
Regression Machine Learning Models Used to Predict DFT-Computed NMR Parameters of Zeolites

 , , and
, , and
Abstract
:1. Introduction
2. Methods and Computational Details
2.1. Kernel-Ridge Regression
2.2. Gradient Boosting Regression
2.3. SOAP Descriptors
2.4. DFT Computational Details
3. Results and Discussion
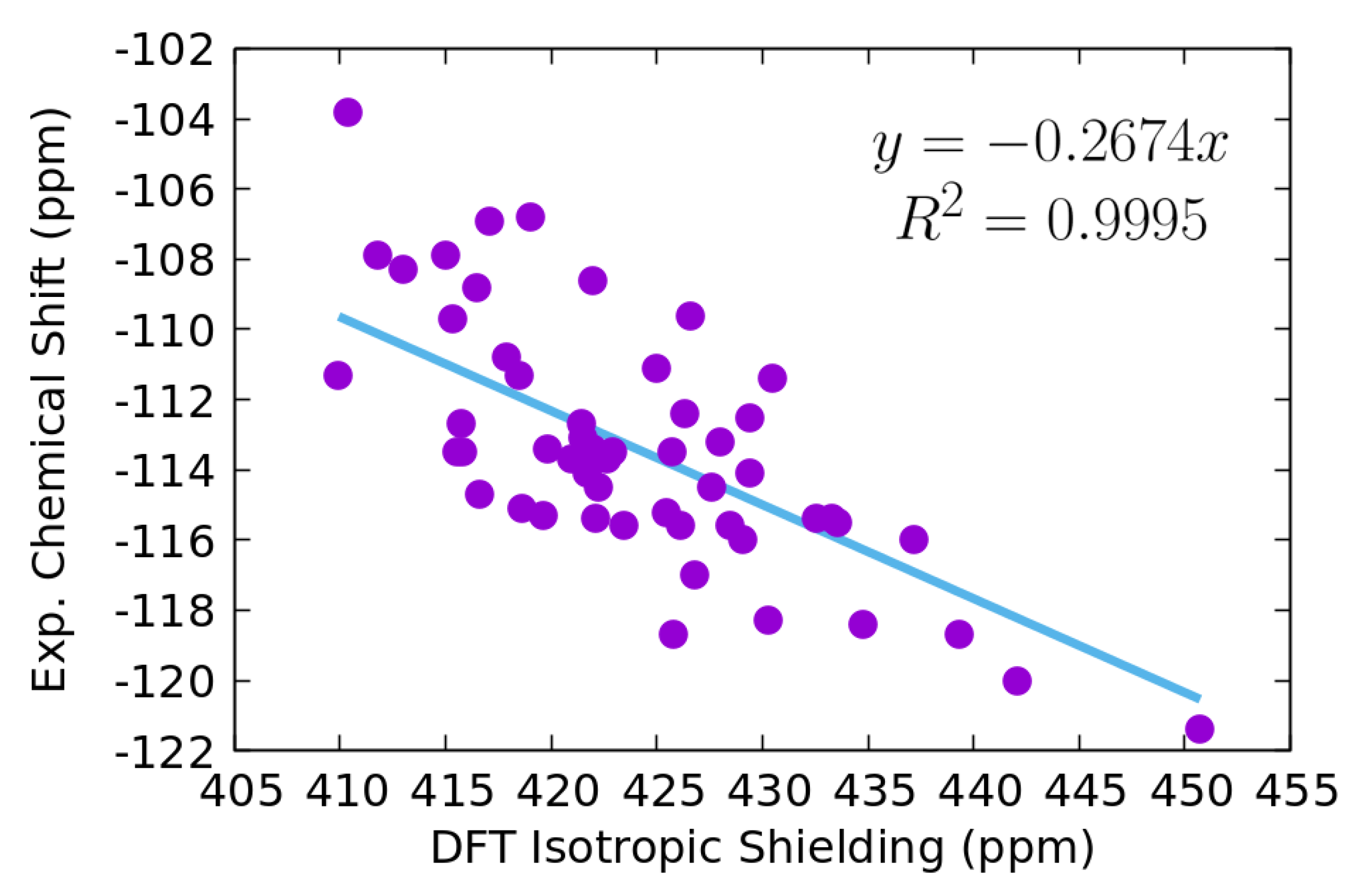
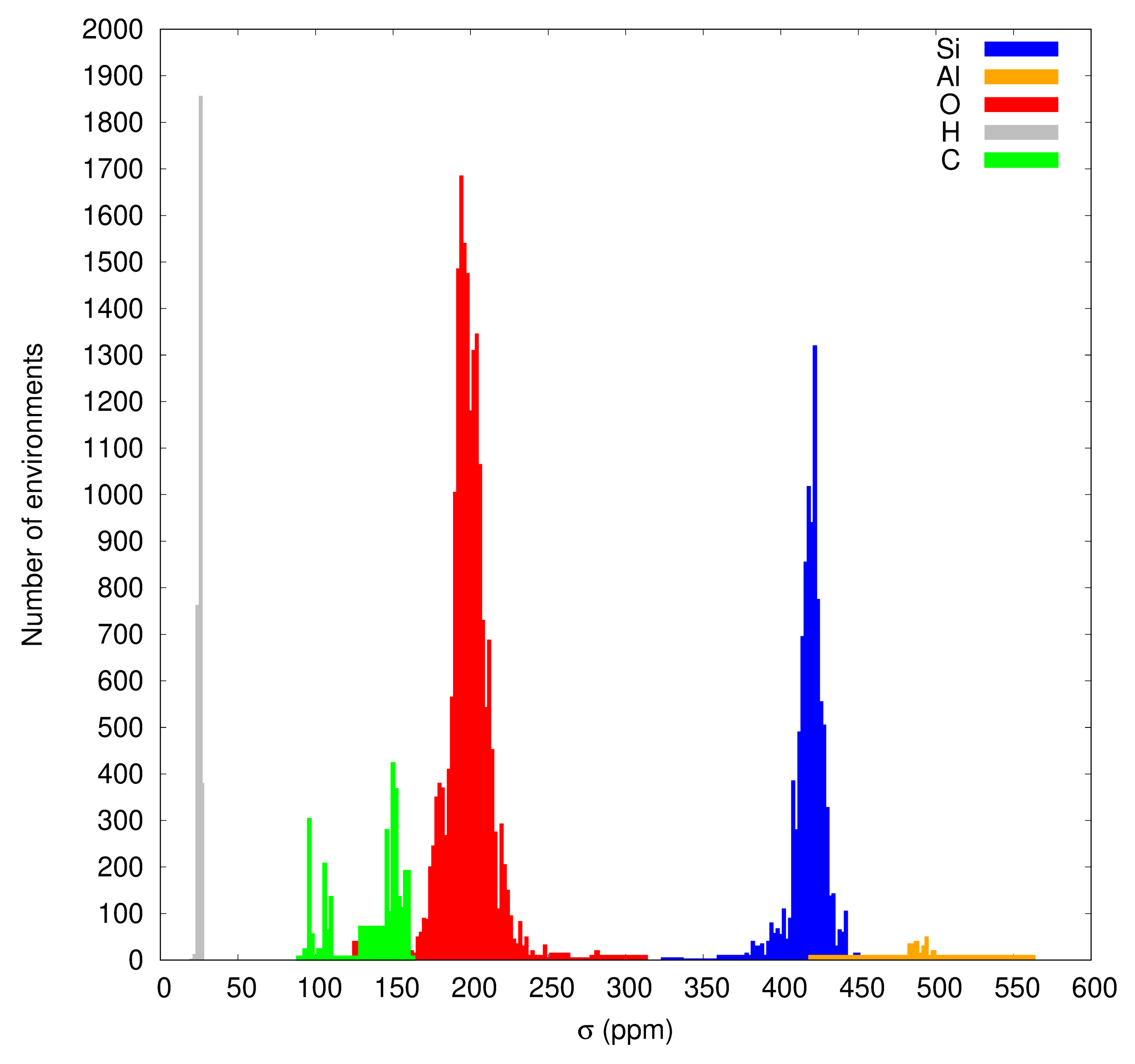
3.1. Data Set and DFT Isotropic Shielding
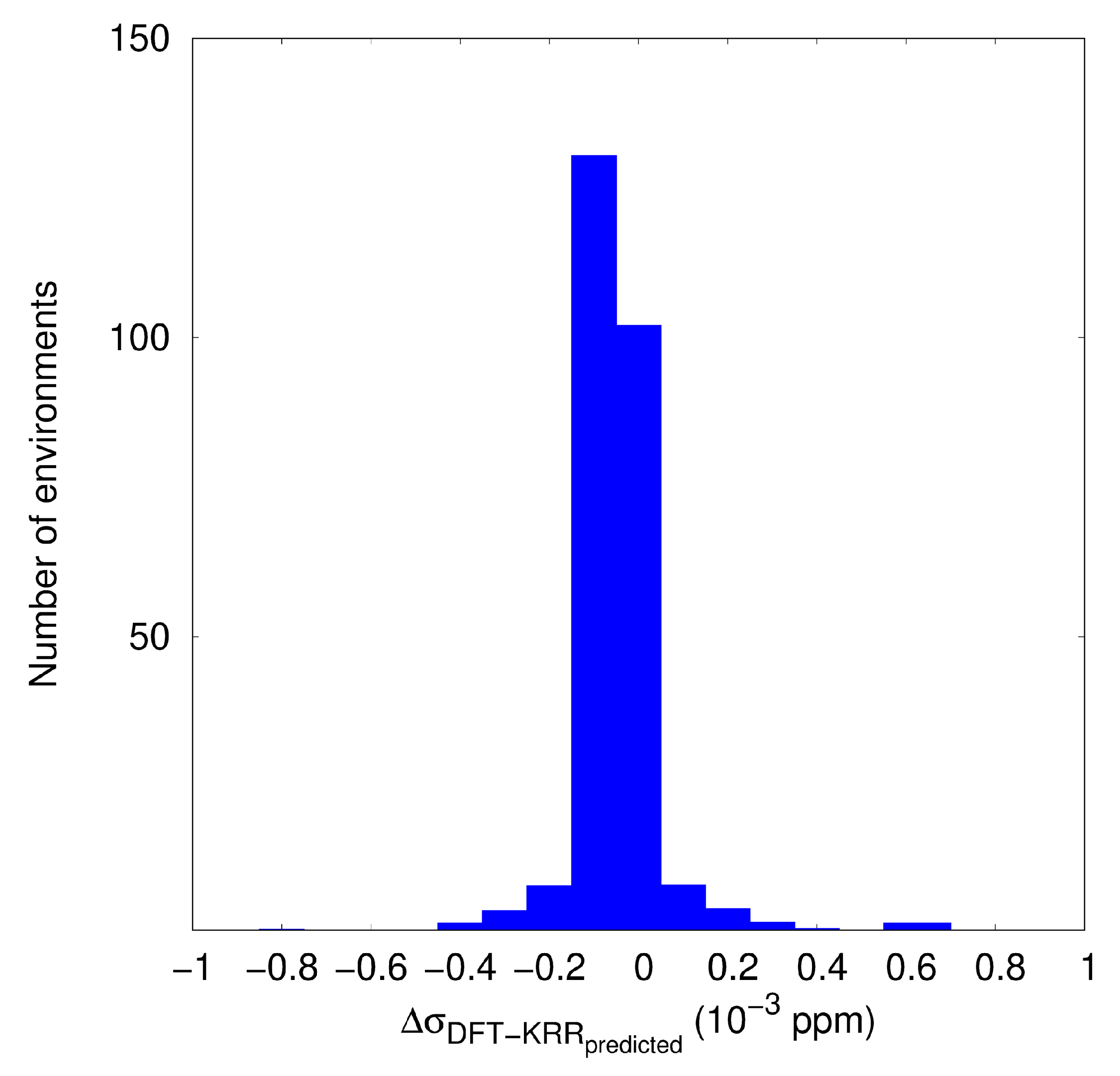
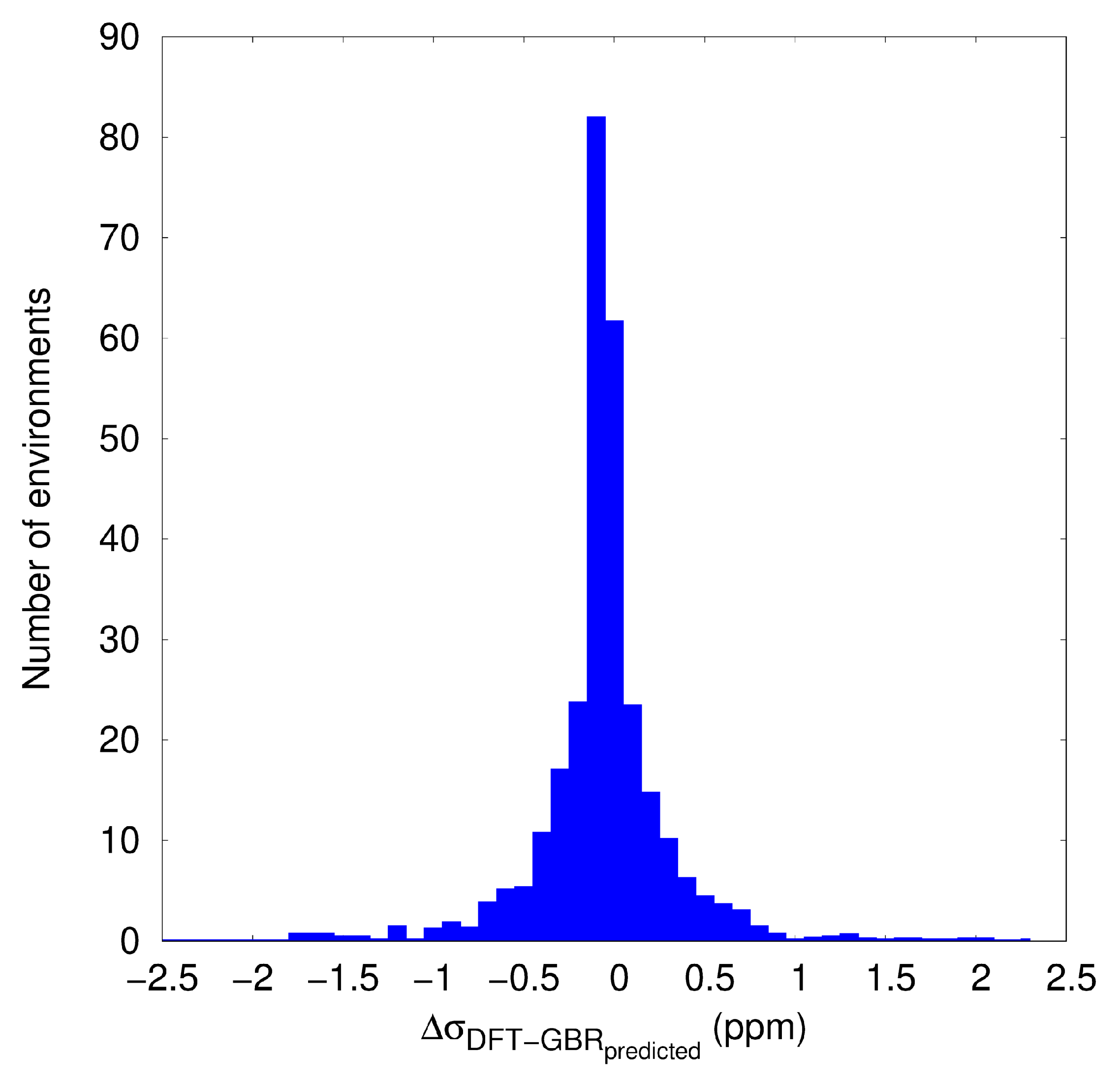
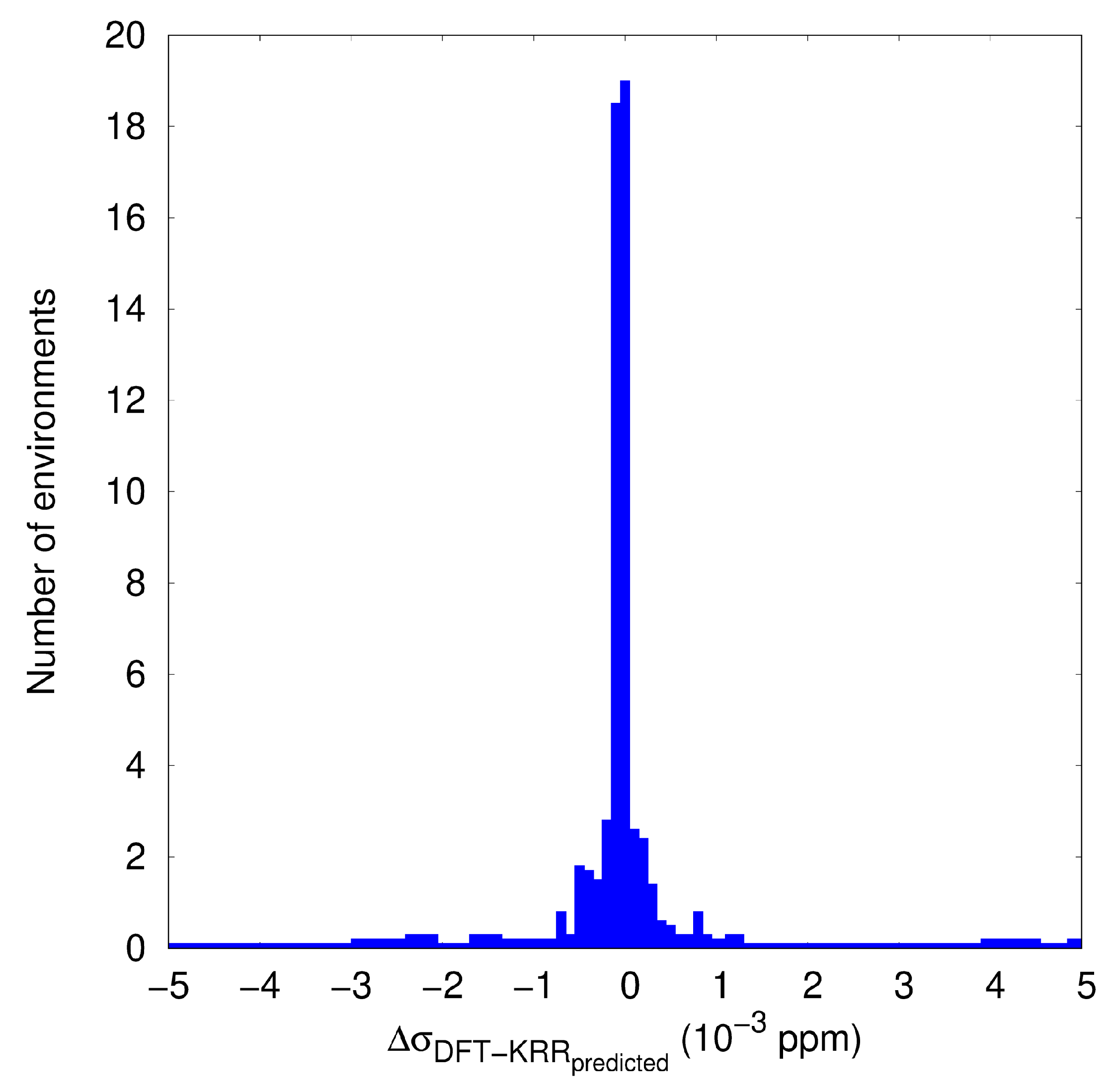
3.2. KRR and GBR Models to Predict NMR Isotropic Shielding
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cuny, J.; Xie, Y.; Pickard, C.J.; Hassanali, A.A. Ab Initio Quality NMR Parameters in Solid-State Materials Using a High-Dimensional Neural-Network Representation. J. Chem. Theory Comput. 2016, 12, 765–773. [Google Scholar] [CrossRef] [Green Version]
- Chaker, Z.; Salanne, M.; Delaye, J.M.; Charpentier, T. NMR shifts in aluminosilicate glasses via machine learning. Phys. Chem. Chem. Phys. 2019, 21, 21709–21725. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Li, J.; Bennett, K.C.; Ganoe, B.; Stauch, T.; Head-Gordon, M.; Hexemer, A.; Ushizima, D.; Head-Gordon, T. Multiresolution 3D-DenseNet for Chemical Shift Prediction in NMR Crystallography. J. Phys. Chem. Lett. 2019, 10, 4558–4565. [Google Scholar] [CrossRef] [PubMed]
- Paruzzo, F.M.; Hofstetter, A.; Musil, F.; De, S.; Ceriotti, M.; Emsley, L. Chemical shifts in molecular solids by machine learning. Nat. Commun. 2018, 9, 4501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rupp, M.; Ramakrishnan, R.; von Lilienfeld, O.A. Machine Learning for Quantum Mechanical Properties of Atoms in Molecules. J. Phys. Chem. Lett. 2015, 6, 3309–3313. [Google Scholar] [CrossRef] [Green Version]
- Gerrard, W.; Bratholm, L.A.; Packer, M.J.; Mulholland, A.J.; Glowacki, D.R.; Butts, C.P. IMPRESSION—Prediction of NMR parameters for 3-dimensional chemical structures using machine learning with near quantum chemical accuracy. Chem. Sci. 2020, 11, 508–515. [Google Scholar] [CrossRef] [Green Version]
- Unzueta, P.A.; Greenwell, C.S.; Beran, G.J.O. Predicting Density Functional Theory-Quality Nuclear Magnetic Resonance Chemical Shifts via Δ-Machine Learning. J. Chem. Theory Comput. 2021, 17, 826–840. [Google Scholar] [CrossRef]
- Cordova, M.; Balodis, M.; Hofstetter, A.; Paruzzo, F.; Nilsson Lill, S.O.; Eriksson, E.S.E.; Berruyer, P.; Simões de Almeida, B.; Quayle, M.J.; Norberg, S.T.; et al. Structure determination of an amorphous drug through large-scale NMR predictions. Nat. Commun. 2021, 12, 2964. [Google Scholar] [CrossRef]
- Aguilera-Segura, S.M.; Dragún, D.; Gaumard, R.; Di Renzo, F.; Malkin Ondík, I.; Mineva, T. Thermal fluctuations and conformational effects on NMR parameters in β-O-4 lignin dimer from QM/MM and machine learning approaches. Phys. Chem. Chem. Phys. 2022, 24, 8820–8831. [Google Scholar] [CrossRef]
- Charpentier, T. The PAW/GIPAW approach for computing NMR parameters: A new dimension added to NMR study of solids. Solid State Nucl. Magn. Reson. 2011, 40, 1–20. [Google Scholar] [CrossRef]
- Dib, E.; Mineva, T.; Alonso, B. Chapter Three—Recent Advances in 14N Solid-State NMR Annu. Rep. NMR Spectrosc. 2016, 87, 175–235. [Google Scholar] [CrossRef]
- Jonas, E.; Kuhn, S.; Schlörer, N. Prediction of chemical shift in NMR: A review. Magn. Reson. Chem. 2021, in press. [CrossRef] [PubMed]
- Arun, K.; Langmead, C.J. Structure based chemical shift prediction usgin random forests non-linear regression. In Proceedings of the 4th Asia-Pacific Bioinformatics Conference, Taipei, Taiwan, 13–16 February 2006; Series on Advances in Bioinformatics and Computational Biology. World Scientific Publishing Co.: Singapore, 2005; Volume 3, pp. 317–326. [Google Scholar] [CrossRef] [Green Version]
- Han, B.; Liu, Y.; Ginzinger, S.W.; Wishart, D.S. SHIFTX2: Significantly improved protein chemical shift prediction. J. Biomol. NMR 2011, 50, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, P.; Zhang, J.; Peng, Q.; Zhang, J.; Glezakou, V.A. General Protocol for the Accurate Prediction of Molecular 13C/1H NMR Chemical Shifts via Machine Learning Augmented DFT. J. Chem. Inf. Model. 2020, 60, 3746–3754. [Google Scholar] [CrossRef]
- Gao, P.; Zhang, J.; Sun, Y.; Yu, J. Toward Accurate Predictions of Atomic Properties via Quantum Mechanics Descriptors Augmented Graph Convolutional Neural Network: Application of This Novel Approach in NMR Chemical Shifts Predictions. J. Phys. Chem. Lett. 2020, 11, 9812–9818. [Google Scholar] [CrossRef]
- Haghighatlari, M.; Li, J.; Heidar-Zadeh, F.; Liu, Y.; Guan, X.; Head-Gordon, T. Learning to Make Chemical Predictions: The Interplay of Feature Representation, Data, and Machine Learning Methods. Chem 2020, 6, 1527–1542. [Google Scholar] [CrossRef]
- Li, J.; Bennett, K.C.; Liu, Y.; Martin, M.V.; Head-Gordon, T. Accurate prediction of chemical shifts for aqueous protein structure on “Real World” data. Chem. Sci. 2020, 11, 3180–3191. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.; Chakraborty, S.; Ramakrishnan, R. Revving up 13C NMR shielding predictions across chemical space: Benchmarks for atoms-in-molecules kernel machine learning with new data for 134 kilo molecules. Mach. Learn. Sci. Technol. 2021, 2, 035010. [Google Scholar] [CrossRef]
- Gerrard, W.; Yiu, C.; Butts, C.P. Prediction of 15N chemical shifts by machine learning. Magn. Reson. Chem. 2021, in press. [CrossRef]
- Beygelzimer, A.; Hazan, E.; Kale, S.; Luo, H. Online gradient boosting. In Proceedings of the Advances in Neural Information Processing Systems 28: Annual Conference on Neural Information Processing Systems 2015, Montreal, QC, Canada, 7–12 December 2015; pp. 2458–2466. [Google Scholar]
- Biau, G.; Cadre, B.; Rouvière, L. Accelerated gradient boosting. Mach. Learn. 2019, 108, 971–992. [Google Scholar] [CrossRef] [Green Version]
- Guan, Y.; Shree Sowndarya, S.V.; Gallegos, L.C.; John, P.C.S.; Paton, R.S. Real-time prediction of 1H and 13C chemical shifts with DFT accuracy using a 3D graph neural network. Chem. Sci. 2021, 12, 12012–12026. [Google Scholar] [CrossRef] [PubMed]
- Jonas, E.; Kuhn, S. Rapid prediction of NMR spectral properties with quantified uncertainty. J. Cheminform. 2019, 11, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, K.; Xu, X.; Kikuchi, J. Improved Prediction of Carbonless NMR Spectra by the Machine Learning of Theoretical and Fragment Descriptors for Environmental Mixture Analysis. Anal. Chem. 2021, 93, 6901–6906. [Google Scholar] [CrossRef] [PubMed]
- Baerlocher, C.; McCusker, L. Database of Zeolite Structures. Available online: http://www.iza-structure.org/databases/ (accessed on 28 March 2022).
- Hölderich, W.; Hesse, M.; Näumann, F. Zeolites: Catalysts for Organic Syntheses. Angew. Chem. Int. Ed. Engl. 1988, 27, 226–246. [Google Scholar] [CrossRef]
- Cejka, J.; van Bekkum, H.; Corma, A.; Schueth, F. Introduction to Zeolite Molecular Sieves. In Introduction to Zeolite Molecular Sieves, John Wiley and Sons: Hoboken, NJ, USA, 2007, 3rd ed.; John Wiley and Sons: Hoboken, NJ, USA, 2007; p. 1094. [Google Scholar]
- Evans, J.D.; Coudert, F.X. Predicting the Mechanical Properties of Zeolite Frameworks by Machine Learning. Chem. Mater. 2017, 29, 7833–7839. [Google Scholar] [CrossRef]
- Gu, Y.; Liu, Z.; Yu, C.; Gu, X.; Xu, L.; Gao, Y.; Ma, J. Zeolite Adsorption Isotherms Predicted by Pore Channel and Local Environmental Descriptors: Feature Learning on DFT Binding Strength. J. Phys. Chem. C 2020, 124, 9314–9328. [Google Scholar] [CrossRef]
- Helfrecht, B.A.; Semino, R.; Pireddu, G.; Auerbach, S.M.; Ceriotti, M. A new kind of atlas of zeolite building blocks. J. Chem. Phys. 2019, 151, 154112. [Google Scholar] [CrossRef] [Green Version]
- Kwak, S.J.; Kim, H.S.; Park, N.; Park, M.J.; Lee, W.B. Recent progress on Al distribution over zeolite frameworks: Linking theories and experiments. Korean J. Chem. Eng. 2021, 38, 1117–1128. [Google Scholar] [CrossRef]
- Welling, M. Kernel ridge Regression. Available online: https://web2.qatar.cmu.edu/~gdicaro/10315-Fall19/additional/welling-notes-on-kernel-ridge.pdf (accessed on 28 March 2022).
- An, S.; Liu, W.; Venkatesh, S. Fast cross-validation algorithms for least squares support vector machine and kernel ridge regression. Pattern Recognit. 2007, 40, 2154–2162. [Google Scholar] [CrossRef]
- ML-CSC-tutorial. Available online: https://github.com/fullmetalfelix/ML-CSC-tutorial (accessed on 24 March 2022).
- Freund, Y.; Schapire, R.E. A Decision-Theoretic Generalization of On-Line Learning and an Application to Boosting. J. Comput. Syst. Sci. 1997, 55, 119–139. [Google Scholar] [CrossRef] [Green Version]
- Freund, Y.; Schapire, R.E. A Short Introduction to Boosting. In Proceedings of the Sixteenth International Joint Conference on Artificial Intelligence (IJCAI ’99), Stockholm, Sweden, 31 July–6 August 1999; Morgan Kaufmann Publishers Inc.: San Francisco, CA, USA, 1999; pp. 1401–1406. [Google Scholar]
- Natekin, A.; Knoll, A. Gradient boosting machines, a tutorial. Front. Neurorobot. 2013, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Z.; Lin, D.; Lau, T.; Wu, M. Gradient Boosting Machine: A Survey. arXiv 2019, arXiv:1908.06951. [Google Scholar] [CrossRef]
- Hastie, T.; Tibshirani, R.; Friedman, J. Boosting and Additive Trees. In The Elements of Statistical Learning: Data Mining, Inference, and Prediction; Springer: New York, NY, USA, 2009; pp. 337–387. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Himanen, L.; Jäger, M.O.; Morooka, E.V.; Federici Canova, F.; Ranawat, Y.S.; Gao, D.Z.; Rinke, P.; Foster, A.S. DScribe: Library of descriptors for machine learning in materials science. Comput. Phys. Commun. 2020, 247, 106949. [Google Scholar] [CrossRef]
- Larsen, A.H.; Mortensen, J.J.; Blomqvist, J.; Castelli, I.E.; Christensen, R.; Dułak, M.; Friis, J.; Groves, M.N.; Hammer, B.; Hargus, C.; et al. The atomic simulation environment—A Python library for working with atoms. J. Phys. Condens. Matter 2017, 29, 273002. [Google Scholar] [CrossRef] [Green Version]
- Bahn, S.R.; Jacobsen, K.W. An object-oriented scripting interface to a legacy electronic structure code. Comput. Sci. Eng. 2002, 4, 56–66. [Google Scholar] [CrossRef] [Green Version]
- Dovesi, R.; Erba, A.; Orlando, R.; Zicovich-Wilson, C.M.; Civalleri, B.; Maschio, L.; Rérat, M.; Casassa, S.; Baima, J.; Salustro, S.; et al. Quantum-mechanical condensed matter simulations with CRYSTAL. WIRES Comput. Mol. Sci. 2018, 8, e1360. [Google Scholar] [CrossRef]
- Gatti, C.; Saunders, V.R.; Roetti, C. Crystal field effects on thetopological properties of the electron density in molecular crystals: The case of urea. J. Chem. Phys. 1994, 101, 10686–10696. [Google Scholar] [CrossRef]
- Catti, M.; Valerio, G.; Dovesi, R.; Causà, M. Quantum-mechanical calculation of the solid-state equilibrium MgO + α-Al2O3 ⇄ MgAl2O4 (spinel) versus pressure. Phys. Rev. B 1994, 49, 14179–14187. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Dib, E.; Mineva, T.; Gaveau, P.; Véron, E.; Sarou-Kanian, V.; Fayon, F.; Alonso, B. Probing Disorder in Al-ZSM-5 Zeolites by 14N NMR Spectroscopy. J. Phys. Chem. C 2017, 121, 15831–15841. [Google Scholar] [CrossRef]
- Mineva, T.; Dib, E.; Gaje, A.; Petitjean, H.; Bantignies, J.L.; Alonso, B. Zeolite Structure Direction: Identification, Strength and Involvement of Weak CHO Hydrogen Bonds. Chem. Phys. Chem. 2020, 21, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Al-Nahari, S.; Ata, K.; Mineva, T.; Alonso, B. Ubiquitous Presence of Intermolecular CHO Hydrogen Bonds in As-synthesized Host-Guest Zeolite Materials. ChemistrySelect 2021, 6, 9728–9734. [Google Scholar] [CrossRef]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef]
- Giannozzi, P.; Andreussi, O.; Brumme, T.; Bunau, O.; Nardelli, M.B.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Cococcioni, M.; et al. Advanced capabilities for materials modelling with Quantum ESPRESSO. J. Phys. Condens. Matter 2017, 29, 465901. [Google Scholar] [CrossRef] [Green Version]
- Quantum Espresso. Available online: https://www.quantum-espresso.org (accessed on 28 March 2022).
- Pickard, C.J.; Mauri, F. All-electron magnetic response with pseudopotentials: NMR chemical shifts. Phys. Rev. B 2001, 63, 245101. [Google Scholar] [CrossRef] [Green Version]
- Yates, J.R.; Pickard, C.J.; Mauri, F. Calculation of NMR chemical shifts for extended systems using ultrasoft pseudopotentials. Phys. Rev. B 2007, 76, 024401. [Google Scholar] [CrossRef]
- Quantum Espresso Pseudopotentials. Available online: http://www.quantum-espresso.org/pseudopotentials (accessed on 28 March 2022).
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Chao, K.J.; Lin, J.C.; Wang, Y.; Lee, G. Single crystal structure refinement of TPA ZSM-5 zeolite. Zeolites 1986, 6, 35–38. [Google Scholar] [CrossRef]
- Yokomori, Y.; Idaka, S. The structure of TPA-ZSM-5 with Si/Al = 23. Microporous Mesoporous Mater. 1999, 28, 405–413. [Google Scholar] [CrossRef]
- Dib, E.; Mineva, T.; Veron, E.; Sarou-Kanian, V.; Fayon, F.; Alonso, B. ZSM-5 Zeolite: Complete Al Bond Connectivity and Implications on Structure Formation from Solid-State NMR and Quantum Chemistry Calculations. J. Phys. Chem. Lett. 2018, 9, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Fabbiani, M.; Al-Nahari, S.; Piveteau, L.; Dib, E.; Veremeienko, V.; Gaje, A.; Dumitrescu, D.G.; Gaveau, P.; Mineva, T.; Massiot, D.; et al. Host–Guest Silicalite-1 Zeolites: Correlated Disorder and Phase Transition Inhibition by a Small Guest Modification. Chem. Mater. 2022, 34, 366–387. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Machine Learning Models | ||||||
|---|---|---|---|---|---|---|
| Parameters | KRR (All) | GBR (All) | AVG (All) | KRR (Si) | GBR (Si) | AVG (Si) |
| Training time (s) | 3796.4 | 49.0 | - | 136.7 | 12.2 | - |
| Prediction time (s) | 1900.8 | 0.6 | - | 74.2 | 0.02 | - |
| MAE (ppm) | 0.023 | 0.226 | 0.116 | 0.037 | 0.057 | 0.046 |
| STD AE (ppm) | 0.524 | 0.538 | 0.236 | 0.490 | 0.054 | 0.246 |
| MSE (ppm) | 0.275 | 0.341 | 0.069 | 0.241 | 0.006 | 0.062 |
| STD SE (ppm) | 12.669 | 8.158 | 1.285 | 5.304 | 0.011 | 1.341 |
| RMSE (ppm) | 0.524 | 0.584 | 0.262 | 0.491 | 0.008 | 0.250 |
| R | 0.999 | 0.999 | - | 0.999 | 0.997 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaumard, R.; Dragún, D.; Pedroza-Montero, J.N.; Alonso, B.; Guesmi, H.; Malkin Ondík, I.; Mineva, T. Regression Machine Learning Models Used to Predict DFT-Computed NMR Parameters of Zeolites. Computation 2022, 10, 74. https://0-doi-org.brum.beds.ac.uk/10.3390/computation10050074
Gaumard R, Dragún D, Pedroza-Montero JN, Alonso B, Guesmi H, Malkin Ondík I, Mineva T. Regression Machine Learning Models Used to Predict DFT-Computed NMR Parameters of Zeolites. Computation. 2022; 10(5):74. https://0-doi-org.brum.beds.ac.uk/10.3390/computation10050074
Chicago/Turabian StyleGaumard, Robin, Dominik Dragún, Jesús N. Pedroza-Montero, Bruno Alonso, Hazar Guesmi, Irina Malkin Ondík, and Tzonka Mineva. 2022. "Regression Machine Learning Models Used to Predict DFT-Computed NMR Parameters of Zeolites" Computation 10, no. 5: 74. https://0-doi-org.brum.beds.ac.uk/10.3390/computation10050074