Neurophysiology and Psychopathology Underlying PTSD and Recent Insights into the PTSD Therapies—A Comprehensive Review

 , , , and
, , , and
Abstract
: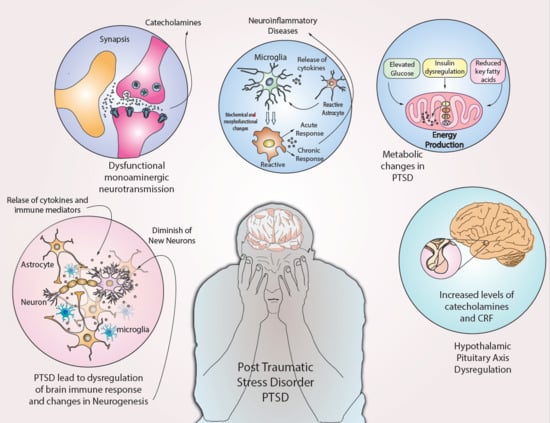
{kind=link}
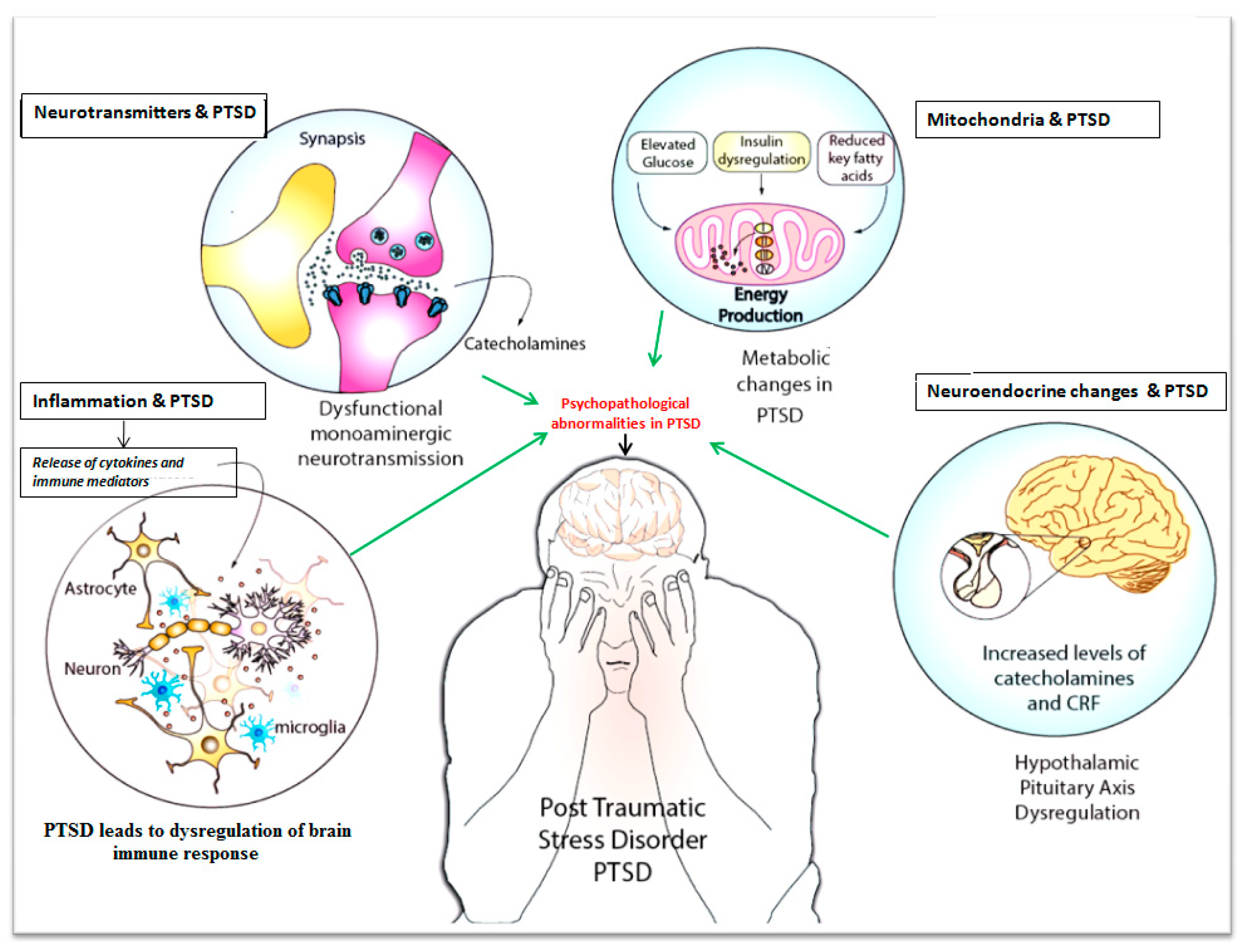{kind=link}
1. Introduction
- Directly experiences the traumatic event;
- Witnesses the traumatic event in person;
- Learns that the traumatic event occurred to a close family member or friend (with the actual or threatened death being either violent or accidental); or
- Exposed extensively to aversive traumatic events but specifically not through television, pictures or media.
Diagnosis and PTSD
2. Genetics and PTSD
3. Inflammation and PTSD
4. Neurotransmitter Genomics and PTSD
5. Metabolic Changes and PTSD
6. Neuroendocrine Disturbances and PTSD
7. Mitochondria and PTSD
8. Neurogenesis and PTSD
9. Premature Aging and PTSD
10. PTSD Therapeutic Modalities—Recent Advances
11. Conclusions
12. Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| PPARγ | peroxisome proliferator-activated receptors; |
| RXR | retinoid X receptor; |
| COX | cyclooxygenase; |
| nAChR | nicotinic acetylcholinergic receoptor; |
| ACE | angiotensin converting enzyme |
References
- Bisson, J.I.; Cosgrove, S.; Lewis, C.; Roberts, N.P. Post-traumatic stress disorder. BMJ 2015, 351. [Google Scholar] [CrossRef] [PubMed]
- Stein, D.J.; McLaughlin, K.A.; Koenen, K.C.; Atwoli, L.; Friedman, M.J.; Hill, E.D.; Maercker, A.; Petukhova, M.; Shahly, V.; Van Ommeren, M.; et al. DSM-5 and ICD-11 definitions of posttraumatic stress disorder: Investigating “narrow” and “broad” approaches. Depress. Anxiety 2014, 31, 494–505. [Google Scholar] [CrossRef] [PubMed]
- Iribarren, J.; Prolo, P.; Neagos, N.; Chiappelli, F. Post-Traumatic Stress Disorder: Evidence-Based Research for the Third Millennium. Evid. Based Complement. Altern. Med. 2005, 2, 503–512. [Google Scholar] [CrossRef] [Green Version]
- Rosenfield, P.J.; Stratyner, A.; Tufekcioglu, S.; Karabell, S.; McKelvey, J.; Litt, L. Complex PTSD in ICD-11. J. Psychiatr. Pr. 2018, 24, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Erjavec, G.N.; Konjevod, M.; Perkovic, M.N.; Strac, D.S.; Tudor, L.; Barbas, C.; Grune, T.; Zarkovic, N.; Pivac, N. Short overview on metabolomic approach and redox changes in psychiatric disorders. Redox Biol. 2017, 14, 178–186. [Google Scholar] [CrossRef]
- Sherin, J.E.; Nemeroff, C.B. Post-traumatic stress disorder: The neurobiological impact of psychological trauma. Dialog. Clin. Neurosci. 2011, 13, 263–278. [Google Scholar]
- Rosen, R.L.; Levy-Carrick, N.; Reibman, J.; Xu, N.; Shao, Y.; Liu, M.; Ferri, L.; Kazeros, A.; Caplan-Shaw, C.E.; Pradhan, D.R. Elevated C-reactive protein and posttraumatic stress pathology among survivors of the 9/11 World Trade Center attacks. J. Psychiatr. Res. 2017, 89, 14–21. [Google Scholar] [CrossRef]
- Hauger, R.L.; Olivares-Reyes, J.A.; Dautzenberg, F.M.; Lohr, J.B.; Braun, S.; Oakley, R.H. Molecular and cell signaling targets for PTSD pathophysiology and pharmacotherapy. Neuropharmacology 2012, 62, 705–714. [Google Scholar] [CrossRef] [Green Version]
- Friedman, M.J.; Charney, D.S.; Deutch, A.Y. Neurobiological and Clinical Consequences of Stress: From Normal Adaptation to Post-Traumatic Stress Disorder; Lippincott Williams Wilkins Publishers: Philadelphia, PA, USA, 1995. [Google Scholar]
- Antelman, S.M.; Brown, T.S. Hippocampal lesions and shuttlebox avoidance behavior: A fear hypothesis. Physiol. Behav. 1972, 9, 15–20. [Google Scholar] [CrossRef]
- Lyons, D.M.; Yang, C.; Sawyer-Glover, A.M.; Moseley, M.E.; Schatzberg, A.F. Early life stress and inherited variation in monkey hippocampal volumes. Arch. Gen. Psychiatry 2001, 58, 1145. [Google Scholar] [CrossRef] [Green Version]
- Orr, S.P.; Metzger, L.J.; Lasko, N.B.; Macklin, M.L.; Peri, T.; Pitman, R.K. De novo conditioning in trauma-exposed individuals with and without posttraumatic stress disorder. J. Abnorm. Psychol. 2000, 109, 290. [Google Scholar] [CrossRef] [PubMed]
- Gilbertson, M.W.; Shenton, M.E.; Ciszewski, A.; Kasai, K.; Lasko, N.B.; Orr, S.P.; Pitman, R.K. Smaller hippocampal volume predicts pathologic vulnerability to psychological trauma. Nat. Neurosci. 2002, 5, 1242–1247. [Google Scholar] [CrossRef] [Green Version]
- Rabinak, C.A.; Angstadt, M.; Welsh, R.C.; Kenndy, A.E.; Lyubkin, M.; Martis, B.; Phan, K.L. Altered Amygdala Resting-State Functional Connectivity in Post-Traumatic Stress Disorder. Front. Psychiatry 2011, 2, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morey, R.A.; Gold, A.L.; LaBar, K.S.; Beall, S.K.; Brown, V.M.; Haswell, C.C.; Nasser, J.D.; Wagner, H.R.; McCarthy, G.; Workgroup, F.T.M.-A.M. Amygdala Volume Changes in Posttraumatic Stress Disorder in a Large Case-Controlled Veterans Group. Arch. Gen. Psychiatry 2012, 69, 1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitman, R.K.M.; Rasmusson, A.M.; Koenen, K.C.; Shin, L.M.; Orr, S.P.; Gilbertson, M.W.; Milad, M.R.; Liberzon, I. Biological studies of post-traumatic stress disorder. Nat. Rev. Neurosci. 2012, 13, 769–787. [Google Scholar] [CrossRef]
- Sripada, R.K.; King, A.P.; Garfinkel, S.; Wang, X.; Sripada, C.S.; Welsh, R.C.; Liberzon, I. Altered resting-state amygdala functional connectivity in men with posttraumatic stress disorder. J. Psychiatry Neurosci. 2012, 37, 241–249. [Google Scholar] [CrossRef] [Green Version]
- Knx, J. Trauma and defences: Their roots in relationship: An overview. J. Anal. Psychol. 2003, 48, 207–233. [Google Scholar] [CrossRef]
- Brewin, C.R.; Gregory, J.D.; Lipton, M.; Burgess, N. Intrusive images in psychological disorders: Characteristics, neural mechanisms, and treatment implications. Psychol. Rev. 2010, 117, 210–232. [Google Scholar] [CrossRef] [Green Version]
- Ehlers, A.; Hackmann, A.; Steil, R.; Clohessy, S.; Wenninger, K.; Winter, H. The nature of intrusive memories after trauma: The warning signal hypothesis. Behav. Res. Ther. 2002, 40, 995–1002. [Google Scholar] [CrossRef]
- Hackmann, A.; Ehlers, A.; Speckens, A.; Clark, D.M. Characteristics and content of intrusive memories in PTSD and their changes with treatment. J. Trauma. Stress 2004, 17, 231–240. [Google Scholar] [CrossRef]
- Badura-Brack, A.S.; Heinrichs-Graham, E.; McDermott, T.J.; Becker, K.M.; Ryan, T.J.; Khanna, M.M.; Wilson, T.W. Resting-State Neurophysiological Abnormalities in Posttraumatic Stress Disorder: A Magnetoencephalography Study. Front. Hum. Neurosci. 2017, 11, 205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amen, D.G.; Raji, C.A.; Willeumier, K.; Taylor, D.; Tarzwell, R.; Newberg, A.; Henderson, T.A. Functional Neuroimaging Distinguishes Posttraumatic Stress Disorder from Traumatic Brain Injury in Focused and Large Community Datasets. PLoS ONE 2015, 10, e0129659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanius, R.A.; Williamson, P.C.; Bluhm, R.L.; Densmore, M.; Boksman, K.; Neufeld, R.W.; Gati, J.S.; Menon, R.S. Functional connectivity of dissociative responses in posttraumatic stress disorder: A functional magnetic resonance imaging investigation. Biol. Psychiatry 2005, 57, 873–884. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, C.; Barreto, G.E.; Iarkov, A.; Tarasov, V.V.; Aliev, G.; Moran, V. Cotinine: A Therapy for Memory Extinction in Post-traumatic Stress Disorder. Mol. Neurobiol. 2018, 55, 6700–6711, Erratum in 2018, 55, 6712. [Google Scholar] [CrossRef]
- Stevens, J.S.; Jovanovic, T. Role of social cognition in post-traumatic stress disorder: A review and meta-analysis. Genes Brain Behav. 2018, 18, e12518. [Google Scholar] [CrossRef] [Green Version]
- Amir, M.; Kaplan, Z.; Neumann, L.; Sharabani, R.; Shani, N.; Buskila, D. Posttraumatic stress disorder, tenderness and fibromyalgia. J. Psychosom. Res. 1997, 42, 607–613. [Google Scholar] [CrossRef]
- Sherman, J.J.; Turk, D.C.; Okifuji, A. Prevalence and Impact of Posttraumatic Stress Disorder-Like Symptoms on Patients With Fibromyalgia Syndrome. Clin. J. Pain 2000, 16, 127–134. [Google Scholar] [CrossRef]
- Engel, C.C.; Liu, X.; McCarthy, B.D.; Miller, R.F.; Ursano, R.J. Relationship of Physical Symptoms to Posttraumatic Stress Disorder Among Veterans Seeking Care for Gulf War-Related Health Concerns. Psychosom. Med. 2000, 62, 739–745. [Google Scholar] [CrossRef]
- Schwartz, A.C.; Bradley, R.; Penza, K.M.; Sexton, M.; Jay, D.; Haggard, P.J.; Garlow, S.J.; Ressler, K.J. Pain medication use among patients with posttraumatic stress disorder. J. Psychosom. Res. 2006, 47, 136–142. [Google Scholar] [CrossRef] [Green Version]
- Mellon, S.H.; Gautam, A.; Hammamieh, R.; Jett, M.; Wolkowitz, O.M. Metabolism, Metabolomics, and Inflammation in Posttraumatic Stress Disorder. Boil. Psychiatry 2018, 83, 866–875. [Google Scholar] [CrossRef] [Green Version]
- Metzger, L.J.; Orr, S.P.; Berry, N.J.; Ahern, C.E.; Lasko, N.B.; Pitman, R.K. Physiologic reactivity to startling tones in women with posttraumatic stress disorder. J. Abnorm. Psychol. 1999, 108, 347. [Google Scholar] [CrossRef] [PubMed]
- Pollard, H.B.; Shivakumar, C.; Starr, J.; Eidelman, O.; Jacobowitz, D.M.; Dalgard, C.L.; Srivastava, M.; Wilkerson, M.D.; Stein, M.B.; Ursano, R.J. “Soldier’s heart”: A genetic basis for elevated cardiovascular disease risk associated with post-traumatic stress disorder. Front. Mol. Neurosci. 2016, 9, 87. [Google Scholar] [CrossRef] [Green Version]
- Stein, M.B.; Chen, C.-Y.; Ursano, R.J.; Cai, T.; Gelernter, J.; Heeringa, S.G.; Jain, S.; Jensen, K.P.; Maihofer, A.X.; Mitchell, C.; et al. Genome-wide Association Studies of Posttraumatic Stress Disorder in 2 Cohorts of US Army Soldiers. JAMA Psychiatry 2016, 73, 695–704. [Google Scholar] [CrossRef] [PubMed]
- Xie, P.; Kranzler, H.R.; Yang, C.; Zhao, H.; Farrer, L.A.; Gelernter, J. Genome-wide Association Study Identifies New Susceptibility Loci for Posttraumatic Stress Disorder. Biol. Psychiatry 2013, 74, 656–663. [Google Scholar] [CrossRef] [Green Version]
- Rytwinski, N.K.; Scur, M.; Feeny, N.C.; Youngstrom, E. The Co-Occurrence of Major Depressive Disorder Among Individuals With Posttraumatic Stress Disorder: A Meta-Analysis. J. Trauma. Stress 2013, 26, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Pitman, R.K.; Orr, S.P.; Forgue, D.F.; De Jong, J.B.; Claiborn, J.M. Psychophysiologic Assessment of Posttraumatic Stress Disorder Imagery in Vietnam Combat Veterans. Arch. Gen. Psychiatry 1987, 44, 970–975. [Google Scholar] [CrossRef] [PubMed]
- Stein, M.B.; Jang, K.L.; Taylor, S.; A Vernon, P.; Livesley, W.J. Genetic and Environmental Influences on Trauma Exposure and Posttraumatic Stress Disorder Symptoms: A Twin Study. Am. J. Psychiatry 2002, 159, 1675–1681. [Google Scholar] [CrossRef]
- Miller, M.W.; Wolf, E.J.; Logue, M.W.; Baldwin, C.T. The retinoid-related orphan receptor alpha (RORA) gene and fear-related psychopathology. J. Affect. Disord. 2013, 151, 702–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Logue, M.W.; Baldwin, C.; Guffanti, G.; Melista, E.; Wolf, E.J.; Reardon, A.F.; Uddin, M.; Wildman, D.; Galea, S.; Koenen, K.C.; et al. A genome-wide association study of post-traumatic stress disorder identifies the retinoid-related orphan receptor alpha (RORA) gene as a significant risk locus. Mol. Psychiatry 2012, 18, 937–942. [Google Scholar] [CrossRef] [Green Version]
- Amstadter, A.B.; A Sumner, J.; Acierno, R.; Ruggiero, K.J.; Koenen, K.C.; Kilpatrick, D.G.; Galea, S.; Gelernter, J. Support for association of RORA variant and post traumatic stress symptoms in a population-based study of hurricane exposed adults. Mol. Psychiatry 2013, 18, 1148–1149. [Google Scholar] [CrossRef] [Green Version]
- A Maddox, S.; Kilaru, V.; Shin, J.; Jovanovic, T.; Almli, L.; Dias, B.G.; Norrholm, S.D.; Fani, N.; Michopoulos, V.; Ding, Z.; et al. Estrogen-dependent association of HDAC4 with fear in female mice and women with PTSD. Mol. Psychiatry 2017, 23, 658–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaudry, H.; Falluel-Morel, A.; Bourgault, S.; Basille, M.; Burel, D.; Wurtz, O.; Fournier, A.; Chow, B.K.C.; Hashimoto, H.; Galas, L.; et al. Pituitary Adenylate Cyclase-Activating Polypeptide and Its Receptors: 20 Years after the Discovery. Pharmacol. Rev. 2009, 61, 283–357. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Mandel, H.; Levingston, C.A.; Young, M.R.I. An exploratory approach demonstrating immune skewing and a loss of coordination among cytokines in plasma and saliva of Veterans with combat-related PTSD. Hum. Immunol. 2016, 77, 652–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hori, H.; Kim, Y. Inflammation and post-traumatic stress disorder. Psychiatry Clin. Neurosci. 2019, 73, 143–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furtado, M.; Katzman, M.A. Examining the role of neuroinflammation in major depression. Psychiatry Res. 2015, 229, 27–36. [Google Scholar] [CrossRef]
- Kim, Y.-K.; Amidfar, M.; Won, E. A review on inflammatory cytokine-induced alterations of the brain as potential neural biomarkers in post-traumatic stress disorder. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2019, 91, 103–112. [Google Scholar] [CrossRef]
- O’Mahony, S.; Marchesi, J.R.; Scully, P.; Codling, C.; Ceolho, A.-M.; Quigley, E.M.; Cryan, J.F.; Dinan, T.G. Early Life Stress Alters Behavior, Immunity, and Microbiota in Rats: Implications for Irritable Bowel Syndrome and Psychiatric Illnesses. Biol. Psychiatry 2009, 65, 263–267. [Google Scholar] [CrossRef]
- Garcia-Rodenas, C.L.; Bergonzelli, G.E.; Nutten, S.; Schumann, A.; Cherbut, C.; Turini, M.; Ornstein, K.; Rochat, F.; Corth??sy-Theulaz, I. Nutritional Approach to Restore Impaired Intestinal Barrier Function and Growth After Neonatal Stress in Rats. J. Pediatr. Gastroenterol. Nutr. 2006, 43, 16–24. [Google Scholar] [CrossRef]
- Pace, T.W.W.; Heim, C.M. A short review on the psychoneuroimmunology of posttraumatic stress disorder: From risk factors to medical comorbidities. Brain, Behav. Immun. 2011, 25, 6–13. [Google Scholar] [CrossRef]
- Zuany-Amorim, C.; Sawicka, E.; Manlius, C.; Le Moine, A.; Brunet, L.R.; Kemeny, D.M.; Bowen, G.; Rook, G.A.W.; Walker, C. Suppression of airway eosinophilia by killed Mycobacterium vaccae-induced allergen-specific regulatory T-cells. Nat. Med. 2002, 8, 625–629. [Google Scholar] [CrossRef]
- Hemmings, S.M.; Malan-Muller, S.; van den Heuvel, L.L.; Demmitt, B.A.; Stanislawski, M.A.; Smith, D.G.; Bohr, A.D.; Stamper, C.E.; Hyde, E.R.; Morton, J.T. The microbiome in posttraumatic stress disorder and trauma-exposed controls: An exploratory study. Psychosom. Med. 2017, 79, 936. [Google Scholar] [CrossRef] [PubMed]
- Ventrano, V.A. Histologic Analysis of Cortical Tissue from Patients with Post Traumatic Stress Disorder and Chronic Traumatic Encephalopathy; Boston University: Boston, MA, USA, 2017. [Google Scholar]
- Levin, S.G.; Godukhin, O.V. Modulating effect of cytokines on mechanisms of synaptic plasticity in the brain. Biochemistry 2017, 82, 264–274. [Google Scholar] [CrossRef] [PubMed]
- Passos, I.C.; Vasconcelos-Moreno, M.P.; Costa, L.G.; Kunz, M.; Brietzke, E.; Quevedo, J.; Salum, G.; Magalhães, P.V.; Kapczinski, F.; Kauer-Sant’Anna, M. Inflammatory markers in post-traumatic stress disorder: A systematic review, meta-analysis, and meta-regression. Lancet Psychiatry 2015, 2, 1002–1012. [Google Scholar] [CrossRef]
- Michopoulos, V.; Rothbaum, A.O.; Jovanovic, T.; Almli, L.; Bradley, B.; Rothbaum, B.O.; Gillespie, C.; Ressler, K.J. Association of CRP genetic variation and CRP level with elevated PTSD symptoms and physiological responses in a civilian population with high levels of trauma. Am. J. Psychiatry 2014, 172, 353–362. [Google Scholar] [CrossRef] [Green Version]
- Heidt, T.; Sager, H.B.; Courties, G.; Dutta, P.; Iwamoto, Y.; Zaltsman, A.; Mühlen, C.V.Z.; Bode, C.; Fricchione, G.L.; Denninger, J.; et al. Chronic variable stress activates hematopoietic stem cells. Nat. Med. 2014, 20, 754–758. [Google Scholar] [CrossRef] [Green Version]
- Thachil, J. Platelets in Inflammatory Disorders: A Pathophysiological and Clinical Perspective. Semin. Thromb. Hemost. 2015, 41, 572–581. [Google Scholar] [CrossRef]
- Lindqvist, D.; Mellon, S.H.; Dhabhar, F.S.; Yehuda, R.; Grenon, S.M.; Flory, J.D.; Bierer, L.M.; Abu-Amara, D.; Coy, M.; Makotkine, I.; et al. Increased circulating blood cell counts in combat-related PTSD: Associations with inflammation and PTSD severity. Psychiatry Res. Neuroimaging 2017, 258, 330–336. [Google Scholar] [CrossRef]
- Cornelis, M.C.; Nugent, N.R.; Amstadter, A.B.; Koenen, K.C. Genetics of Post-Traumatic Stress Disorder: Review and Recommendations for Genome-Wide Association Studies. Curr. Psychiatry Rep. 2010, 12, 313–326. [Google Scholar] [CrossRef] [Green Version]
- Bernardy, N.C.; Friedman, M.J. Psychopharmacological Strategies in the Management of Posttraumatic Stress Disorder (PTSD): What Have We Learned? Curr. Psychiatry Rep. 2015, 17, 20. [Google Scholar] [CrossRef]
- Domschke, K.; Tidow, N.; Schwarte, K.; Deckert, J.; Lesch, K.-P.; Arolt, V.; Zwanzger, P.; Baune, B.T. Serotonin transporter gene hypomethylation predicts impaired antidepressant treatment response. Int. J. Neuropsychopharmacol. 2014, 17, 1167–1176. [Google Scholar] [CrossRef] [Green Version]
- Solovieff, N.; Roberts, A.L.; Ratanatharathorn, A.; Haloosim, M.; De Vivo, I.; King, A.P.; Liberzon, I.; Aiello, A.; Uddin, M.; E Wildman, D.; et al. Genetic Association Analysis of 300 Genes Identifies a Risk Haplotype in SLC18A2 for Post-traumatic Stress Disorder in Two Independent Samples. Neuropsychopharmacology 2014, 39, 1872–1879. [Google Scholar] [CrossRef]
- Tseilikman, V.; Dremencov, E.; Tseilikman, O.; Pavlovicova, M.; Lacinova, L.; Jezova, D. Role of glucocorticoid- and monoamine-metabolizing enzymes in stress-related psychopathological processes. Stress 2019, 23, 1–12. [Google Scholar] [CrossRef]
- Zhang, L.; Li, H.; Su, T.; Barker, J.; Maric, D.; Fullerton, C.; Webster, M.; Hough, C.; Li, X.; Ursano, R.J. p11 is up-regulated in the forebrain of stressed rats by glucocorticoid acting via two specific glucocorticoid response elements in the p11 promoter. Neuroscience 2008, 153, 1126–1134. [Google Scholar] [CrossRef]
- Somvanshi, P.R.; Mellon, S.H.; Flory, J.D.; Abu-Amara, P.D.; Wolkowitz, O.M.; Yehuda, R.; Jett, M.; Hood, L.; Marmar, C.; Doyle 3rd, F.J. Immunometabolic Cross-Talk and Regulation of Endocrine and Metabolic Functions: Mechanistic inferences on metabolic dysfunction in posttraumatic stress disorder from an integrated model and multiomic analysis: Role of glucocorticoid receptor sensitivity. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E879. [Google Scholar] [CrossRef] [PubMed]
- Ryder, A.L.; Azcarate, P.M.; Cohen, B.E. PTSD and Physical Health. Curr. Psychiatry Rep. 2018, 20, 116. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.-C.; Koenen, K.C.; Galea, S.; Aiello, A.E.; Soliven, R.; Wildman, D.E.; Uddin, M. Molecular Variation at the SLC6A3 Locus Predicts Lifetime Risk of PTSD in the Detroit Neighborhood Health Study. PLoS ONE 2012, 7, e39184. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, L. Hypothalamic-Pituitary-Adrenocortical Axis: Neuropsychiatric Aspects. Compr. Physiol. 2014, 4, 715–738. [Google Scholar] [CrossRef] [PubMed]
- Nelson, E.C.; Agrawal, A.; Pergadia, M.L.; Lynskey, M.T.; Todorov, A.A.; Wang, J.C.; Todd, R.D.; Martin, N.G.; Heath, A.C.; Goate, A.; et al. Association of childhood trauma exposure and GABRA2 polymorphisms with risk of posttraumatic stress disorder in adults. Mol. Psychiatry 2009, 14, 234–235. [Google Scholar] [CrossRef]
- Sabban, E.; Alaluf, L.G.; Serova, L.I. Potential of neuropeptide Y for preventing or treating post-traumatic stress disorder. Neuropeptides 2016, 56, 19–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theal, R.; Tay, V.X.P.; Hickman, I.J. Conflicting relationship between dietary intake and metabolic health in PTSD: A systematic review. Nutr. Res. 2018, 54, 12–22. [Google Scholar] [CrossRef] [Green Version]
- Konjevod, M.; Tudor, L.; Strac, D.S.; Erjavec, G.N.; Barbas, C.; Zarkovic, N.; Perkovic, M.N.; Uzun, S.; Kozumplik, O.; Lauc, G.; et al. Metabolomic and glycomic findings in posttraumatic stress disorder. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2019, 88, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Mocking, R.J.T.; Assies, J.; Ruhé, H.G.; Schene, A.H. Focus on fatty acids in the neurometabolic pathophysiology of psychiatric disorders. J. Inherit. Metab. Dis. 2018, 41, 597–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, M.W.; Sadeh, N. Traumatic stress, oxidative stress and post-traumatic stress disorder: Neurodegeneration and the accelerated-aging hypothesis. Mol. Psychiatry 2014, 19, 1156–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauser, P.S.; Narayanaswami, V.; Ryan, R.O. Apolipoprotein E: From lipid transport to neurobiology. Prog. Lipid Res. 2011, 50, 62–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, L.A.; Zuloaga, D.G.; Bidiman, E.; Marzulla, T.; Weber, S.; Wahbeh, H.; Raber, J. ApoE2 Exaggerates PTSD-Related Behavioral, Cognitive, and Neuroendocrine Alterations. Neuropsychopharmacology 2015, 40, 2443–2453. [Google Scholar] [CrossRef] [Green Version]
- Yehuda, R. Status of Glucocorticoid Alterations in Post-traumatic Stress Disorder. Ann. N. Y. Acad. Sci. 2009, 1179, 56–69. [Google Scholar] [CrossRef]
- Somvanshi, P.R.; Mellon, S.H.; Flory, J.D.; Abu-Amara, D.; PTSD Systems Biology Consortium; Wolkowitz, O.M.; Yehuda, R.; Jett, M.; Hood, L.; Marmar, C.; et al. Mechanistic inferences on metabolic dysfunction in PTSD from an integrated model and multi-omic analysis: Role of glucocorticoid receptor sensitivity. Am. J. Physiol. Metab. 2019, 317, E879–E898. [Google Scholar] [CrossRef]
- Vasileva, L.; Ivanovska, M.V.; Murdjeva, M.A.; Saracheva, K.E.; Georgiev, M.I. Immunoregulatory natural compounds in stress-induced depression: An alternative or an adjunct to conventional antidepressant therapy? Food Chem. Toxicol. 2019, 127, 81–88. [Google Scholar] [CrossRef]
- Heim, C.; Newport, D.J.; Mletzko, T.; Miller, A.H.; Nemeroff, C.B. The link between childhood trauma and depression: Insights from HPA axis studies in humans. Psychoneuroendocrinology 2008, 33, 693–710. [Google Scholar] [CrossRef]
- Cacabelos, R.; Torrellas, C.; Fernández-Novoa, L.; Aliev, G. Neuroimmune Crosstalk in CNS Disorders: The Histamine Connection. Curr. Pharm. Des. 2016, 22, 819–848. [Google Scholar] [CrossRef]
- Bale, T.L.; Vale, W.W. CRF and CRF receptors: Role in stress responsivity and other behaviors. Annu. Rev. Pharmacol. Toxicol. 2004, 44, 525–557. [Google Scholar] [CrossRef] [PubMed]
- Grigoriadis, D.E. The corticotropin-releasing factor receptor: A novel target for the treatment of depression and anxiety-related disorders. Expert Opin. Ther. Targets 2005, 9, 651–684. [Google Scholar] [CrossRef] [PubMed]
- Hauger, R.L.; Risbrough, V.; Brauns, O.; Dautzenberg, F.M. Corticotropin releasing factor (CRF) receptor signaling in the central nervous system: New molecular targets. CNS Neurol. Disord.Drug Targets 2006, 5, 453–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holsboer, F.; Ising, M. Central CRH system in depression and anxiety—Evidence from clinical studies with CRH1 receptor antagonists. Eur. J. Pharmacol. 2008, 583, 350–357. [Google Scholar] [CrossRef]
- Banerjee, S.B.; Morrison, F.G.; Ressler, K.J. Genetic approaches for the study of PTSD: Advances and challenges. Neurosci. Lett. 2017, 649, 139–146. [Google Scholar] [CrossRef] [Green Version]
- Castro-Vale, I.; Van Rossum, E.F.C.; Machado, J.C.; Mota-Cardoso, R.; Carvalho, D. Genetics of glucocorticoid regulation and posttraumatic stress disorder—What do we know? Neurosci. Biobehav. Rev. 2016, 63, 143–157. [Google Scholar] [CrossRef]
- Yehuda, R. Is the glucocorticoid receptor a therapeutic target for the treatment of PTSD. Psychoneuroendocrinology 2015, 61, 2. [Google Scholar] [CrossRef]
- Hauer, D.; Weis, F.; Papassotiropoulos, A.; Schmoeckel, M.; Beiras-Fernandez, A.; Lieke, J.; Kaufmann, I.; Kirchhoff, F.; Vogeser, M.; Roozendaal, B.; et al. Relationship of a common polymorphism of the glucocorticoid receptor gene to traumatic memories and posttraumatic stress disorder in patients after intensive care therapy. Crit. Care Med. 2011, 39, 643–650. [Google Scholar] [CrossRef]
- Palma-Gudiel, H.; Córdova-Palomera, A.; Leza, J.; Fañanás, L. Glucocorticoid receptor gene (NR3C1) methylation processes as mediators of early adversity in stress-related disorders causality: A critical review. Neurosci. Biobehav. Rev. 2015, 55, 520–535. [Google Scholar] [CrossRef] [Green Version]
- Hawn, S.E.; Sheerin, C.M.; Lind, M.J.; Hicks, T.A.; Marraccini, M.E.; Bountress, K.; Bacanu, S.-A.; Nugent, N.R.; Amstadter, A.B. GxE effects of FKBP5 and traumatic life events on PTSD: A meta-analysis. J. Affect. Disord. 2019, 243, 455–462. [Google Scholar] [CrossRef]
- Binder, E.B. The role of FKBP5, a co-chaperone of the glucocorticoid receptor in the pathogenesis and therapy of affective and anxiety disorders. Psychoneuroendocrinology 2009, 34, S186–S195. [Google Scholar] [CrossRef] [PubMed]
- Koenen, K.C.; Widom, C.S. A prospective study of sex differences in the lifetime risk of posttraumatic stress disorder among abused and neglected children grown up. J. Trauma. Stress 2009, 22, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Xie, P.; Kranzler, H.R.; Poling, J.; Stein, M.B.; Anton, R.F.; Farrer, L.A.; Gelernter, J. Interaction of FKBP5 with Childhood Adversity on Risk for Post-Traumatic Stress Disorder. Neuropsychopharmacology 2010, 35, 1684–1692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Licznerski, P.; Duric, V.; Banasr, M.; Alavian, K.N.; Ota, K.T.; Kang, H.J.; Jonas, E.A.; Ursano, R.J.; Krystal, J.H.; Duman, R.S.; et al. Decreased SGK1 Expression and Function Contributes to Behavioral Deficits Induced by Traumatic Stress. PLoS Biol. 2015, 13, e1002282. [Google Scholar] [CrossRef] [Green Version]
- Preston, G.; Kirdar, F.; Kozicz, T. The role of suboptimal mitochondrial function in vulnerability to post-traumatic stress disorder. J. Inherit. Metab. Dis. 2018, 41, 585–596. [Google Scholar] [CrossRef]
- Du, J.; Wang, Y.; Hunter, R.G.; Wei, Y.; Blumenthal, R.; Falke, C.; Khairova, R.; Zhou, R.; Yuan, P.; Machado-Vieira, R.; et al. Dynamic regulation of mitochondrial function by glucocorticoids. Proc. Natl. Acad. Sci. USA 2009, 106, 3543–3548. [Google Scholar] [CrossRef] [Green Version]
- Mellon, S.H.; Bersani, F.S.; Lindqvist, D.; Hammamieh, R.; Donohue, D.; Dean, K.; Jett, M.; Yehuda, R.; Flory, J.; Reus, V.I.; et al. Metabolomic analysis of male combat veterans with post traumatic stress disorder. PLoS ONE 2019, 14, e0213839. [Google Scholar] [CrossRef] [Green Version]
- Kao, C.-Y.; He, Z.; Henes, K.; Asara, J.M.; Webhofer, C.; Filiou, M.D.; Khaitovich, P.; Wotjak, C.T.; Turck, C.W. Fluoxetine Treatment Rescues Energy Metabolism Pathway Alterations in a Posttraumatic Stress Disorder Mouse Model. Mol. Neuropsychiatry 2016, 2, 46–59. [Google Scholar] [CrossRef]
- Flaquer, A.; Baumbach, C.; Ladwig, K.-H.; Kriebel, J.; Waldenberger, M.; Grallert, H.; Baumert, J.; Meitinger, T.; Kruse, J.; Peters, A.; et al. Mitochondrial genetic variants identified to be associated with posttraumatic stress disorder. Transl. Psychiatry 2015, 5, e524. [Google Scholar] [CrossRef] [Green Version]
- Manoli, I.; Alesci, S.; Blackman, M.R.; Su, Y.A.; Rennert, O.M.; Chrousos, G.P. Mitochondria as key components of the stress response. Trends Endocrinol. Metab. 2007, 18, 190–198. [Google Scholar] [CrossRef]
- Manji, H.; Kato, T.; Di Prospero, N.A.; Ness, S.; Beal, M.F.; Krams, M.; Chen, G. Impaired mitochondrial function in psychiatric disorders. Nat. Rev. Neurosci. 2012, 13, 293–307. [Google Scholar] [CrossRef] [PubMed]
- Bersani, F.S.; Morley, C.; Lindqvist, D.; Epel, E.S.; Picard, M.; Yehuda, R.; Flory, J.; Bierer, L.M.; Makotkine, I.; Abu-Amara, D.; et al. Mitochondrial DNA copy number is reduced in male combat veterans with PTSD. Prog. Neuro-Psychopharmacol. Boil. Psychiatry 2016, 64, 10–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andero, R.; Ressler, K.J. Fear extinction and BDNF: Translating animal models of PTSD to the clinic. Genes Brain Behav. 2012, 11, 503–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammamieh, R.; Chakraborty, N.; Gautam, A.; Muhie, S.; Yang, R.; Donohue, D.E.; Kumar, R.; Daigle, B.J.; Zhang, Y.; A Amara, D.; et al. Whole-genome DNA methylation status associated with clinical PTSD measures of OIF/OEF veterans. Transl. Psychiatry 2017, 7, e1169. [Google Scholar] [CrossRef]
- O’Donovan, A.; Epel, E.; Lin, J.; Wolkowitz, O.; Cohen, B.; Maguen, S.; Metzler, T.; Lenoci, M.; Blackburn, E.; Neylan, T.C. Childhood Trauma Associated with Short Leukocyte Telomere Length in Posttraumatic Stress Disorder. Biol. Psychiatry 2011, 70, 465–471. [Google Scholar] [CrossRef] [Green Version]
- Amstadter, A.B.; Koenen, K.C.; Ruggiero, K.J.; Acierno, R.; Galea, S.; Kilpatrick, D.G.; Gelernter, J. Variant in RGS2 moderates posttraumatic stress symptoms following potentially traumatic event exposure. J. Anxiety Disord. 2009, 23, 369–373. [Google Scholar] [CrossRef] [Green Version]
- Shalev, A.Y.; Friedman, M.J.; Foa, E.B.; Keane, T.M. Integration and summary. In Effective Treatments for PTSD: Practice Guidelines from the International Society for Traumatic Stress Studies; The Guilford Press: New York, NY, USA, 2000; pp. 60–83. [Google Scholar]
- Bradley, R.; Greene, J.; Russ, E.; Dutra, L.; Westen, D. A Multidimensional Meta-Analysis of Psychotherapy for PTSD. Am. J. Psychiatry 2005, 162, 214–227. [Google Scholar] [CrossRef] [Green Version]
- Kozaric-Kovacic, D.; Hercigonja, D.K.; Grubisić-Ilić, M. Posttraumatic stress disorder and depression in soldiers with combat experiences. Croat. Med J. 2001, 42, 165–170. [Google Scholar]
- Kozarić-Kovačić, D.; Kocijan-Hercigonja, D. Assessment of post-traumatic stress disorder and comorbidity. Mil. Med. 2001, 166, 677–680. [Google Scholar] [CrossRef] [Green Version]
- Hamner, M.; Robert, S.; Frueh, B.C. Treatment-resistant posttraumatic stress disorder: Strategies for intervention. CNS Spectr. 2004, 9, 740–752. [Google Scholar] [CrossRef]
- Davidson, J.R.; Malik, M.L.; Sutherland, S.N. Response characteristics to antidepressants and placebo in post-traumatic stress disorder. Int. Clin. Psychopharmacol. 1997, 12, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Lichtblau, N.; Schmidt, F.M.; Schumann, R.; Kirkby, K.C.; Himmerich, H. Cytokines as biomarkers in depressive disorder: Current standing and prospects. Int. Rev. Psychiatry 2013, 25, 592–603. [Google Scholar] [CrossRef] [PubMed]
- Chung, M.Y.; Min, K.H.; Jun, Y.J.; Kim, S.S.; Kim, W.C.; Jun, E.M. Efficacy and tolerability of mirtazapine and sertraline in Korean veterans with posttraumatic stress disorder: A randomized open label trial. Hum. Psychopharmacol. Clin. Exp. 2004, 19, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Pivac, N.; Kozaric-Kovacic, D.; Muck-Seler, D. Olanzapine versus fluphenazine in an open trial in patients with psychotic combat-related post-traumatic stress disorder. Psychopharmacology 2003, 175, 1. [Google Scholar] [CrossRef] [PubMed]
- Hurley, E.A. Pharmacotherapy to Blunt Memories of Sexual Violence: What’s a Feminist to Think? Hypatia 2010, 25, 527–552. [Google Scholar] [CrossRef]
- Maier, S.F. Stressor Controllability and Stress-Induced Analgesia. Ann. N. Y. Acad. Sci. 1986, 467, 55–72. [Google Scholar] [CrossRef]
- Fanselow, M.S. Conditioned Fear-Induced Opiate Analgesia: A Competing Motivational State Theory of Stress Analgesia. Ann. N. Y. Acad. Sci. 1986, 467, 40–54. [Google Scholar] [CrossRef] [PubMed]
- Pitman, R.K.; Van Der Kolk, B.A.; Orr, S.P.; Greenberg, M.S. Naloxone-Reversible Analgesic Response to Combat-Related Stimuli in Posttraumatic Stress Disorder. Arch. Gen. Psychiatry 1990, 47, 541–544. [Google Scholar] [CrossRef]
- Asnis, G.M.; Kohn, S.R.; Henderson, M.; Brown, N.L.; Asnis, G.M. SSRIs versus Non-SSRIs in Post-traumatic Stress Disorder. Drugs 2004, 64, 383–404. [Google Scholar] [CrossRef]
- Cameron, L.P.; Benson, C.J.; DeFelice, B.C.; Fiehn, O.; Olson, D.E. Chronic, Intermittent Microdoses of the Psychedelic N,N-Dimethyltryptamine (DMT) Produce Positive Effects on Mood and Anxiety in Rodents. ACS Chem. Neuroscience 2019, 10, 3261–3270. [Google Scholar] [CrossRef] [Green Version]
- Malikowska, N.; Fijałkowski, Ł.; Nowaczyk, A.; Popik, P.; Sałat, K. Antidepressant-like activity of venlafaxine and clonidine in mice exposed to single prolonged stress–A model of post-traumatic stress disorder. Pharmacodynamic and molecular docking studies. Brain Res. 2017, 1673, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Barreto, G.E.; Aliev, G.; Moran, V. Ginkgo biloba as an Alternative Medicine in the Treatment of Anxiety in Dementia and other Psychiatric Disorders. Curr. Drug Metab. 2017, 18, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Ashley-Koch, A.; Garrett, M.E.; Gibson, J.; Liu, Y.; Dennis, M.F.; Kimbrel, N.A.; Beckham, J.C.; Hauser, M.A. Genome-wide association study of posttraumatic stress disorder in a cohort of Iraq-Afghanistan era veterans. J. Affect. Disord. 2015, 184, 225–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liberzon, I.; King, A.P.; Ressler, K.J.; Almli, L.; Zhang, P.; Ma, S.T.; Cohen, G.H.; Tamburrino, M.B.; Calabrese, J.R.; Galea, S. Interaction of theADRB2Gene Polymorphism With Childhood Trauma in Predicting Adult Symptoms of Posttraumatic Stress Disorder. JAMA Psychiatry 2014, 71, 1174–1182. [Google Scholar] [CrossRef] [Green Version]
- Raevsky, O.A.; Mukhametov, A.; Grigorev, V.Y.; Ustyugov, A.; Tsay, S.-C.; Hwu, J.R.; Yarla, N.S.; Tarasov, V.V.; Aliev, G.; Bachurin, S.O.; et al. Applications of Multi-Target Computer-Aided Methodologies in Molecular Design of CNS Drugs. Curr. Med. Chem. 2019, 25, 5293–5314. [Google Scholar] [CrossRef]
- Cacabelos, R.; Reddy, V.P.; Aliev, G. Editorial (Thematic Issue: Neurodegeneration, Oxidative Stress, Metabolic Syndrome, Drug Design and Development: Clinical Implications). CNS Neurol. Disord. Drug Targets 2016, 15, 126. [Google Scholar] [CrossRef]
- Aliev, G.; Barreto, G.E.; Cacabelos, R. Editorial: Genomics and Epigenomics of Tumor and Aging Cells. Curr. Genom. 2017, 18, 375–377. [Google Scholar] [CrossRef] [Green Version]
- Barreto, G.E.; Ávila-Rodriguez, M.F.; Foitzick, M.; Aliev, G.; Moran, V. Advances in Medicinal Plants with Effects on Anxiety Behavior Associated to Mental and Health Conditions. Curr. Med. Chem. 2017, 24, 411–423. [Google Scholar] [CrossRef]
- Ustyugov, A.A.; Shevtsova, E.; Ashraf, G.M.; Tarasov, V.V.; Bachurin, S.O.; Aliev, G. New Therapeutic Property of Dimebon as a Neuroprotective Agent. Curr. Med. Chem. 2019, 25, 5315–5326. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aliev, G.; Beeraka, N.M.; Nikolenko, V.N.; Svistunov, A.A.; Rozhnova, T.; Kostyuk, S.; Cherkesov, I.; Gavryushova, L.V.; Chekhonatsky, A.A.; Mikhaleva, L.M.; et al. Neurophysiology and Psychopathology Underlying PTSD and Recent Insights into the PTSD Therapies—A Comprehensive Review. J. Clin. Med. 2020, 9, 2951. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm9092951
Aliev G, Beeraka NM, Nikolenko VN, Svistunov AA, Rozhnova T, Kostyuk S, Cherkesov I, Gavryushova LV, Chekhonatsky AA, Mikhaleva LM, et al. Neurophysiology and Psychopathology Underlying PTSD and Recent Insights into the PTSD Therapies—A Comprehensive Review. Journal of Clinical Medicine. 2020; 9(9):2951. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm9092951
Chicago/Turabian StyleAliev, Gjumrakch, Narasimha M. Beeraka, Vladimir N. Nikolenko, Andrey A. Svistunov, Tatyana Rozhnova, Svetlana Kostyuk, Igor Cherkesov, Liliya V. Gavryushova, Andrey A. Chekhonatsky, Liudmila M. Mikhaleva, and et al. 2020. "Neurophysiology and Psychopathology Underlying PTSD and Recent Insights into the PTSD Therapies—A Comprehensive Review" Journal of Clinical Medicine 9, no. 9: 2951. https://0-doi-org.brum.beds.ac.uk/10.3390/jcm9092951