Incongruencies in Vaccinia Virus Phylogenetic Trees


Abstract
:1. Introduction
2. Experimental Section
2.1. Retrieval of Genome Sequences and Alignment
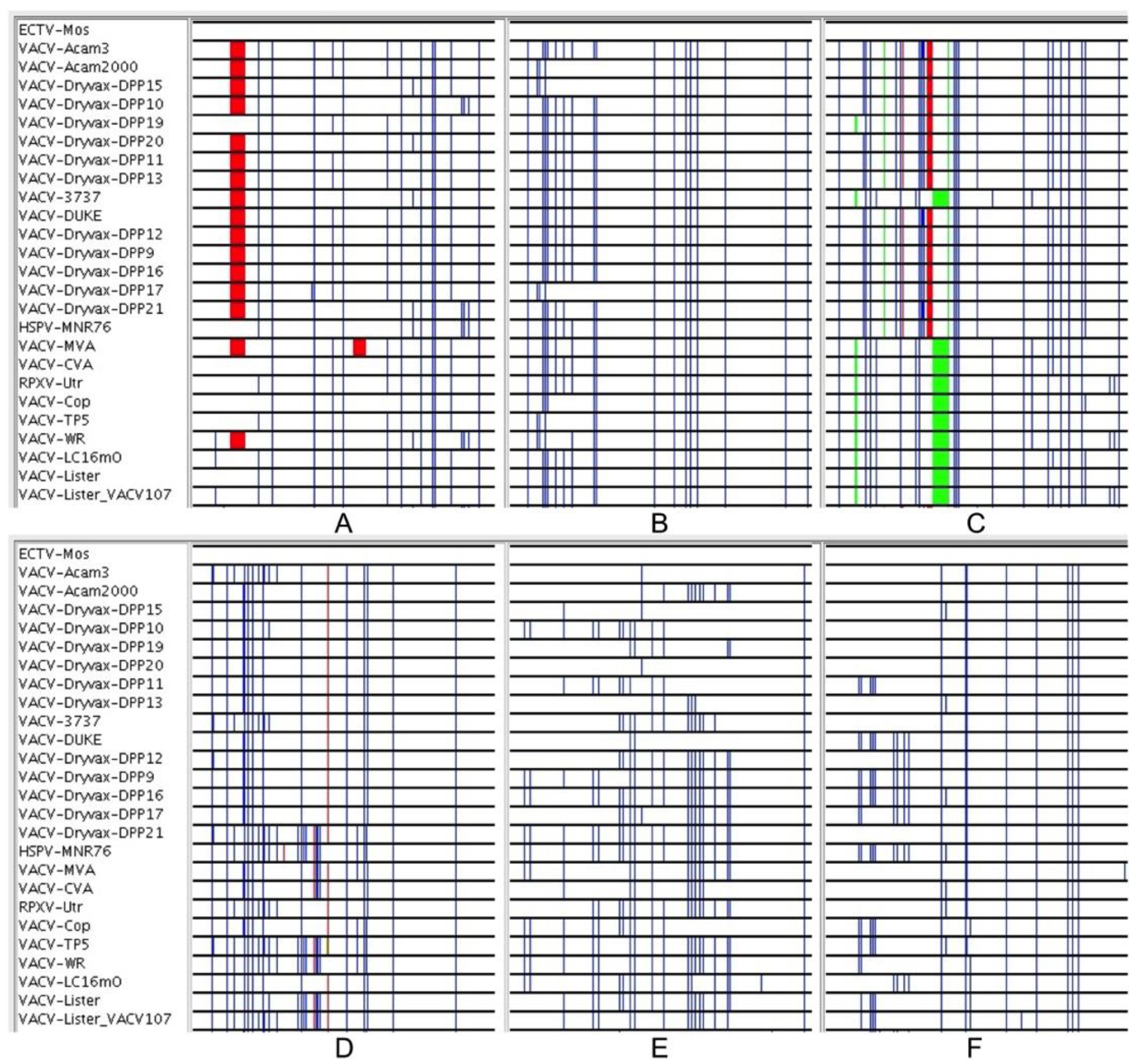
2.2. Visual Examination of the Multiple Sequence Alignment
2.3. Phylogenetic Tree Construction
2.4. Generation of Artificial Multiple Sequence Alignments
2.5. Use of Python Scripts for Further MSA Manipulation and Analysis
3. Results and Discussion
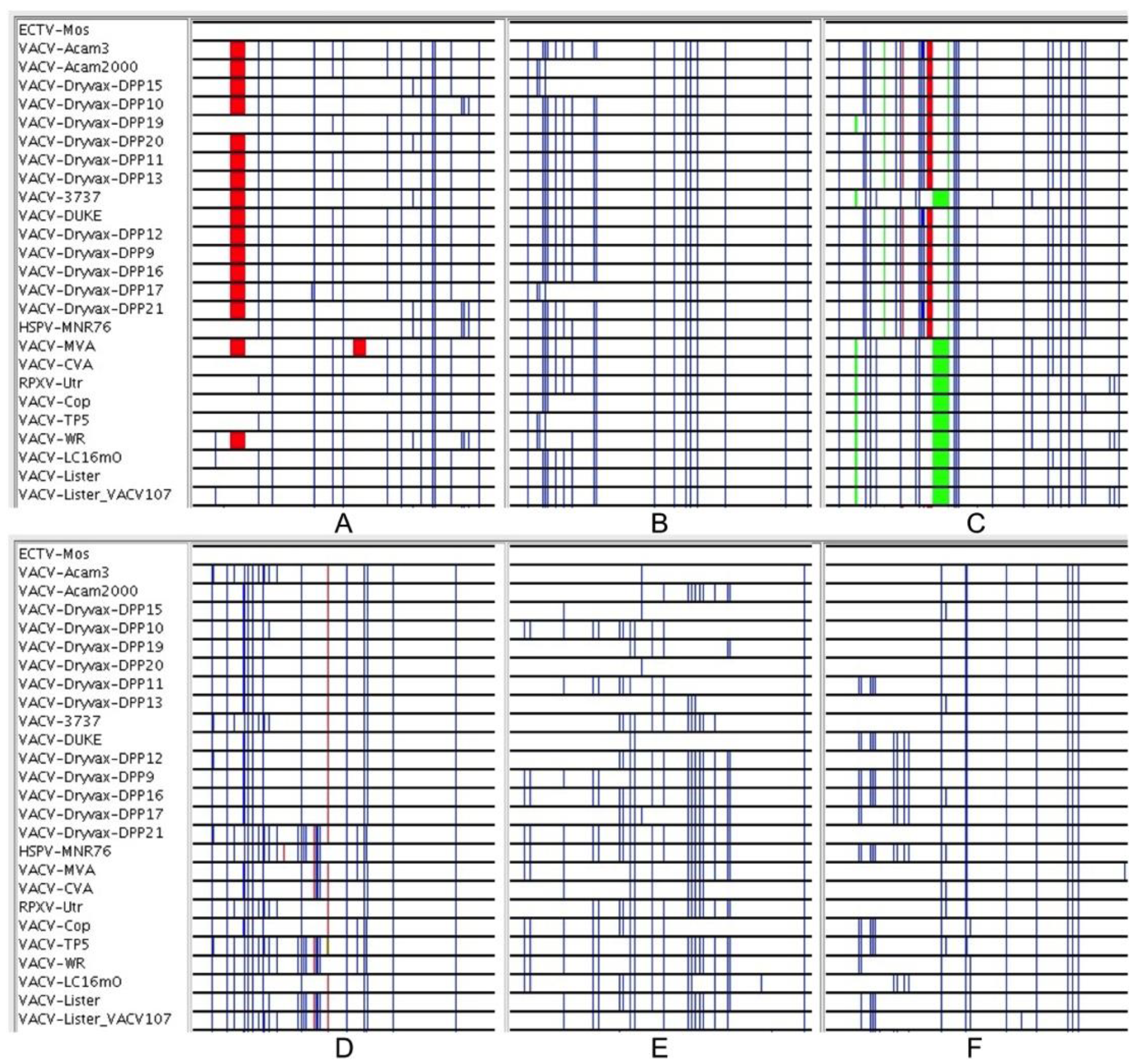
3.1. Development of New Sequence Analysis Tools
3.2. Genomic Comparison of VACV Strains


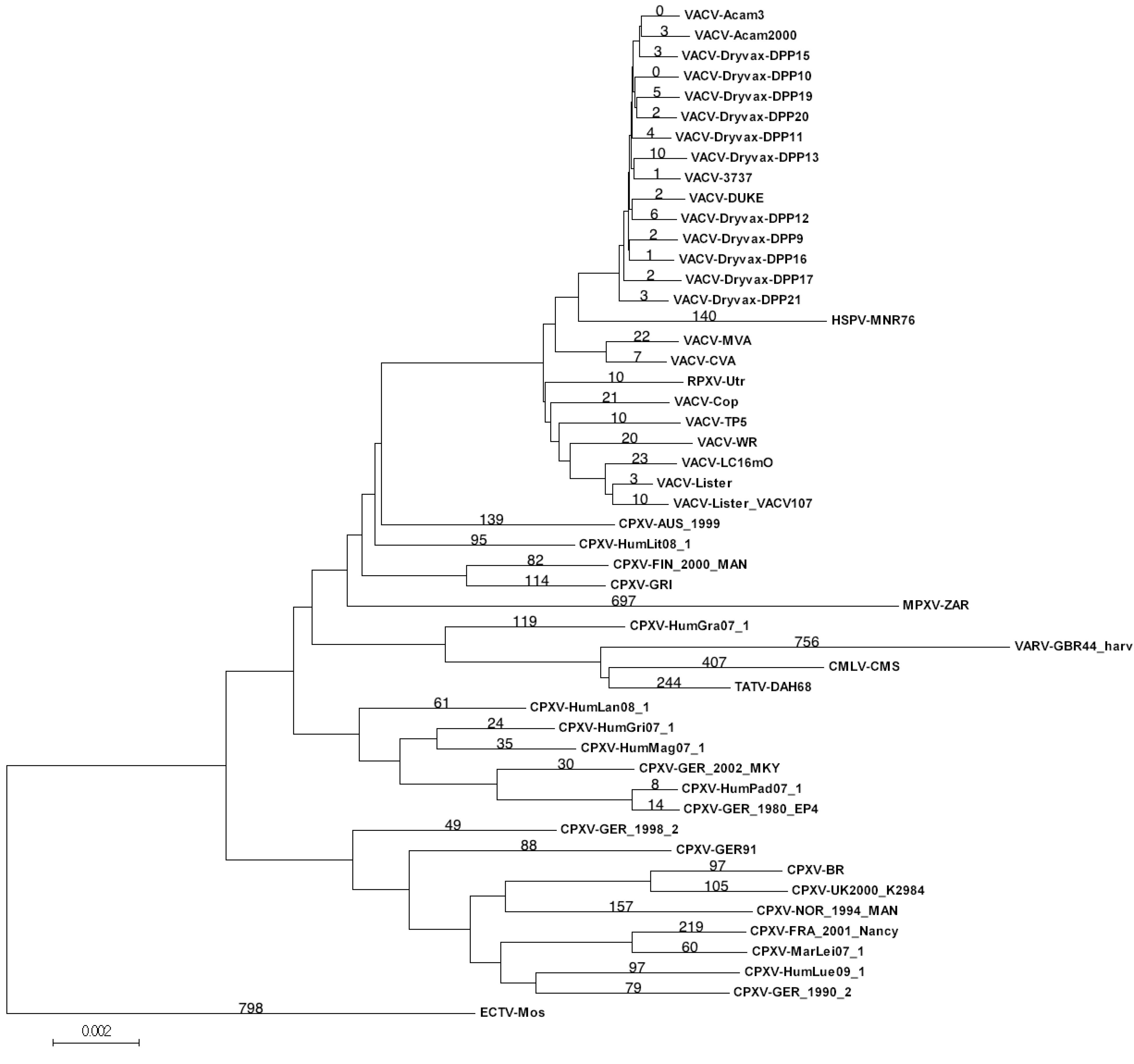{kind=link}
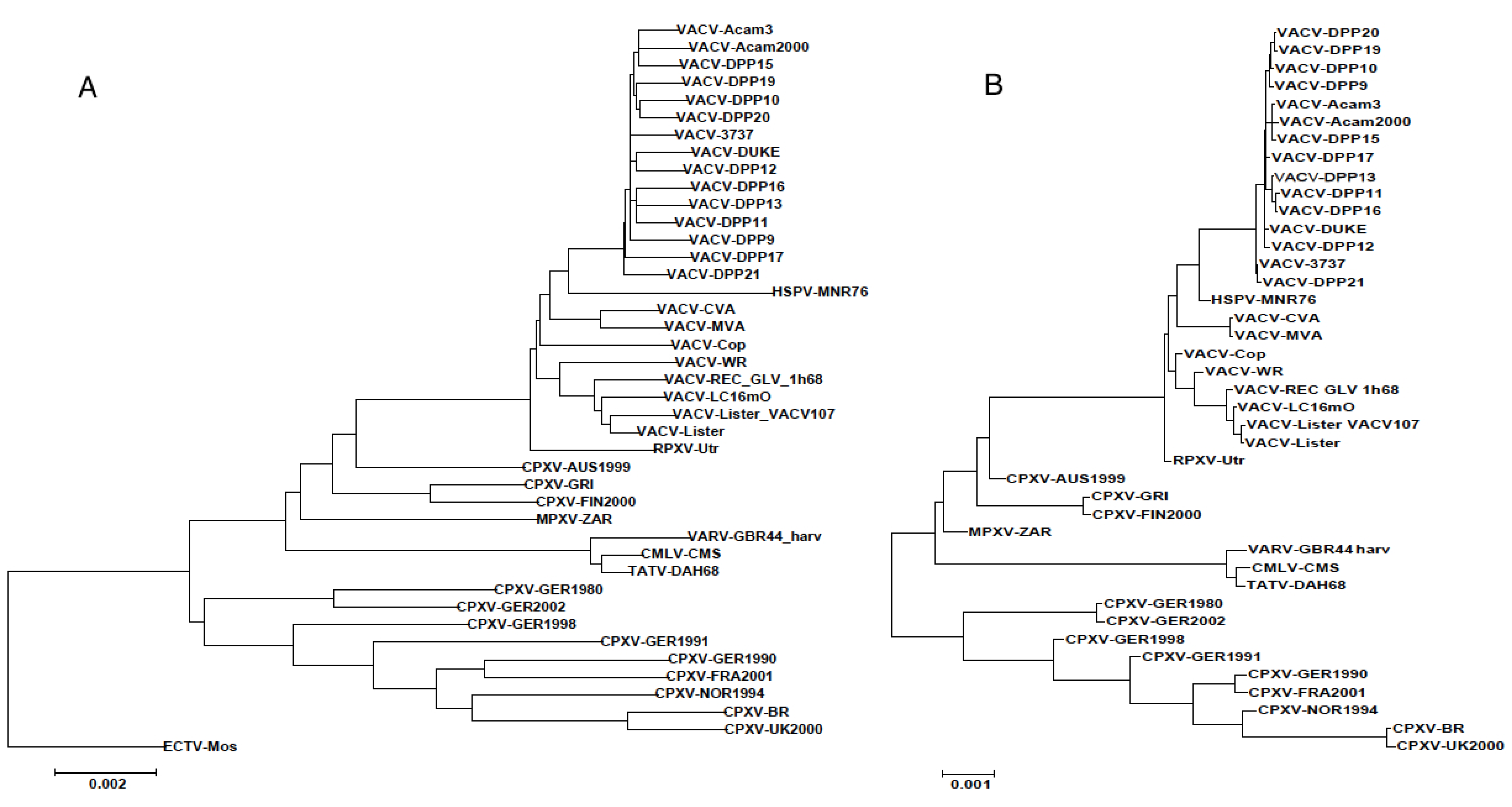{kind=link}
{kind=link}
| Virus: VACV-Dryvax- | Unique positions: Dryvax set | Unique positions: VACV set |
|---|---|---|
| DPP9 | 17 | 2 |
| DPP10 | 16 | 1 |
| DPP11 | 8 | 4 |
| DPP12 | 17 | 6 |
| DPP13 | 29 | 11 |
| DPP15 | 14 | 5 |
| DPP16 | 12 | 1 |
| DPP17 | 23 | 4 |
| DPP19 | 22 | 7 |
| DPP20 | 8 | 2 |
| DPP21 | 27 | 3 |
| Acam3 | 14 | 0 |
| Acam2000 | 34 | 3 |
| 3737 | 29 | 4 |
| DUKE | 25 | 4 |
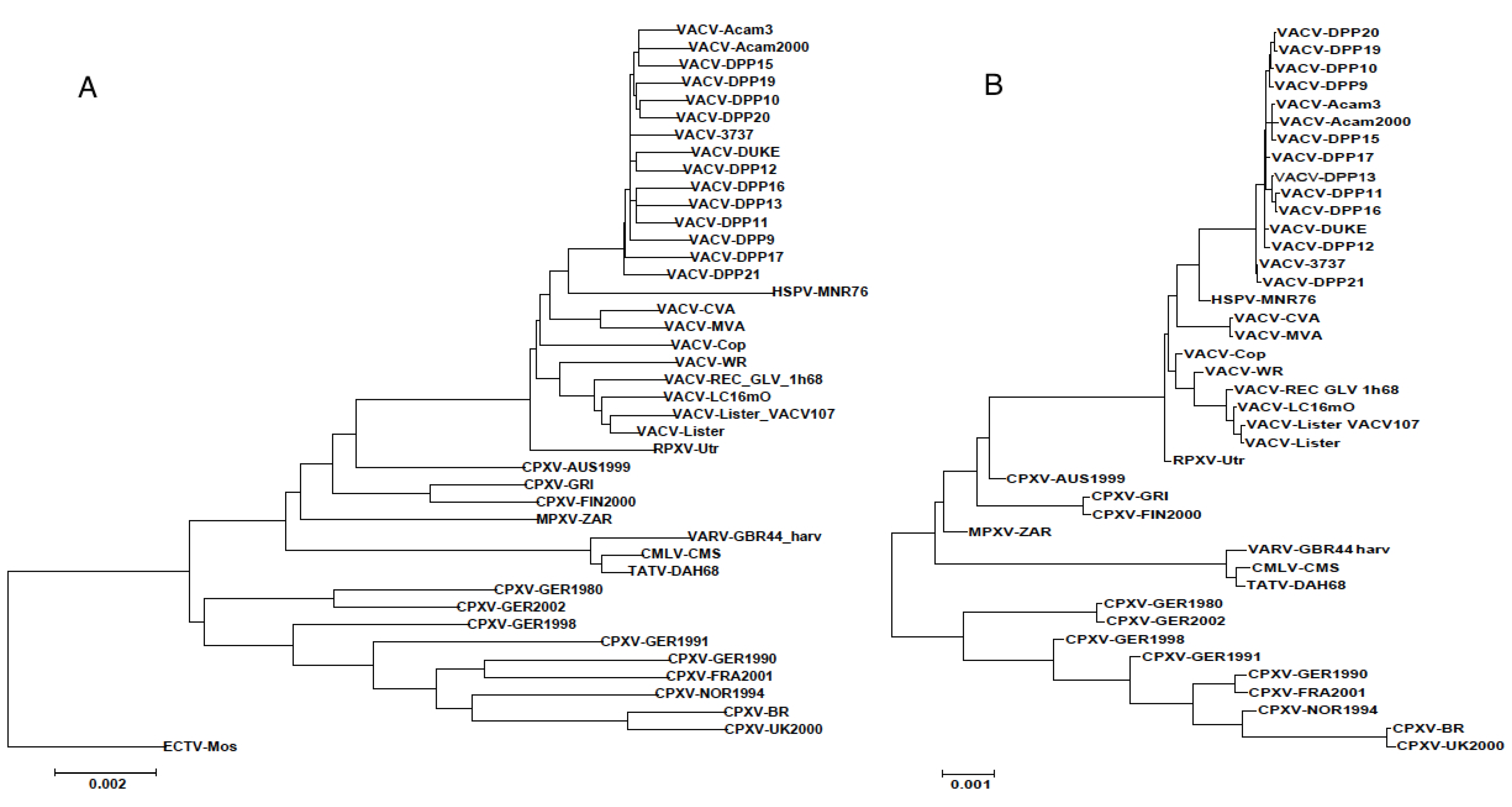
3.3. Sequence Variation in VACV Core Region
| Position | Tolerated Virus Set |
|---|---|
| 5685 | CPXV-AUS_1999, CPXV-HumLit08, CPXV-FIN_2000_MAN, CPXV-GRI, MPXV-ZAR |
| 5759 | CPXV-AUS_1999, CPXV-HumLit08, CPXV-FIN_2000_MAN, CPXV-GRI, MPXV-ZAR |
| 5789 | CPXV-AUS_1999, CPXV-HumLit08, CPXV-FIN_2000_MAN, CPXV-GRI, MPXV-ZAR |
| 5807 | CPXV-AUS_1999, CPXV-HumLit08, CPXV-FIN_2000_MAN, CPXV-GRI, MPXV-ZAR |
| 5820 | CPXV-AUS_1999, CPXV-HumLit08, CPXV-FIN_2000_MAN, CPXV-GRI, MPXV-ZAR |
| 5839 | CPXV-AUS_1999, CPXV-HumLit08, CPXV-FIN_2000_MAN, CPXV-GRI, MPXV-ZAR |
| 13452 | CPXV-HumLit08_1, VARV-GBR44_harv, CMLV-CMS, TATV-DAH68, CPXV-HumPad07_1, CPXV-GER_1980_EP4 |
| 13513 | CPXV-HumLit08_1, VARV-GBR44_harv, CMLV-CMS, TATV-DAH68, CPXV-HumPad07_1, CPXV-GER_1980_EP4 |
| 13514 | CPXV-HumLit08_1, VARV-GBR44_harv, CMLV-CMS, TATV-DAH68, CPXV-HumPad07_1, CPXV-GER_1980_EP4 |
| 13516 | CPXV-HumLit08_1, VARV-GBR44_harv, CMLV-CMS, TATV-DAH68, CPXV-HumPad07_1, CPXV-GER_1980_EP4 |
| 13554 | CPXV-HumLit08_1, VARV-GBR44_harv, CMLV-CMS, TATV-DAH68, CPXV-HumPad07_1, CPXV-GER_1980_EP4 |
| 19369 | CPXV-AUS_1999, CPXV-HumLit08_1, CPXV-FIN_2000_MAN, CPXV-GRI, MPXV-ZAR, CPXV-HumLan08_1 |
| 19378 | CPXV-AUS_1999, CPXV-HumLit08_1, CPXV-FIN_2000_MAN, CPXV-GRI, MPXV-ZAR, CPXV-HumLan08_1 |
| 19380 | CPXV-AUS_1999, CPXV-HumLit08_1, CPXV-FIN_2000_MAN, CPXV-GRI, MPXV-ZAR, CPXV-HumLan08_1 |
| 19387 | CPXV-AUS_1999, CPXV-HumLit08_1, CPXV-FIN_2000_MAN, CPXV-GRI, MPXV-ZAR, CPXV-HumLan08_1 |
| 59334 | CPXV-AUS_1999, CPXV-HumLit08_1, CPXV-FIN_2000_MAN, CPXV-GRI |
| 59343 | CPXV-AUS_1999, CPXV-HumLit08_1, CPXV-FIN_2000_MAN, CPXV-GRI |
| 59444 | CPXV-AUS_1999, CPXV-HumLit08_1, CPXV-FIN_2000_MAN, CPXV-GRI |
| Position | Tolerated Virus Set |
|---|---|
| 61423 | VACV-Dryvax-DPP19, HSPV-MNR76, RPXV-Utr, VACV-Cop, VACV-TP5, VACV-WR, CPXV-AUS_1999 |
| 61426 | VACV-Dryvax-DPP19, HSPV-MNR76, RPXV-Utr, VACV-Cop, VACV-TP5, VACV-WR, CPXV-AUS_1999 |
| 61429 | VACV-Dryvax-DPP19, HSPV-MNR76, RPXV-Utr, VACV-Cop, VACV-TP5, VACV-WR, CPXV-AUS_1999 |
| 61435 | VACV-Dryvax-DPP19, HSPV-MNR76, RPXV-Utr, VACV-Cop, VACV-TP5, VACV-WR, CPXV-AUS_1999 |
| 61436 | VACV-Dryvax-DPP19, HSPV-MNR76, RPXV-Utr, VACV-Cop, VACV-TP5, VACV-WR, CPXV-AUS_1999 |
| 61438 | VACV-Dryvax-DPP19, HSPV-MNR76, RPXV-Utr, VACV-Cop, VACV-TP5, VACV-WR, CPXV-AUS_1999 |
| 87456 | VACV-Acam2000, -MVA, -CVA, -Cop, -WR, -TP5, CPXV-HumMag07_1, -HumGri07_1, -HumLan08_1, -FIN_2000_MAN |
| 87459 | VACV-Acam2000, -MVA, -CVA, -Cop, -WR, -TP5, CPXV-HumMag07_1, -HumGri07_1, -HumLan08_1, -FIN_2000_MAN |
| 87463 | VACV-Acam2000, -MVA, -CVA, -Cop, -WR, -TP5, CPXV-HumMag07_1, -HumGri07_1, -HumLan08_1, -FIN_2000_MAN |
| 87465 | VACV-Acam2000, -MVA, -CVA, -Cop, -WR, -TP5, CPXV-HumMag07_1, -HumGri07_1, -HumLan08_1, -FIN_2000_MAN |
| 66854 | VACV-Cop, CPXV-HumLit08_1, CPXV-HumPad07_1, CPXV-BR, CPXV-UK2000_K2984 |
| 66856 | VACV-Cop, CPXV-HumLit08_1, CPXV-HumPad07_1, CPXV-BR, CPXV-UK2000_K2984 |
3.4. Core Region SNPs Shared by the Variola Clade and More Distantly Related Viruses
| CPXV-GER91 | MPXV-ZAR | CPXV-HumLit08_1 | CPXV-HumLue09_1 |
|---|---|---|---|
| 21074 | 52718 | 21693 | 95965 |
| 21092 | 52745 | 21702 | 96055 |
| 21093 | 52747 | 21705 | 96061 |
| 21116 | 52756 | 45848 | - |
| 21125 | 52760 | - | - |
| 21128 | 52766 | - | - |
| 21140 | 52769 | - | - |
| 21141 | 60513 | - | - |
| 21149 | 66305 | - | - |
| 23126 | - | - | - |
| Position | Tolerated Virus Set |
|---|---|
| 5006 | CPXV-HumGri07_1, CPXV-HumMag07_1, CPXV-GER_2002_MKY, CPXV-HumPad07_1, CPXV-GER_1980_EP4 |
| 5011 | CPXV-HumGri07_1, CPXV-HumMag07_1, CPXV-GER_2002_MKY, CPXV-HumPad07_1, CPXV-GER_1980_EP4 |
| 5035 | CPXV-HumGri07_1, CPXV-HumMag07_1, CPXV-GER_2002_MKY, CPXV-HumPad07_1, CPXV-GER_1980_EP4 |
| 5088 | CPXV-HumGri07_1, CPXV-HumMag07_1, CPXV-GER_2002_MKY, CPXV-HumPad07_1, CPXV-GER_1980_EP4 |
| 50919 | CPXV-HumGri07_1, CPXV-HumMag07_1, CPXV-GER_2002_MKY, CPXV-HumPad07_1, CPXV-GER_1980_EP4 |
| 21528 | CPXV-AUS_1999, CPXV-HumLit08_1, CPXV-HumPad07_1, CPXV-GER_1980_EP4, CPXV-MarLei07_1 |
| 21535 | CPXV-AUS_1999, CPXV-HumLit08_1, CPXV-HumPad07_1, CPXV-GER_1980_EP4, CPXV-MarLei07_1 |
| 21539 | CPXV-AUS_1999, CPXV-HumLit08_1, CPXV-HumPad07_1, CPXV-GER_1980_EP4, CPXV-MarLei07_1 |
| 21540 | CPXV-AUS_1999, CPXV-HumLit08_1, CPXV-HumPad07_1, CPXV-GER_1980_EP4, CPXV-MarLei07_1 |
| 21541 | CPXV-AUS_1999, CPXV-HumLit08_1, CPXV-HumPad07_1, CPXV-GER_1980_EP4, CPXV-MarLei07_1 |
| 21543 | CPXV-AUS_1999, CPXV-HumLit08_1, CPXV-HumPad07_1, CPXV-GER_1980_EP4, CPXV-MarLei07_1 |
| 21579 | CPXV-HumLit08_1, CPXV-HumPad07_1, CPXV-GER_1980_EP4, CPXV-MarLei07_1 |
| 21582 | CPXV-HumLit08_1, CPXV-HumPad07_1, CPXV-GER_1980_EP4, CPXV-MarLei07_1 |
| 21585 | CPXV-HumLit08_1, CPXV-HumPad07_1, CPXV-GER_1980_EP4, CPXV-MarLei07_1 |
| 21587 | CPXV-HumLit08_1, CPXV-HumPad07_1, CPXV-GER_1980_EP4, CPXV-MarLei07_1 |
| 21645 | CPXV-HumLit08_1, CPXV-HumPad07_1, CPXV-GER_1980_EP4, CPXV-MarLei07_1 |
| 21648 | CPXV-HumLit08_1, CPXV-HumPad07_1, CPXV-GER_1980_EP4, CPXV-MarLei07_1 |
| 98836 | HSPV-76, VACV-WR, CPXV-AUS_1999, CPXV-HumLit08_1, MPXV-ZAR, CPXV-FIN_2000_MAN, CPXV-GRI, CPXV-GER91 |
| 98839 | HSPV-76, VACV-WR, CPXV-AUS_1999, CPXV-HumLit08_1, MPXV-ZAR, CPXV-FIN_2000_MAN, CPXV-GRI, CPXV-GER91 |
| 98845 | HSPV-76, VACV-WR, CPXV-AUS_1999, CPXV-HumLit08_1, MPXV-ZAR, CPXV-FIN_2000_MAN, CPXV-GRI, CPXV-GER91 |
3.5. SNPs Associated with the Evolution of VACV-CVA and –MVA
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Upton, C.; Slack, S.; Hunter, A.L.; Ehlers, A.; Roper, R.L. Poxvirus orthologous clusters: Toward defining the minimum essential poxvirus genome. J. Virol. 2003, 77, 7590–7600. [Google Scholar] [CrossRef] [PubMed]
- Gubser, C.; Hué, S.; Kellam, P.; Smith, G.L. Poxvirus genomes: A phylogenetic analysis. J. Gen. Virol. 2004, 85, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Baroudy, B.M.; Venkatesan, S.; Moss, B. Incompletely base-paired flip-flop terminal loops link the two DNA strands of the vaccinia virus genome into one uninterrupted polynucleotide chain. Cell 1982, 28, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Parrino, J.; Graham, B.S. Smallpox vaccines: Past, present, and future. J. Allergy Clin. Immunol. 2006, 118, 1320–1326. [Google Scholar] [CrossRef] [PubMed]
- Verardi, P.H.; Titong, A.; Hagen, C.J. A vaccinia virus renaissance: New vaccine and immunotherapeutic uses after smallpox eradication. Hum. Vaccines Immunother. 2012, 8, 961–970. [Google Scholar] [CrossRef]
- Rosenthal, S.R.; Merchlinsky, M.; Kleppinger, C.; Goldenthal, K.L. Developing new smallpox vaccines. Emerg. Infect. Dis. 2001, 7, 920–926. [Google Scholar] [CrossRef] [PubMed]
- Carroll, D.S.; Emerson, G.L.; Li, Y.; Sammons, S.; Olson, V.; Frace, M.; Nakazawa, Y.; Czerny, C.P.; Tryland, M.; Kolodziejek, J.; et al. Chasing Jenner’s vaccine: Revisiting cowpox virus classification. PLoS One 2011, 6, e23086. [Google Scholar] [CrossRef] [PubMed]
- Dabrowski, P.W.; Radonić, A.; Kurth, A.; Nitsche, A. Genome-wide comparison of cowpox viruses reveals a new clade related to variola virus. PLoS One 2013, 8, e79953. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.L.; Irausquin, S.; Friedman, R. The evolutionary biology of poxviruses. Infect. Genet. Evol. 2010, 10, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Coulson, D.; Upton, C. Characterization of indels in poxvirus genomes. Virus Genes 2011, 42, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Evans, D.H. Genome scale patterns of recombination between co-infecting vaccinia viruses. J. Virol. 2014, 88, 5277–5286. [Google Scholar] [CrossRef] [PubMed]
- Bratke, K.A.; McLysaght, A. Identification of multiple independent horizontal gene transfers into poxviruses using a comparative genomics approach. BMC Evol. Biol. 2008, 8, 67. [Google Scholar] [CrossRef] [PubMed]
- Posada, D.; Crandall, K.A.; Holmes, E.C. Recombination in evolutionary genomics. Annu. Rev. Genet. 2003, 36, 75–97. [Google Scholar] [CrossRef]
- Xing, K.; Deng, R.; Wang, J.; Feng, J.; Huang, M.; Wang, X. Genome-based phylogeny of poxvirus. Intervirology 2006, 49, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Upton, C.; Hazes, B.; Evans, D.H. Genomic analysis of the vaccinia virus strain variants found in Dryvax vaccine. J. Virol. 2011, 85, 13049–13060. [Google Scholar] [CrossRef] [PubMed]
- Brodie, R.; Smith, A.J.; Roper, R.L.; Tcherepanov, V.; Upton, C. Base-By-Base: Single nucleotide-level analysis of whole viral genome alignments. BMC Bioinform. 2004, 5, 96. [Google Scholar] [CrossRef] [Green Version]
- Hillary, W.; Lin, S.-H.; Upton, C. Base-By-Base version 2: Single nucleotide-level analysis of whole viral genome alignments. Microb. Inform. Exp. 2011, 1, 2. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Asimenos, G.; Toh, H. Multiple alignment of DNA sequences with MAFFT. Methods Mol. Biol. 2009, 537, 39–64. [Google Scholar] [PubMed]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- Strope, C.L.; Abel, K.; Scott, S.D.; Moriyama, E.N. Biological sequence simulation for testing complex evolutionary hypotheses: Indel-Seq-Gen version 2.0. Mol. Biol. Evol. 2009, 26, 2581–2593. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Kishino, H.; Yano, T. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 1985, 22, 160–174. [Google Scholar] [CrossRef] [PubMed]
- Blair, C.; Murphy, R.W. Recent trends in molecular phylogenetic analysis: Where to next? J. Hered. 2011, 102, 130–138. [Google Scholar] [CrossRef]
- Paran, N.; Sutter, G. Smallpox vaccines: New formulations and revised strategies for vaccination. Hum. Vaccin. 2009, 5, 824–831. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, B.L.; Langland, J.O.; Kibler, K.V.; Denzler, K.L.; White, S.D.; Holechek, S.A.; Wong, S.; Huynh, T.; Baskin, C.R. Vaccinia virus vaccines: Past, present and future. Antivir. Res. 2009, 84, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Chen, N.; Roper, R.L.; Feng, Z.; Hunter, A.; Danila, M.I.; Lefkowitz, E.J.; Buller, R.M.L.; Upton, C. Complete coding sequences of the rabbitpox virus genome. J. Gen. Virol. 2005, 86, 2969–2977. [Google Scholar] [CrossRef] [PubMed]
- Tulman, E.R.; Delhon, G.; Afonso, C.L.; Lu, Z.; Zsak, L.; Sandybaev, N.T.; Kerembekova, U.Z.; Zaitsev, V.L.; Kutish, G.F.; Rock, D.L. Genome of horsepox virus. J. Virol. 2006, 80, 9244–9258. [Google Scholar] [CrossRef] [PubMed]
- Trindade, G.S.; Emerson, G.L.; Carroll, D.S.; Kroon, E.G.; Damon, I.K. Brazilian vaccinia viruses and their origins. Emerg. Infect. Dis. 2007, 13, 965–972. [Google Scholar] [CrossRef] [PubMed]
- Da Fonseca, F.G.; Trindade, G.S.; Silva, R.L.A.; Bonjardim, C.A.; Ferreira, P.C.P.; Kroon, E.G. Characterization of a vaccinia-like virus isolated in a Brazilian forest. J. Gen. Virol. 2002, 83, 223–228. [Google Scholar] [PubMed]
- Katoh, K.; Toh, H. Recent developments in the MAFFT multiple sequence alignment program. Brief. Bioinform. 2008, 9, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Smithson, C.; Purdy, A.; Verster, A.J.; Upton, C. Prediction of Steps in the Evolution of Variola Virus Host Range. PLoS One 2014, 9, e91520. [Google Scholar] [CrossRef] [PubMed]
- Antoine, G.; Scheiflinger, F.; Dorner, F.; Falkner, F.G. The complete genomic sequence of the modified vaccinia Ankara strain: Comparison with other orthopoxviruses. Biochemistry 1998, 244, 365–396. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smithson, C.; Kampman, S.; Hetman, B.M.; Upton, C. Incongruencies in Vaccinia Virus Phylogenetic Trees. Computation 2014, 2, 182-198. https://0-doi-org.brum.beds.ac.uk/10.3390/computation2040182
Smithson C, Kampman S, Hetman BM, Upton C. Incongruencies in Vaccinia Virus Phylogenetic Trees. Computation. 2014; 2(4):182-198. https://0-doi-org.brum.beds.ac.uk/10.3390/computation2040182
Chicago/Turabian StyleSmithson, Chad, Samantha Kampman, Benjamin M. Hetman, and Chris Upton. 2014. "Incongruencies in Vaccinia Virus Phylogenetic Trees" Computation 2, no. 4: 182-198. https://0-doi-org.brum.beds.ac.uk/10.3390/computation2040182