
In Silico Analysis of Peptide-Based Derivatives Containing Bifunctional Warheads Engaging Prime and Non-Prime Subsites to Covalent Binding SARS-CoV-2 Main Protease (Mpro)

 ,
,  ,
,  ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Computational Details
2.1.1. Protein and Ligand Preparation
2.1.2. Molecular Docking
2.1.3. Molecular Dynamics
2.1.4. Covalent Docking
2.1.5. Physicochemical Properties Evaluation
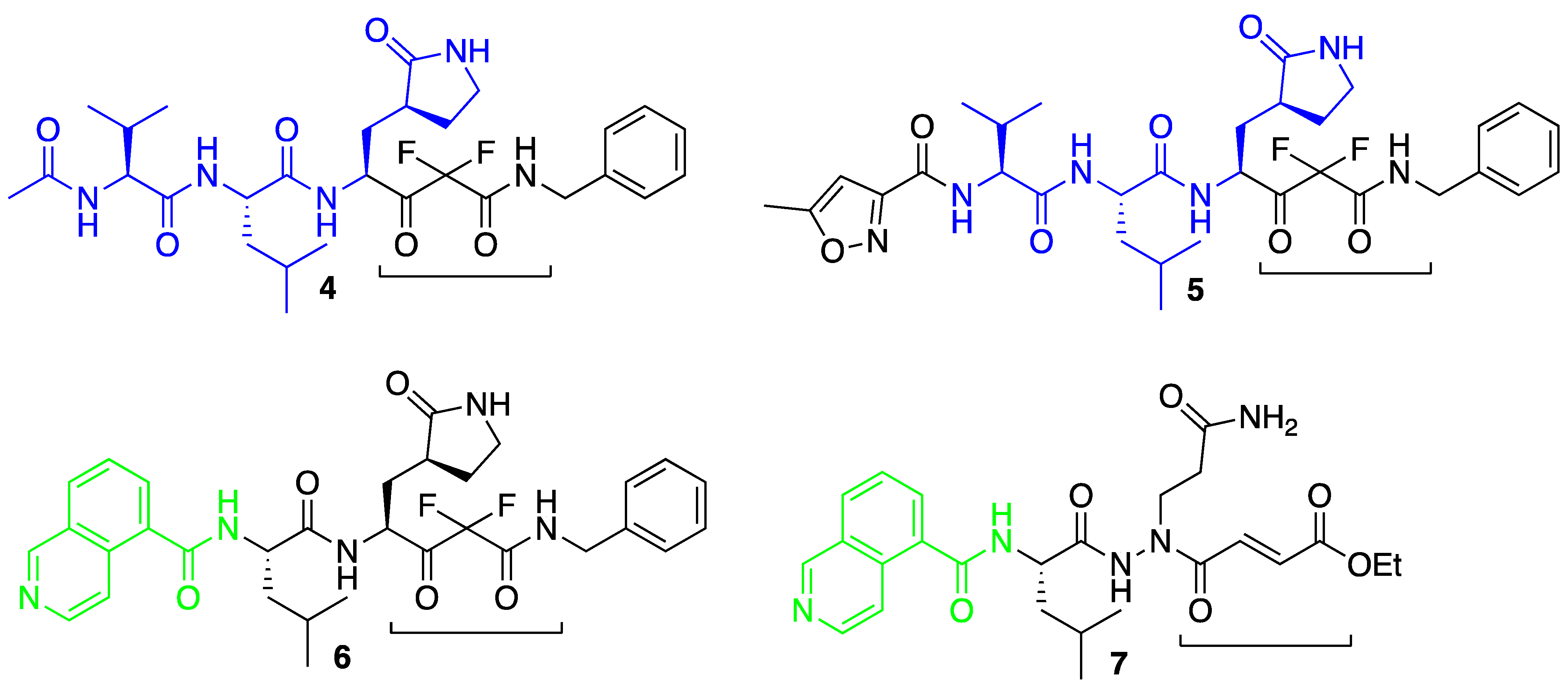
3. Results and Discussion
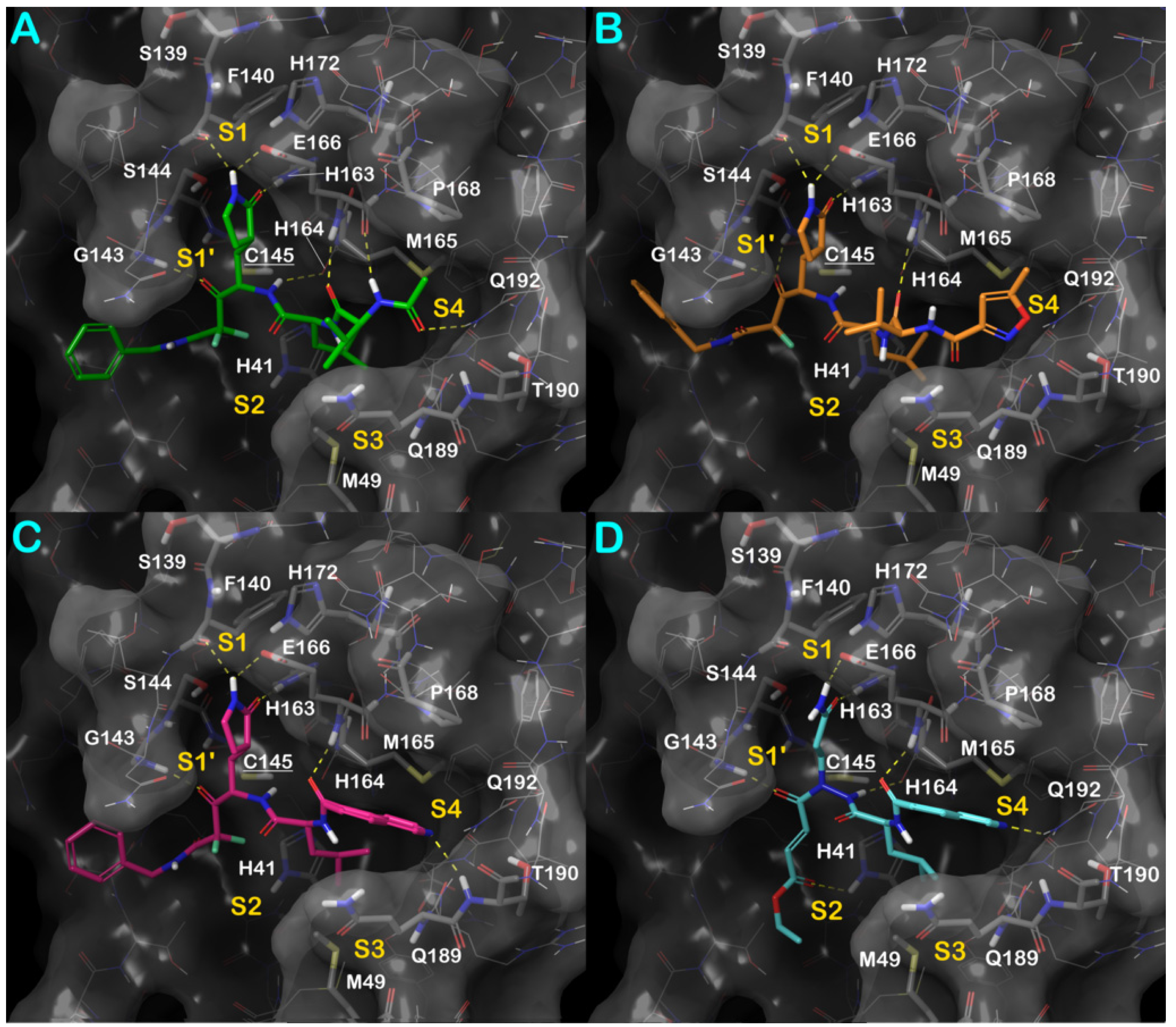
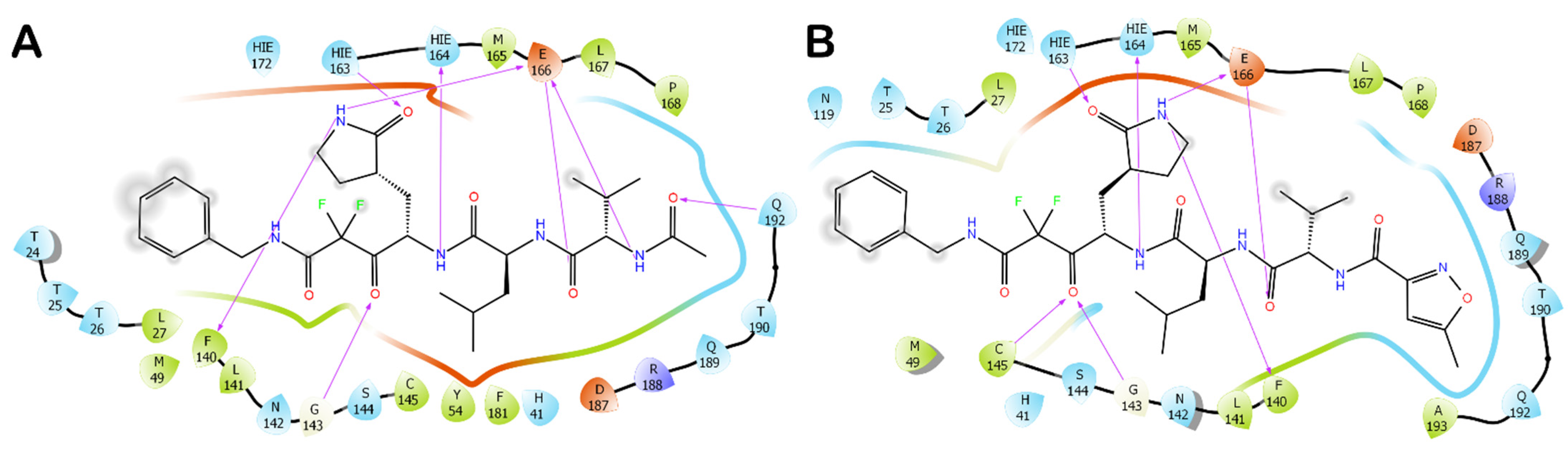
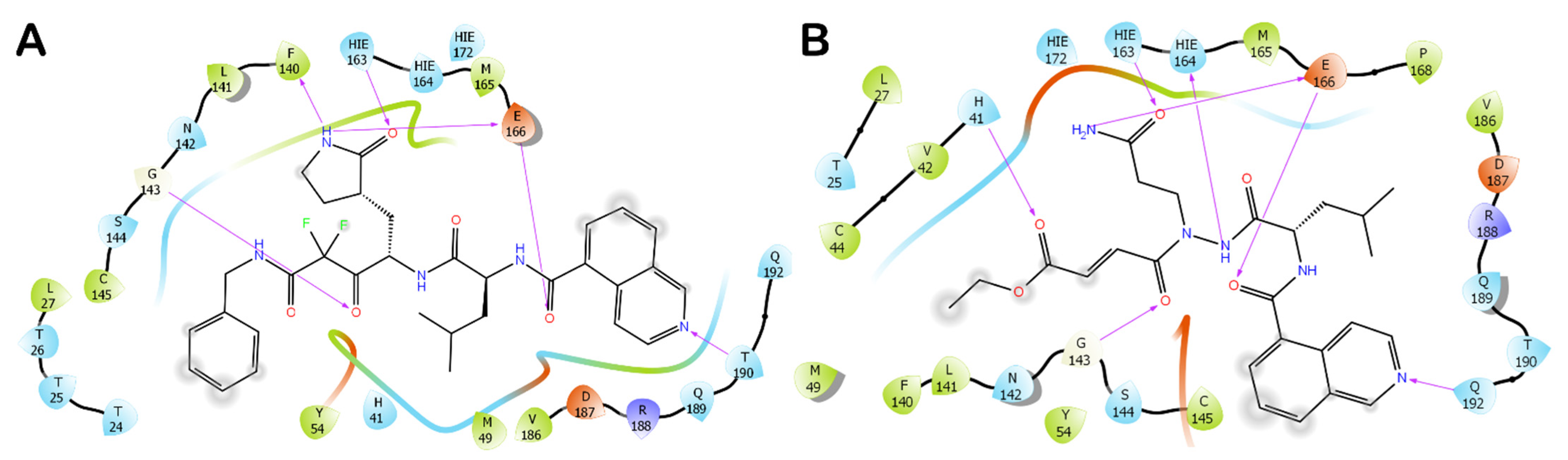
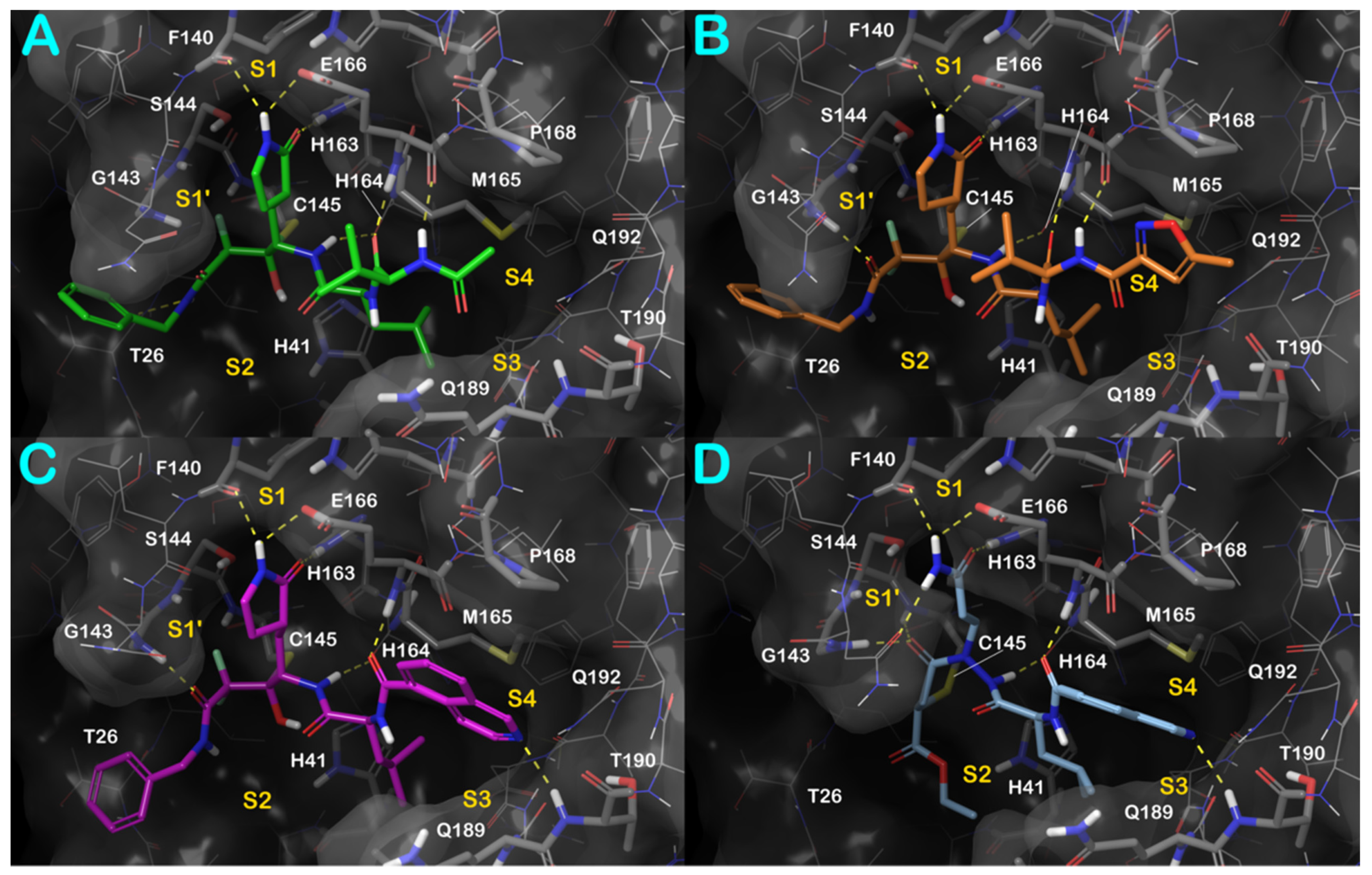
3.1. Molecular Docking Studies
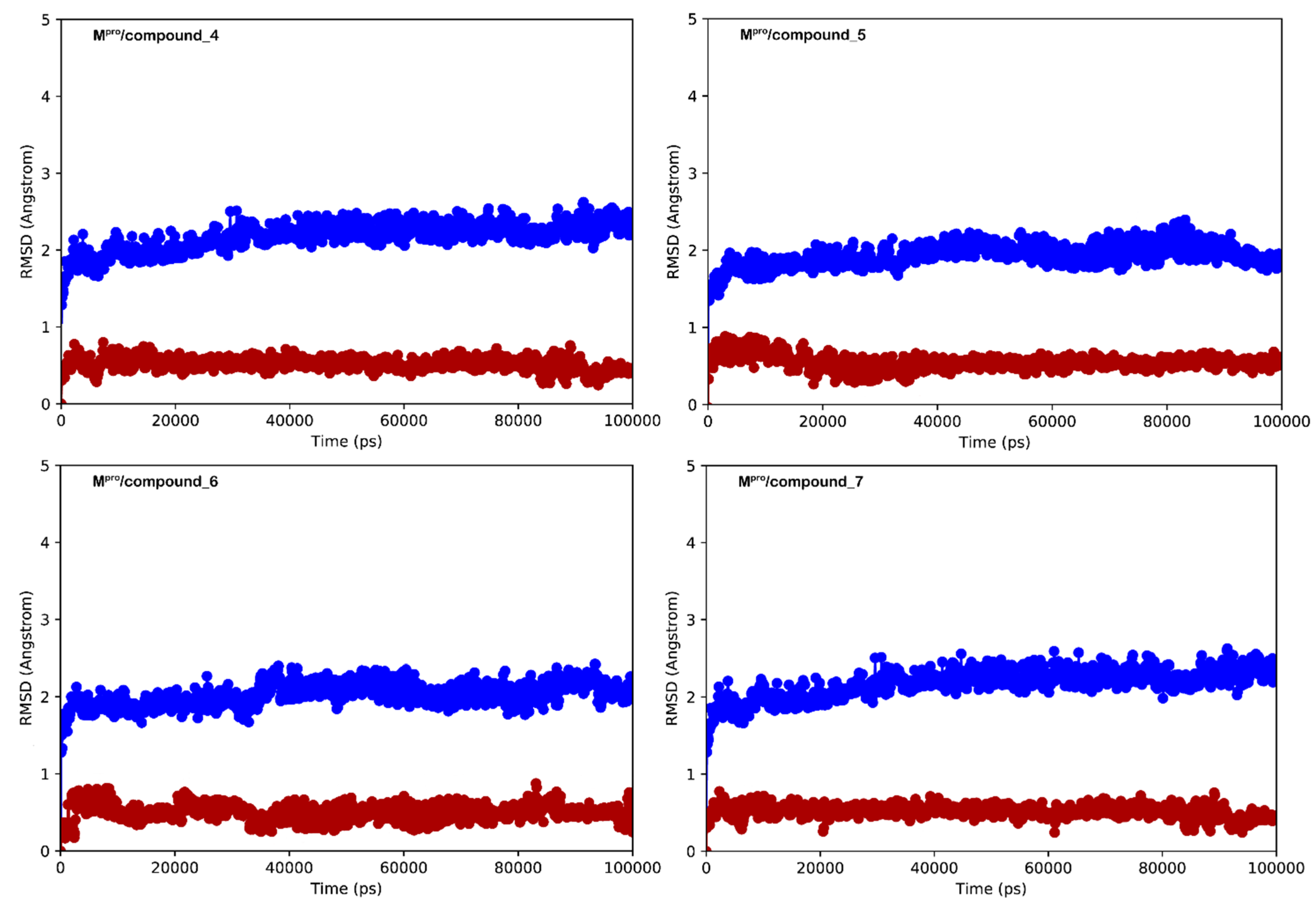
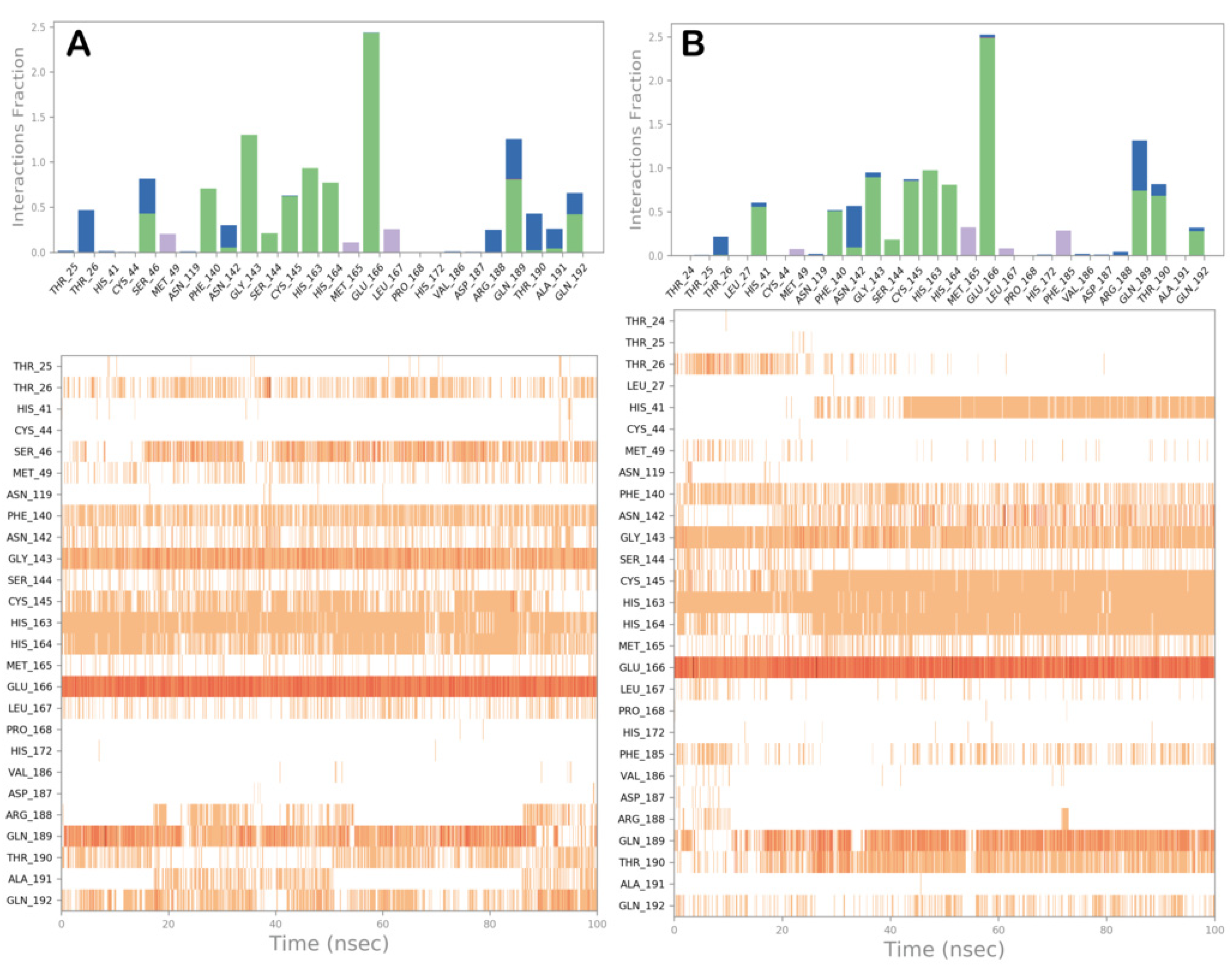
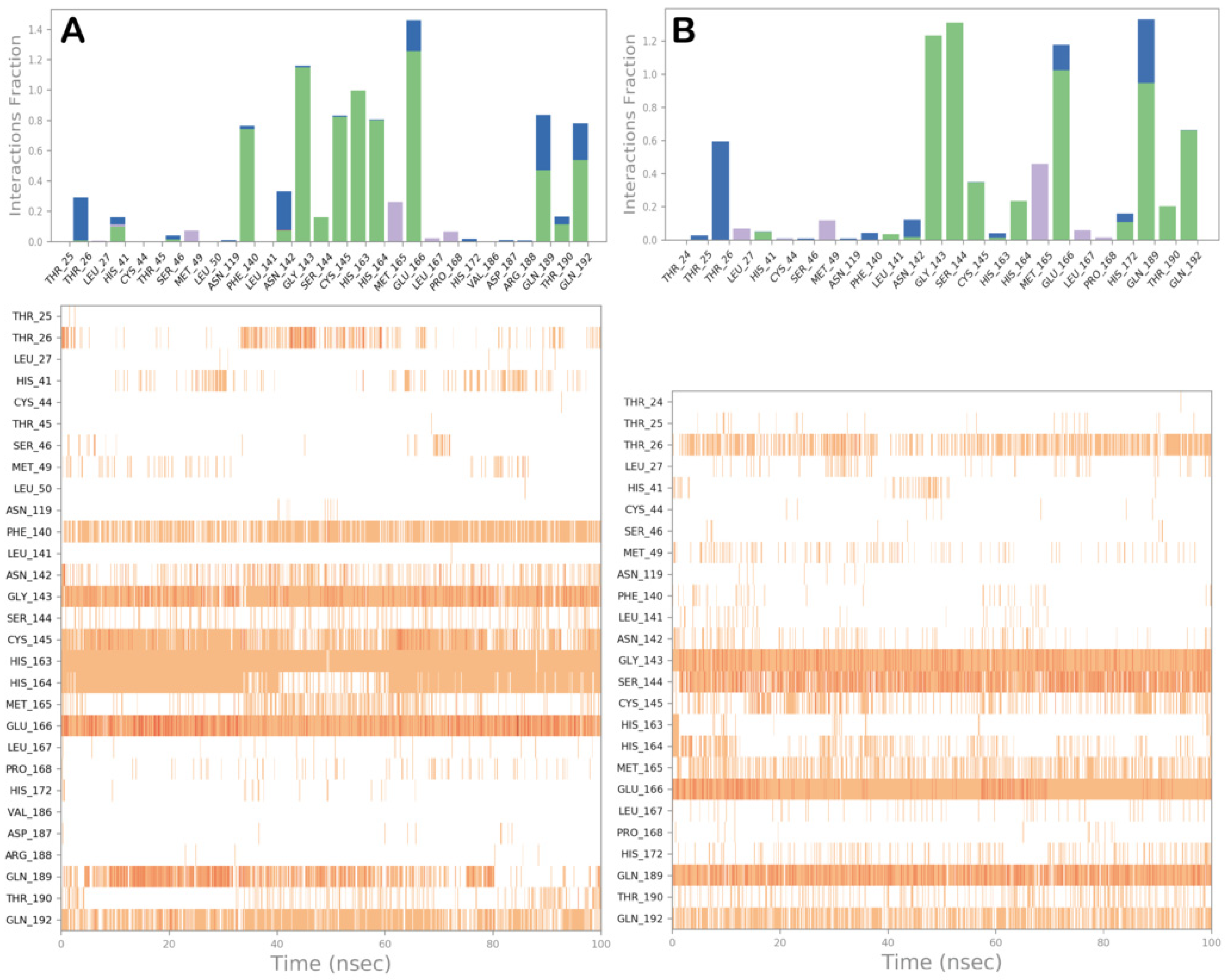
3.2. Molecular Dynamics Simulations
3.3. Covalent Docking Approach
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pollard, C.A.; Morran, M.P.; Nestor-Kalinoski, A.L. The COVID-19 pandemic: A global health crisis. Physiol. Genomics 2020, 52, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Mahase, E. COVID-19: UK becomes first country to authorise antiviral molnupiravir. BMJ 2021, 375, n2697. [Google Scholar] [CrossRef] [PubMed]
- Mahase, E. COVID-19: Pfizer’s paxlovid is 89% effective in patients at risk of serious illness, company reports. BMJ 2021, 375, n2713. [Google Scholar] [CrossRef] [PubMed]
- Painter, W.P.; Holman, W.; Bush, J.A.; Almazedi, F.; Malik, H.; Eraut, N.; Morin, M.J.; Szewczyk, L.J.; Painter, G.R. Human Safety, Tolerability, and Pharmacokinetics of Molnupiravir, a Novel Broad-Spectrum Oral Antiviral Agent with Activity Against SARS-CoV-2. Antimicrob. Agents Chemother. 2021, 65, e02428-20. [Google Scholar] [CrossRef]
- Fischer, W.; Eron, J.J.; Holman, W.; Cohen, M.S.; Fang, L.; Szewczyk, L.J.; Sheahan, T.P.; Baric, R.; Mollan, K.R.; Wolfe, C.R.; et al. Molnupiravir, an Oral Antiviral Treatment for COVID-19. medRxiv 2021. [Google Scholar] [CrossRef]
- Cui, J.; Li, F.; Shi, Z.L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, R.; Perera, L.; Tillekeratne, L.M.V. Potential SARS-CoV-2 main protease inhibitors. Drug Discov. Today 2021, 26, 804–816. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of M(pro) from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Owen, D.R.; Allerton, C.M.N.; Anderson, A.S.; Aschenbrenner, L.; Avery, M.; Berritt, S.; Boras, B.; Cardin, R.D.; Carlo, A.; Coffman, K.J.; et al. An oral SARS-CoV-2 M(pro) inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374, 1586–1593. [Google Scholar] [CrossRef]
- Zhao, Y.; Fang, C.; Zhang, Q.; Zhang, R.; Zhao, X.; Duan, Y.; Wang, H.; Zhu, Y.; Feng, L.; Zhao, J.; et al. Crystal structure of SARS-CoV-2 main protease in complex with protease inhibitor PF-07321332. Protein Cell 2021. [Google Scholar] [CrossRef]
- Narayanan, A.; Narwal, M.; Majowicz, S.A.; Varricchio, C.; Toner, S.A.; Ballatore, C.; Brancale, A.; Murakami, K.S.; Jose, J. Identification of SARS-CoV-2 inhibitors targeting Mpro and PLpro using in-cell-protease assay. Commun. Biol. 2022, 5, 169. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brogi, S.; Giovani, S.; Brindisi, M.; Gemma, S.; Novellino, E.; Campiani, G.; Blackman, M.J.; Butini, S. In silico study of subtilisin-like protease 1 (SUB1) from different Plasmodium species in complex with peptidyl-difluorostatones and characterization of potent pan-SUB1 inhibitors. J. Mol. Graph. Model. 2016, 64, 121–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giovani, S.; Penzo, M.; Brogi, S.; Brindisi, M.; Gemma, S.; Novellino, E.; Savini, L.; Blackman, M.J.; Campiani, G.; Butini, S. Rational design of the first difluorostatone-based PfSUB1 inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 3582–3586. [Google Scholar] [CrossRef]
- Giovani, S.; Penzo, M.; Butini, S.; Brindisi, M.; Gemma, S.; Novellino, E.; Campiani, G.; Blackman, M.J.; Brogi, S. Plasmodium falciparum subtilisin-like protease 1: Discovery of potent difluorostatone-based inhibitors. RSC Adv. 2015, 5, 22431–22448. [Google Scholar] [CrossRef]
- Brogi, S.; Maramai, S.; Brindisi, M.; Chemi, G.; Porcari, V.; Corallo, C.; Gennari, L.; Novellino, E.; Ramunno, A.; Butini, S.; et al. Activation of the Wnt Pathway by Small Peptides: Rational Design, Synthesis and Biological Evaluation. ChemMedChem 2017, 12, 2074–2085. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Testai, L.; Piragine, E.; Piano, I.; Flori, L.; Da Pozzo, E.; Miragliotta, V.; Pirone, A.; Citi, V.; Di Cesare Mannelli, L.; Brogi, S.; et al. The Citrus Flavonoid Naringenin Protects the Myocardium from Ageing-Dependent Dysfunction: Potential Role of SIRT1. Oxid. Med. Cell. Longev. 2020, 2020, 4650207. [Google Scholar] [CrossRef] [Green Version]
- Brogi, S.; Brindisi, M.; Butini, S.; Kshirsagar, G.U.; Maramai, S.; Chemi, G.; Gemma, S.; Campiani, G.; Novellino, E.; Fiorenzani, P.; et al. (S)-2-Amino-3-(5-methyl-3-hydroxyisoxazol-4-yl)propanoic Acid (AMPA) and Kainate Receptor Ligands: Further Exploration of Bioisosteric Replacements and Structural and Biological Investigation. J. Med. Chem. 2018, 61, 2124–2130. [Google Scholar] [CrossRef]
- Frydenvang, K.; Pickering, D.S.; Kshirsagar, G.U.; Chemi, G.; Gemma, S.; Sprogoe, D.; Kaern, A.M.; Brogi, S.; Campiani, G.; Butini, S.; et al. Ionotropic Glutamate Receptor GluA2 in Complex with Bicyclic Pyrimidinedione-Based Compounds: When Small Compound Modifications Have Distinct Effects on Binding Interactions. ACS Chem. Neurosci. 2020, 11, 1791–1800. [Google Scholar] [CrossRef]
- Nickolls, J.; Buck, I.; Garland, M.; Skadron, K. Scalable parallel programming with CUDA. Queue 2008, 6, 40. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Sirous, H.; Chemi, G.; Gemma, S.; Butini, S.; Debyser, Z.; Christ, F.; Saghaie, L.; Brogi, S.; Fassihi, A.; Campiani, G.; et al. Identification of Novel 3-Hydroxy-pyran-4-One Derivatives as Potent HIV-1 Integrase Inhibitors Using in silico Structure-Based Combinatorial Library Design Approach. Front Chem. 2019, 7, 574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brindisi, M.; Ulivieri, C.; Alfano, G.; Gemma, S.; de Asis Balaguer, F.; Khan, T.; Grillo, A.; Chemi, G.; Menchon, G.; Prota, A.E.; et al. Structure-activity relationships, biological evaluation and structural studies of novel pyrrolonaphthoxazepines as antitumor agents. Eur. J. Med. Chem. 2019, 162, 290–320. [Google Scholar] [CrossRef] [PubMed]
- Brogi, S.; Sirous, H.; Calderone, V.; Chemi, G. Amyloid beta fibril disruption by oleuropein aglycone: Long-time molecular dynamics simulation to gain insight into the mechanism of action of this polyphenol from extra virgin olive oil. Food Funct. 2020, 11, 8122–8132. [Google Scholar] [CrossRef]
- Humphreys, D.D.; Friesner, R.A.; Berne, B.J. A Multiple-Time-Step Molecular Dynamics Algorithm for Macromolecules. J. Phys. Chem. 1994, 98, 6885–6892. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [Green Version]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Zhu, K.; Borrelli, K.W.; Greenwood, J.R.; Day, T.; Abel, R.; Farid, R.S.; Harder, E. Docking covalent inhibitors: A parameter free approach to pose prediction and scoring. J. Chem. Inf. Model 2014, 54, 1932–1940. [Google Scholar] [CrossRef]
- Brogi, S.; Fiorillo, A.; Chemi, G.; Butini, S.; Lalle, M.; Ilari, A.; Gemma, S.; Campiani, G. Structural characterization of Giardia duodenalis thioredoxin reductase (gTrxR) and computational analysis of its interaction with NBDHEX. Eur. J. Med. Chem. 2017, 135, 479–490. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Docking Score (kcal/mol) | ΔGbind (kcal/mol) | QPlogP B | QPlogS C | PAINS D |
|---|---|---|---|---|---|
 4 | −10.779 | −123.15 | 1.82 | −3.68 | No |
 5 | −10.027 | −109.41 | 2.32 | −4.68 | No |
 6 | −11.269 | −114.26 | 3.11 | −4.70 | No |
 7 | −9.540 | −114.04 | 1.72 | −3.54 | No |
 2 | −9.976 | −110.55 | 3.23 | −6.04 | No |
 3, N3 | −10.138 | −108.36 | 3.18 | −7.25 | No |
| Compound | Covalent Docking Score (kcal/mol) | Covalent Docking ΔGbind (kcal/mol) | FEP/MD ΔΔGbind (kcal/mol) |
|---|---|---|---|
| 4 | −10.834 | −128.29 | −0.18 ± 0.11 |
| 5 | −10.232 | −119.17 | −0.45 ± 0.21 |
| 6 | −11.681 | −116.49 | −0.73 ± 0.32 |
| 7 | −9.828 | −115.96 | 0.12 ± 0.09 |
| 2 | −10.174 | −113.87 | -- |
| 3,N3 | −10.043 | −114.74 | −0.13 ± 0.12 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brogi, S.; Rossi, S.; Ibba, R.; Butini, S.; Calderone, V.; Campiani, G.; Gemma, S. In Silico Analysis of Peptide-Based Derivatives Containing Bifunctional Warheads Engaging Prime and Non-Prime Subsites to Covalent Binding SARS-CoV-2 Main Protease (Mpro). Computation 2022, 10, 69. https://0-doi-org.brum.beds.ac.uk/10.3390/computation10050069
Brogi S, Rossi S, Ibba R, Butini S, Calderone V, Campiani G, Gemma S. In Silico Analysis of Peptide-Based Derivatives Containing Bifunctional Warheads Engaging Prime and Non-Prime Subsites to Covalent Binding SARS-CoV-2 Main Protease (Mpro). Computation. 2022; 10(5):69. https://0-doi-org.brum.beds.ac.uk/10.3390/computation10050069
Chicago/Turabian StyleBrogi, Simone, Sara Rossi, Roberta Ibba, Stefania Butini, Vincenzo Calderone, Giuseppe Campiani, and Sandra Gemma. 2022. "In Silico Analysis of Peptide-Based Derivatives Containing Bifunctional Warheads Engaging Prime and Non-Prime Subsites to Covalent Binding SARS-CoV-2 Main Protease (Mpro)" Computation 10, no. 5: 69. https://0-doi-org.brum.beds.ac.uk/10.3390/computation10050069