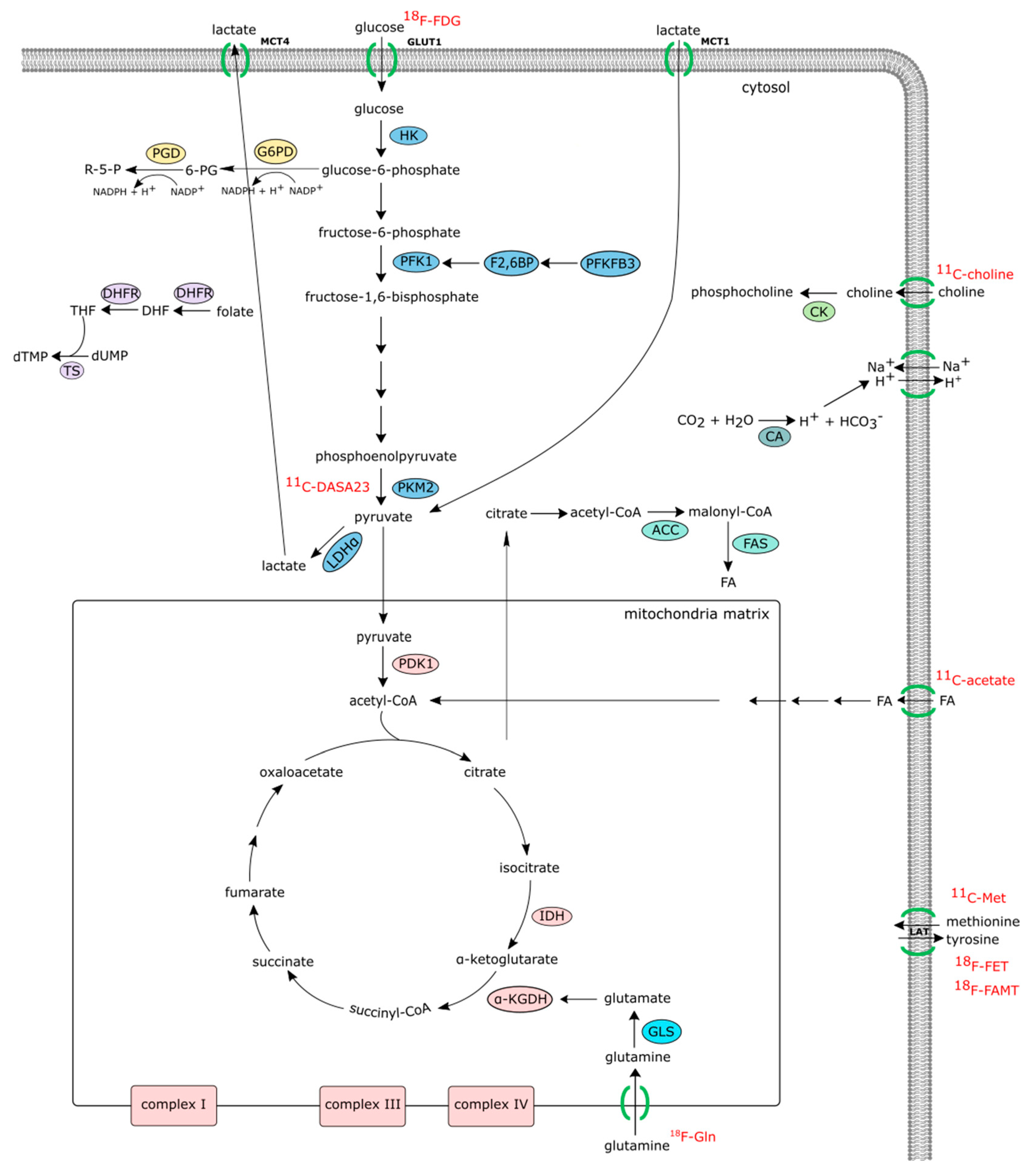{kind=link}
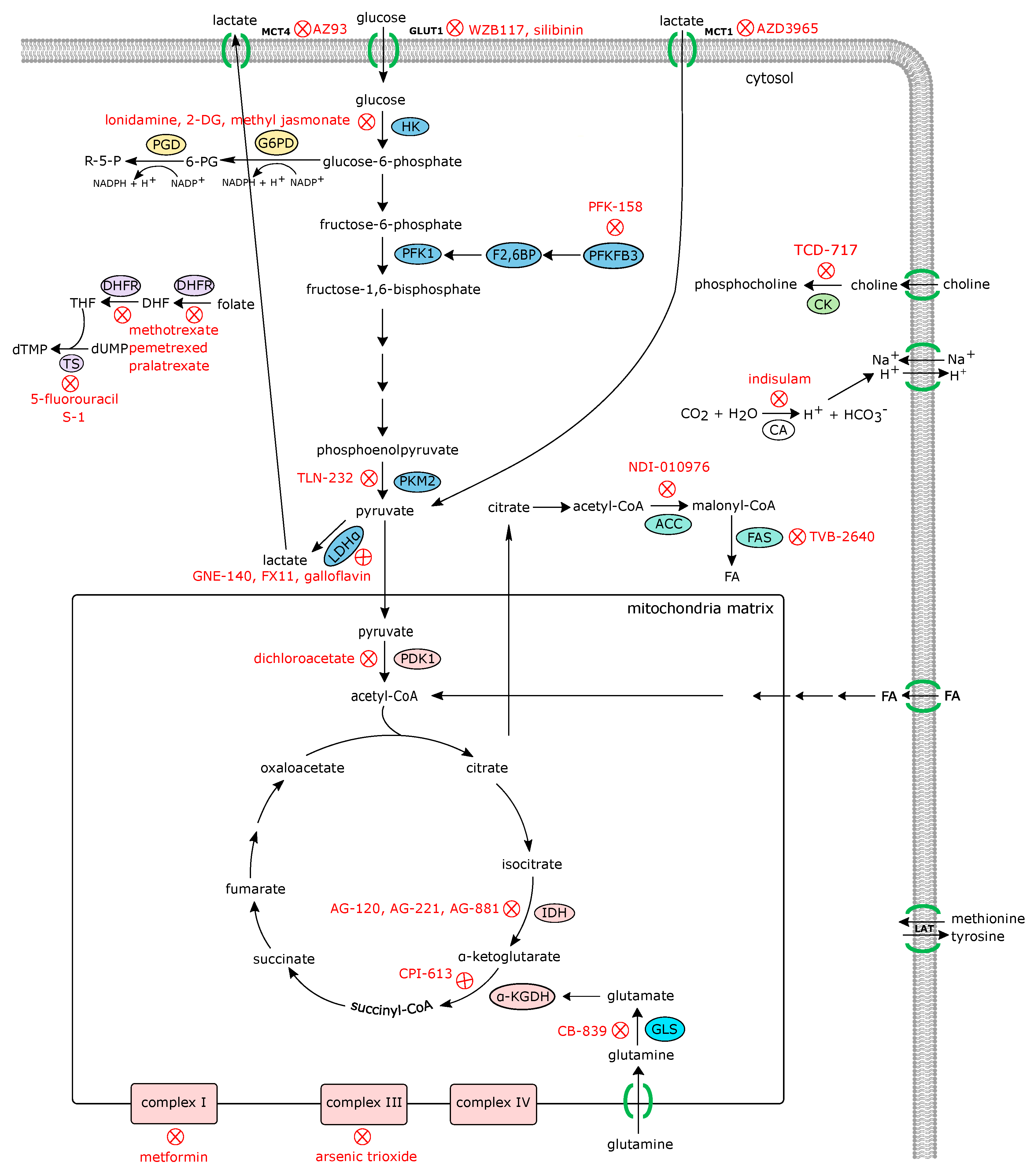{kind=link}
Table 1.
Clinical trials of drugs that target various steps of the glycolytic and mitochondrial metabolic pathways.
Table 1.
Clinical trials of drugs that target various steps of the glycolytic and mitochondrial metabolic pathways.
| Drug | Pathway | Disease | Trial Status | Identifier # |
|---|---|---|---|---|
| Silibinin | glycolysis (glut1) | liver cancer | phase I | NCT01129570 |
| prostate cancer | phase II | NCT02146118 | ||
| hypertension | phase IV | NCT03538327 | ||
| RO7070179 | HIF1α | Hepatocellular carcinoma | Phase I | NCT02564614 |
| lonidamine | glycolysis (HK) | enlarged prostate | phase III | NCT00435448 |
| 2-DG | glycolysis (HK) | advanced solid tumor | phase I | NCT00096707 |
| prostate cancer | phase I/II | NCT00633087 | ||
| PFK-158 | glycolysis (PFKFB3) | advanced solid tumors | phase I | NCT02044861 |
| TLN-232 | glycolysis (PKM2) | melanoma | phase II | NCT00735332 |
| renal cell carcinoma | NCT00422786 | |||
| AZD3965 | lactate uptake (MCT 1) | advanced solid tumor | phase I | NCT01791595 |
| Indisulam | H+ secretion | gastric cancer | phase I/II | NCT00165594 |
| kidney cancer | phase II | NCT00059735 | ||
| Dichloroacetate | PDK1 | head and neck cancer | phase I | NCT01163487 |
| breast, lung cancer | phase II | NCT01029925 | ||
| CPI-613 | aKGDH | small cell lung cancer | phase I | NCT01931787 |
| lymphoma, leukemia | phase II | NCT03793140 | ||
| AG-120 | isocitrate DH | advanced solid tumor | phase I | NCT02073994 |
| leukemia | phase II | NCT03503409 | ||
| cholangiocarcinoma | phase III | NCT02989857 | ||
| AG-221 | isocitrate DH | leukemia | phase I | NCT03728335 |
| leukemia | phase II | NCT03744390 | ||
| advanced solid tumor | phase I/II | NCT02273739 | ||
| AG-881 | isocitrate DH | glioma | phase I | NCT02481154 |
| metformin | Mitochondrial complex I | RA | phase I/II | NCT03686657 |
| prostate cancer | phase II | NCT03137186 | ||
| SLE | phase IV | NCT02741960 | ||
| arsenic trioxide | Mitochondrial complex III | leukemia | phase II | NCT03624270 |
glut1, glucose transporter 1; HK, hexokinase; PFKFB3, 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3; MCT1, monocarboxylate transporter 1; MCT4, monocarboxylate transporter 4; PDK1, pyruvate dehydrogenase kinase 1; α-KGDH, alpha-ketoglutarate dehydrogenase; isocitrate DH, isocitrate dehydrogenase.
Table 2.
Clinical trials of drugs that target other metabolic pathways than glycolysis, involved in the upregulated synthesis, proliferation, and survival of cells that have undergone metabolic rewiring.
Table 2.
Clinical trials of drugs that target other metabolic pathways than glycolysis, involved in the upregulated synthesis, proliferation, and survival of cells that have undergone metabolic rewiring.
| Drug | Pathway | Disease | Trial Status | Identifier |
|---|---|---|---|---|
| CB-839 | glutaminase | advanced solid tumor | phase I | NCT02071862 |
| renal cell carcinoma | phase II | NCT03428217 | ||
| ADI-PEG20 | arginine availability | breast cancer | phase I | NCT01948843 |
| hepatocellular cancer | phase II | NCT00056992 | ||
| hepatocellular cancer | phase III | NCT01287585 | ||
| TVB-2640 | fatty acid synthase | advanced solid tumor | phase I | NCT02223247 |
| NSCLC | phase II | NCT03808558 | ||
| NDI-010976 | acetyl-CoA carboxylase | healthy obese adults | phase I | NCT02876796 |
| TCD-717 | choline kinase | advanced solid tumor | phase I | NCT01215864 |
| epacadostat | indoleamine-2,3-dioxygenase | solid tumor | phase I | NCT03471286 |
| MDS | phase II | NCT01822691 | ||
| indoximod | indoleamine-2,3-dioxygenase | prostate cancer | phase II | NCT01560923 |
| rapamycin | mTOR | thyroid cancer | phase II | NCT00936858 |
| everolimus | mTOR | prostate cancer | phase II | NCT00976755 |
| kidney cancer | phase III | NCT01120249 | ||
| temsirolimus | mTOR | RA | phase II | NCT00076206 |
| leflunomide | Pyrimidine synthetase | approved | ||
| methotrexate | dihydrofolate reductase | approved | ||
| pemetrexed | dihydrofolate reductase | approved | ||
| pralatrexate | dihydrofolate reductase | approved | ||
| 5-fluorouracil | thymidylate synthase | approved | ||
| S-1 | thymidylate synthase | approved | ||
| pentostatin | adenosine deaminase | approved | ||
| 6-mercaptopurine | adenine deaminase | approved | ||
| azathioprine | purine synthesis | approved | ||
| cladribine | adenosine deaminase | approved | ||
| gemcitabine | ribonucleotide reductase | approved | ||
| cytarabine | DNA polymerase/ribonucleotide reductase | approved | ||
| fludarabine | DNA polymerase/ribonucleotide reductase | approved | ||
| hydroxyurea | ribonucleotide reductase | approved |
mTOR, mammalian target of rapamycin; NSCLC, non-small cell lung cancer; MDS, myelodysplasic syndrome.
Table 3.
Preclinical data of drugs that target metabolic pathway.
| Pathway | Animal Model | Effect on Cells | Reference |
|---|---|---|---|
| glycolysis (HK II) | K/BxN | Genetic ablation of HK2 inhibits invasive capacities of FLS and secretion of inflammatory ctytokines. | [75,78], |
| glycolytic inhibitors (2DG, bromopyruvate and ionidamine) | K/BxN, CIA, SKG | Glycolytic inhibitors reduced FLS aggressive phenotype, decrease effector CD4+ cells, and modulated Th17/Treg differentiation. | [60,76,77,78] |
| glycolysis (PFKFB) | CIA | PFKFB3 inhibition reduced FLS migration and invasion, and the production of inflammatory mediators | [86] |
| glycolysis (FPB) | AIA, zymosan | Systemic generation of extracellular adenosine and subsequent activation of adenosine receptor A2a | [87] |
| succinate receptor (SUCNR1) | AIA | Sucnr1 guides dendritic cells into the lymph nodes, leading the expansion of the Th17-cell population | [100] |
| glutaminase 1 (GLS1) | K/BxN | Inhibition or genetic ablation of glutaminase 1 (GLS1) inhibits RA-FLS proliferation | [43] |
| choline kinase | K/BxN | Blocking choline kinase activity limits the proliferative and migratory capacity of FLS | [109] |
| amino acid uptake (BCAT1) metformin | CIA CIA, CAIA, K/BxN | BCAT Inhibition reduces IRG1 and itaconate levels in macrophages. | [138] |
| Metformin modulated Th17/Treg differentiation and osteoclastogenesis. | [122,123,124] |
CIA: collagen-induced arthritis, AIA: antigen-induced arthritis, CAIA: collagen antibody-induced arthritis.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sanchez-Lopez, E.; Cheng, A.; Guma, M. Can Metabolic Pathways Be Therapeutic Targets in Rheumatoid Arthritis? J. Clin. Med. 2019, 8, 753. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm8050753
AMA Style
Sanchez-Lopez E, Cheng A, Guma M. Can Metabolic Pathways Be Therapeutic Targets in Rheumatoid Arthritis? Journal of Clinical Medicine. 2019; 8(5):753. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm8050753
Chicago/Turabian StyleSanchez-Lopez, Elsa, Anyan Cheng, and Monica Guma. 2019. "Can Metabolic Pathways Be Therapeutic Targets in Rheumatoid Arthritis?" Journal of Clinical Medicine 8, no. 5: 753. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm8050753
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.