3.1. Design of Low-Strain Cathode Materials for Solid-State Li-Ion Batteries
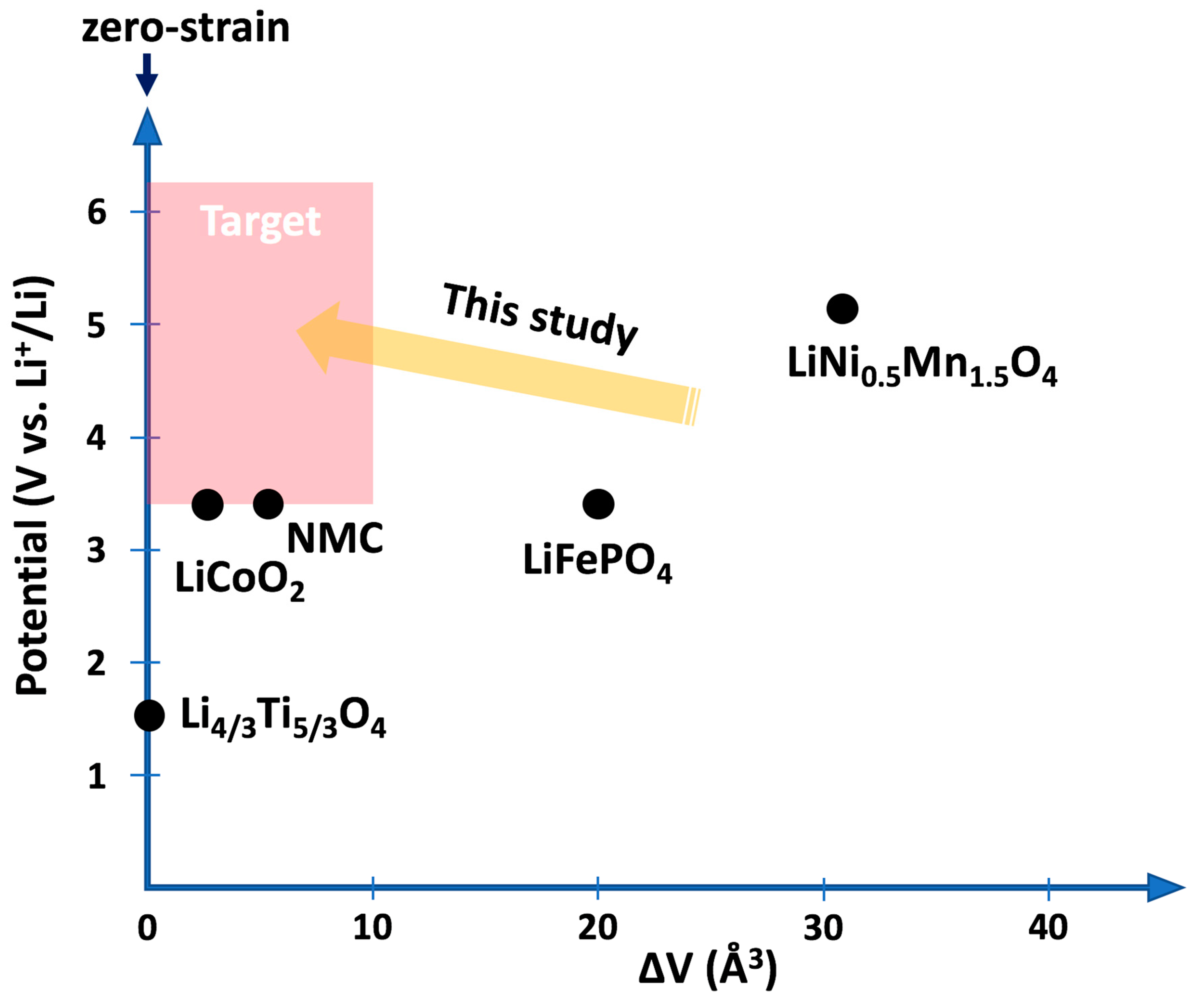
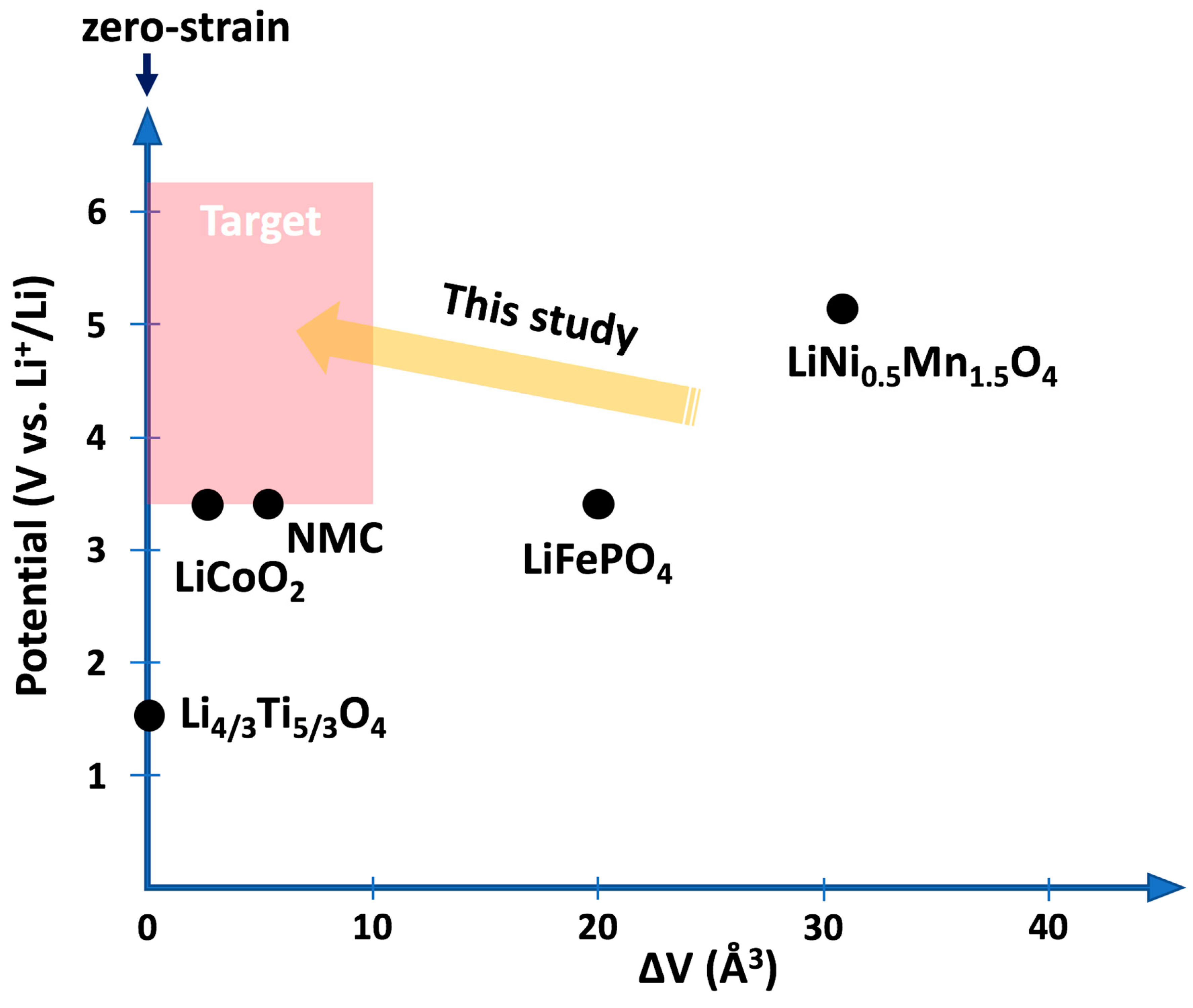
Long-term stability and safety are critical requirements in the search for new battery materials with higher capacities. While the need for safer batteries has initiated the development of solid-state electrolytes, new challenges have emerged regarding the mechanical stability at the interfaces with the electrodes. Since most of the known active materials, employed both as negative and positive electrodes, experience expansion and contraction during operation, cracks form within the electrodes and at the interfaces with the rigid electrolyte causing degradation of the battery performance and limitation of the overall lifetime [
20]. While on the anode side Li
4Ti
5O
12 has been shown to operate with nearly zero strain [
21], a cathode material with similar properties was sought at the time this study was initiated. Candidate electrode materials as given in
Figure 1 show either rather low voltage or large volume change as in the case of the previous benchmark material LiNi
0.5Mn
1.5O
4 [
22]. In the following, we describe how ab initio calculations based on density functional theory can be utilized to investigate a class of transition-metal oxides based on the spinel structure for their suitability as low-strain cathode materials [
23,
24]. Indeed, from these calculations, three low-strain materials have been identified within the class of LiMn
xCr
yMg
zO
4. The most promising materials have been synthesized and characterized by X-ray diffraction and electrochemical techniques. The results are consistent with the ab initio predictions [
23,
24].
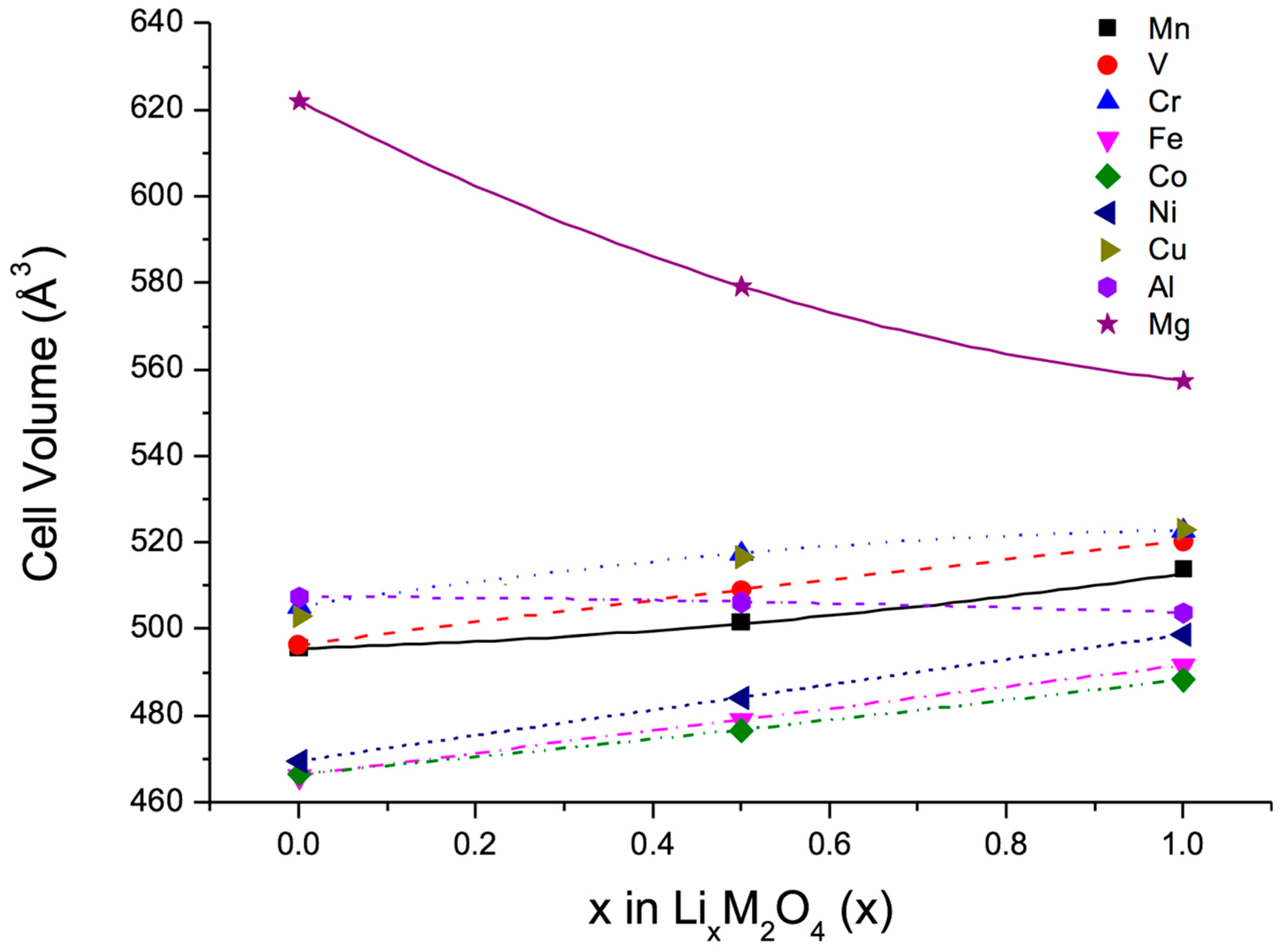
In a first step, a set of calculations was performed to evaluate the volume changes of the spinel-type compounds Li
xM
2O
4, where M = Mg, Al, V, Cr, Mn, Fe, Co, Ni, Cu, on lithiation and delithiation. To this end, three different values for the lithium content were considered, namely, x = 0, 0.5, 1.0. All structures were fully relaxed and different spin-structures were taken into account. In addition, deviations from the cubic symmetry underlying the spinel-structure were allowed for. In a second step, the previous results were used to minimize the volume change of the quasi-ternary compounds LiM
12-y-zM
2yM
3zO
4 in an approach closely following Vegard’s law [
25].
The results of the ab initio calculations for the pure spinel-type compounds are summarized in
Figure 2, which shows for each compound the calculated volume at the three Li concentrations mentioned above. The fact that some of these compounds have not yet been observed in nature was irrelevant, rather the calculations illustrate the intrinsic behaviour of proposed cathodic materials. The lines included in
Figure 2 were obtained from parabolic fits to the data points. Obviously, all transition metals induce a volume increase with increasing lithium concentration, whereas only Al and Mg cause a decrease of the cell volume, which in case of Mg is particularly strong. As a consequence, any quasi-ternary spinel-type compound must contain a non-negligible fraction of Mg to counterbalance the volume increase of the transition-metal spinels on lithiation. Hence, Mg was the first choice to be inserted into the basic cathode material LiMn
2O
4. However, since Mg is electrochemically inactive, Cr was selected as a third component in addition to Mn because of the many oxidation states offered by this element.
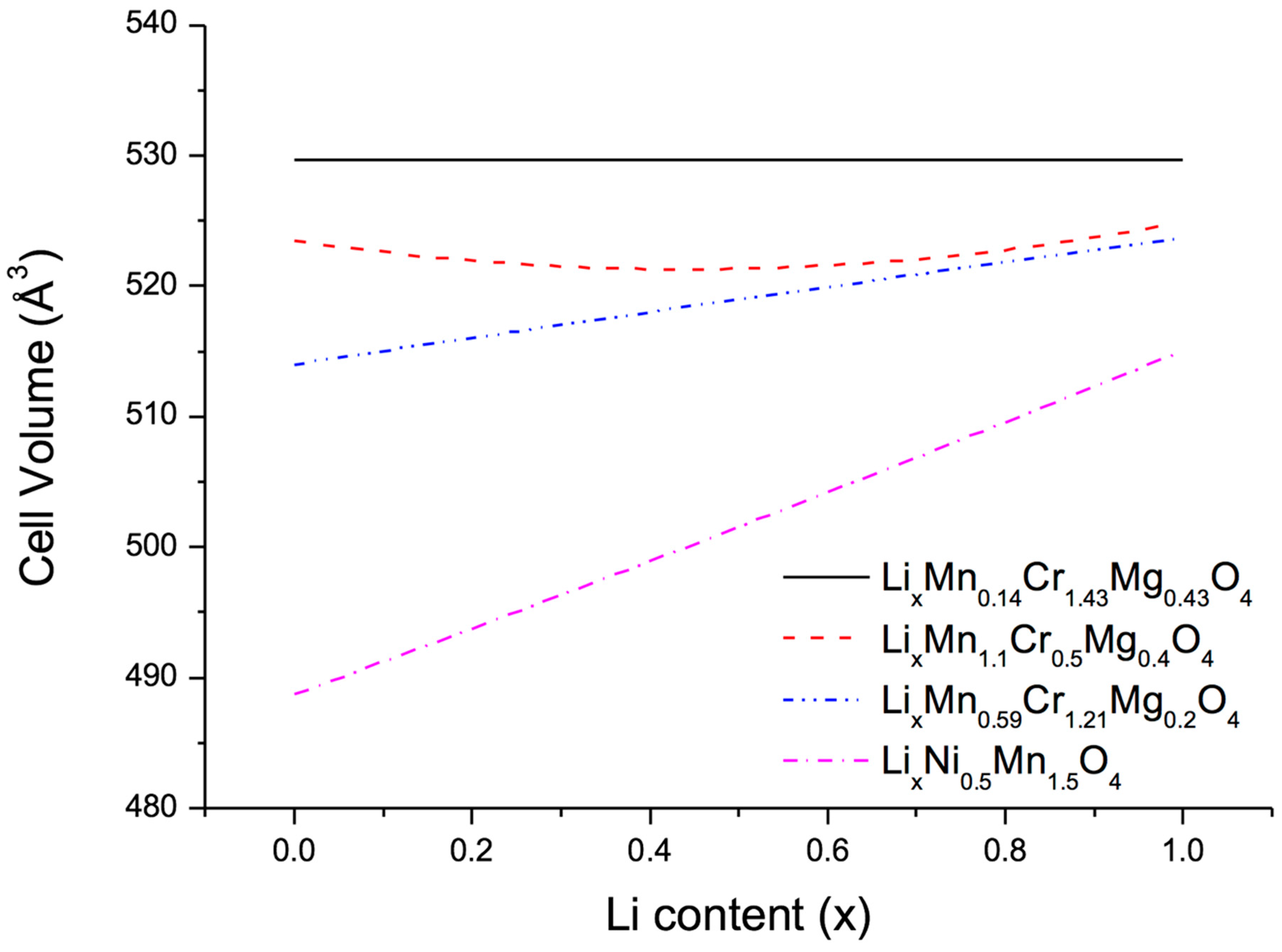
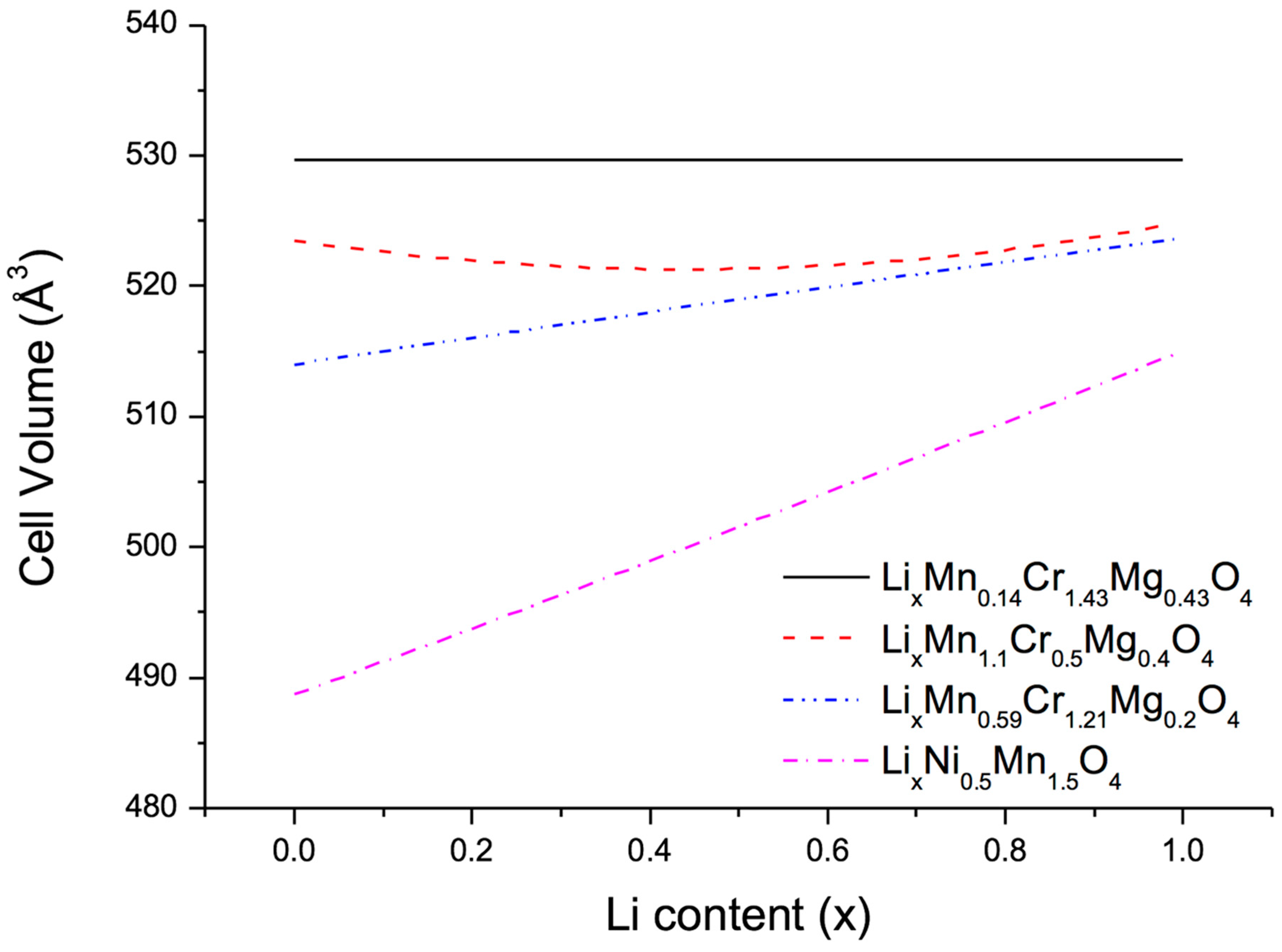
After these preparations the next step consisted in optimizing the relative concentrations at the metal sites of the quasi-ternary compound LiMn
2-y-zCr
yMg
zO
4 in order to minimize the volume change on lithiation and delithiation. To this end, three different approaches were used, namely, (1) full unconstrained optimization to obtain zero-strain behaviour; (2) constrained optimization to find a low-strain material with Mn content 2-y-z larger than 1 in order to ensure structural stability; and (3) constrained optimization to find a low-strain material with Mg content z smaller than 0.2 in order to reduce the amount of highly oxidized Cr necessary to achieve full charge. The results of these three approaches together with the volume change of the benchmark compounds LiNi
0.5Mn
1.5O
4 are displayed in
Figure 3. As expected, unconstrained optimization indeed leads to zero volume change, however, at a very small Mn and large Cr content. More interesting are the two low-strain configurations LiMn
1.1Cr
0.5Mg
0.4O
4 and LiMn
0.59Cr
1.21Mg
0.2O
4 with volume changes of 0.6% (3 Å
3) and 1.6% (8 Å
3), respectively, much lower than the value of 6% (30 Å
3) obtained for LiNi
0.5Mn
1.5O
4.
In order to confirm the validity of the approach and to compare with existing experimental data, the initial optimizations were complemented with calculations of the volume changes of a set of selected compositions, namely, LiNi
0.5Mn
1.5O
4, LiCu
0.25Ni
0.25Mn
1.5O
4, LiCr
0.5Mn
1.5O
4, LiFe
0.5Mn
1.5O
4, LiCo
0.16Mn
1.84O
4 and LiAl
0.15Mn
1.85O
4, which were found in very good agreement with measured values in all cases [
23,
24].
Powder samples of all three optimized compounds were synthesized at Toyota Motors Europe [
23]. However, XRD patterns of the two samples with high Cr concentration revealed the presence of at least two phases, indicating a phase separation induced by the high Cr content. The instability caused by high Cr content is consistent with the fact that LiCr
2O
4 has not been observed. In contrast, LiMn
1.1Cr
0.5Mg
0.4O
4 could be synthesized in almost pure form and was thus used to build electrochemical cells in order to investigate the volume change upon charging and discharging the cells. As a result, ex situ XRD patterns showed a volume change of 0.8% (4 Å
3) in very good agreement with the calculations. In addition, charge curves taken for this system revealed a performance comparable to that of the benchmark material LiNi
0.5Mn
1.5O
4.
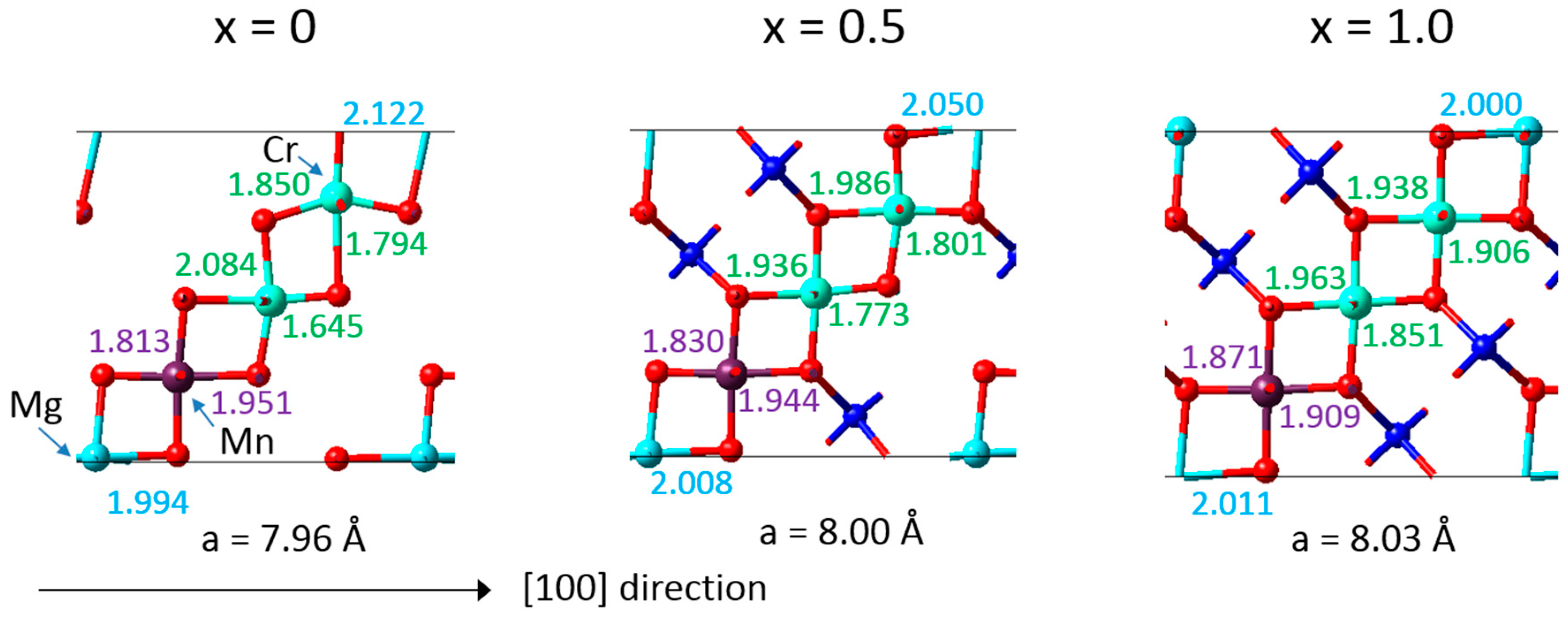
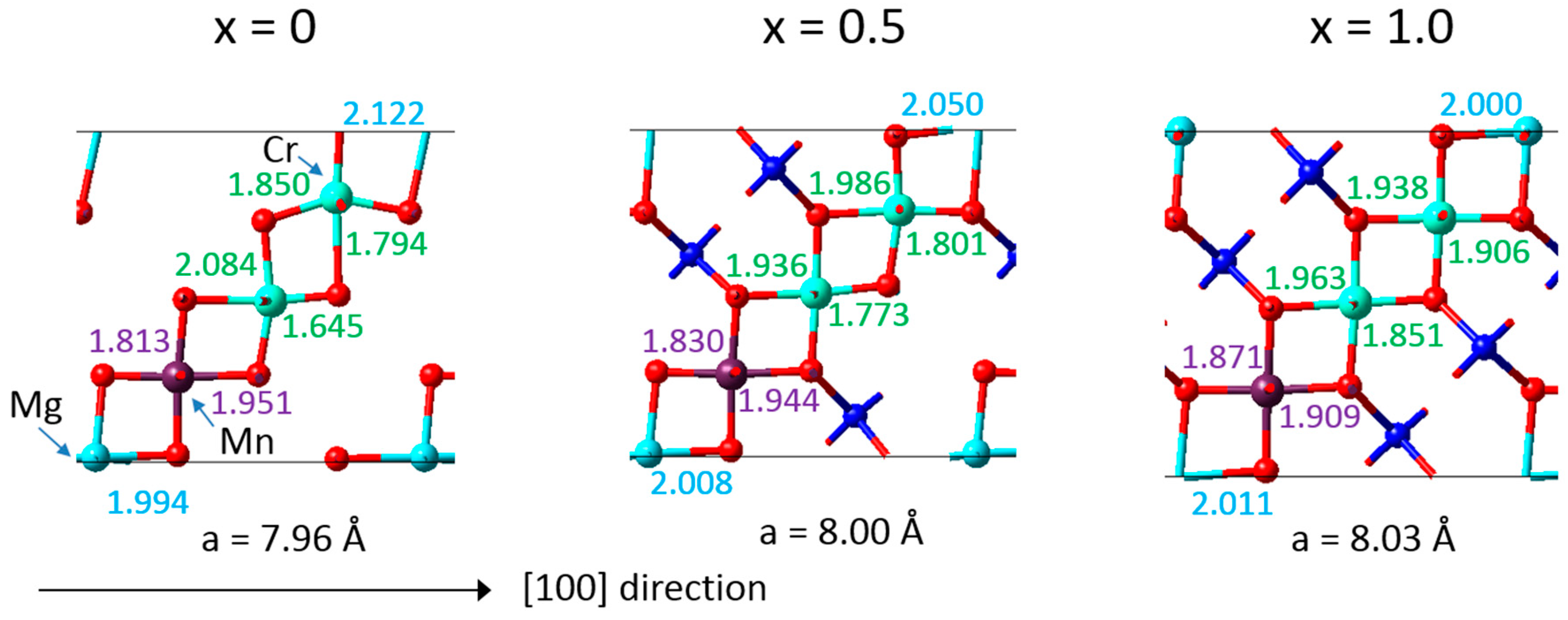
The DFT calculations also provided detailed insight into the mechanisms resulting in a near zero-strain behaviour. A synergistic compensation mechanism underlies the desired property as illustrated in
Figure 4, which includes the bond lengths calculated for an eightfold supercell of Li
xMn
1.125Cr
0.5Mg
0.375O
4. With increasing Li concentration, the Mg-O bonds tend to decrease and the Mn-O bonds remain similar, whereas the Cr-O bonds tend to increase. As a result, the overall volume of the crystal structure changes little upon charging and discharging with Li ions. Furthermore, the microscopic analysis reflects the behaviour of the “pure” compounds as displayed in
Figure 2. This behaviour is also consistent with observations for the zero-strain mechanism for Li
4Ti
5O
12, where local distortions in the crystal structure similarly allow this material to keep the volume nearly unchanged upon lithium insertion [
26].
Here, DFT simulations have been combined with an optimization approach based on Vegard’s law to obtain a spinel-type quasi-ternary compound with small volume change on lithiation and delithiation. The validity of the procedure has been confirmed by comparison to measured volume changes obtained for various compositions. In addition, experiments performed for the best candidate, LiMn1.1Cr0.5Mg0.4O4, found a volume change of 0.8% (4 Å3) in very good agreement with the calculated 0.6% (3 Å3). Finally, analysis of the DFT results provided an explanation for the observed volume changes based on a microscopic compensation mechanism.
3.2. Embrittlement of Cu Micro-Structures
In the race for still smaller structures of semiconductor devices, copper metallization layers have a significant advantage over those made of aluminium, since their much higher electrical and thermal conductivity allows for smaller metal components and less energy consumption. However, long-term stability of the devices requires a high mechanical performance of the metal contacts. Fortunately, the mechanical properties of such microstructures can be controlled and considerably improved by reducing the grain size, which is well known as the Hall-Petch effect [
27,
28]. Indeed, for electrodeposited Cu thin films it has been demonstrated that additives can cause a reduction of the grain size. However, as revealed by electron backscatter diffraction in a scanning electron microscopy (EBSD/SEM), these additives also cause embrittlement of the microstructures at elevated temperatures [
29]. In the present section we describe how ab initio calculations as based on density functional theory can be employed to obtain a deeper understanding of the origin of this failure [
29]. Importantly, additional calculations successfully proposed measures to overcome the detrimental behaviour.
In the experiments reported in Ref. [
29] Cu thin films were synthesized by electrodeposition using two different additive systems in the electrolyte intended to modify the grain size (sample type A and B). EBSD/SEM images of these films are displayed in
Figure 5. Samples of types A and B possess median grain sizes of about 2.7 ± 0.6 μm and 10.1 ± 2.6 μm, respectively. As also shown in
Figure 5, the grain size of sample type A increases with additional annealing at 1073 K.
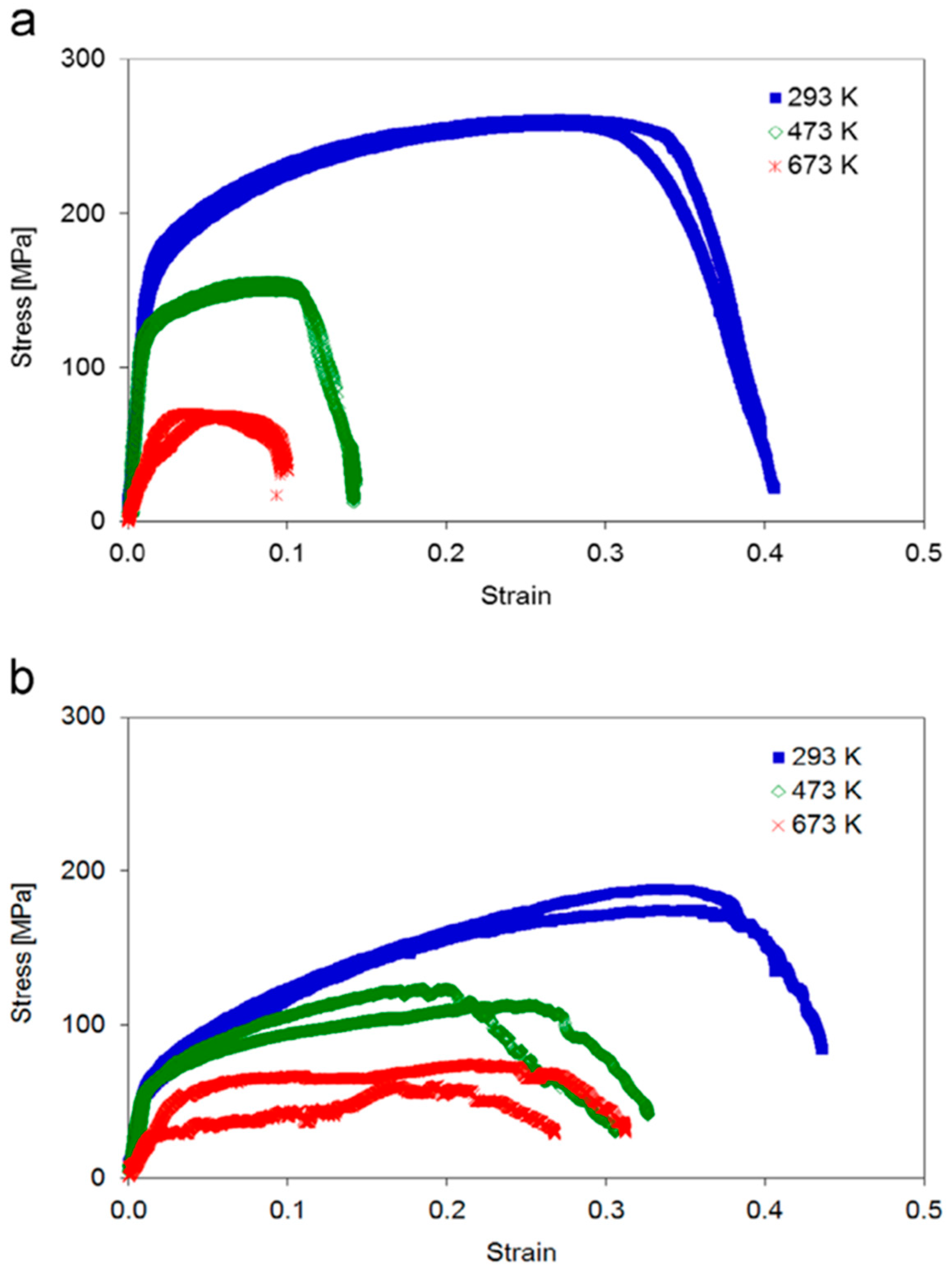
Mechanical testing revealed distinct differences between the two sample types as is obvious from
Figure 6. At room temperature sample type A exhibited considerably higher yield strength and ultimate tensile strength as compared to sample type B, as expected from the Hall-Petch effect. This advantage was lost at elevated temperatures, where sample type A showed a drastic reduction in yield strength, ultimate tensile strength and elongation to fracture. In contrast, sample type B showed only a moderate decrease of these values.
SEM images of sample type B taken at 293 K, 473 K and 673 K clearly showed ductile behaviour with intragranular fracture accompanied by the formation of glide steps at all temperatures. In contrast, sample type A showed strong plastic deformation with several grains involved at 293 K, whereas at higher temperatures cracking occurred preferentially along the grain boundaries indicative of grain boundary embrittlement [
29].
An indication of the origin of the different behaviour of the two sample types was obtained from chemical analysis, specifically from time-of-flight secondary ion mass spectroscopy (TOF-SIMS), which showed a significantly higher content of both S and Cl in sample type A. In addition, the experiments indicated considerable enrichment of these impurities at the grain boundaries [
29].
In order to address the possible detrimental behaviour of S and Cl impurities on the mechanical strength of the microstructure, DFT calculations were performed for models of slabs and grain boundaries, respectively, which were then decorated with impurity atoms at selected sites. All structures were fully relaxed yielding energy minimum configurations representing mechanical equilibrium for these atomistic systems.
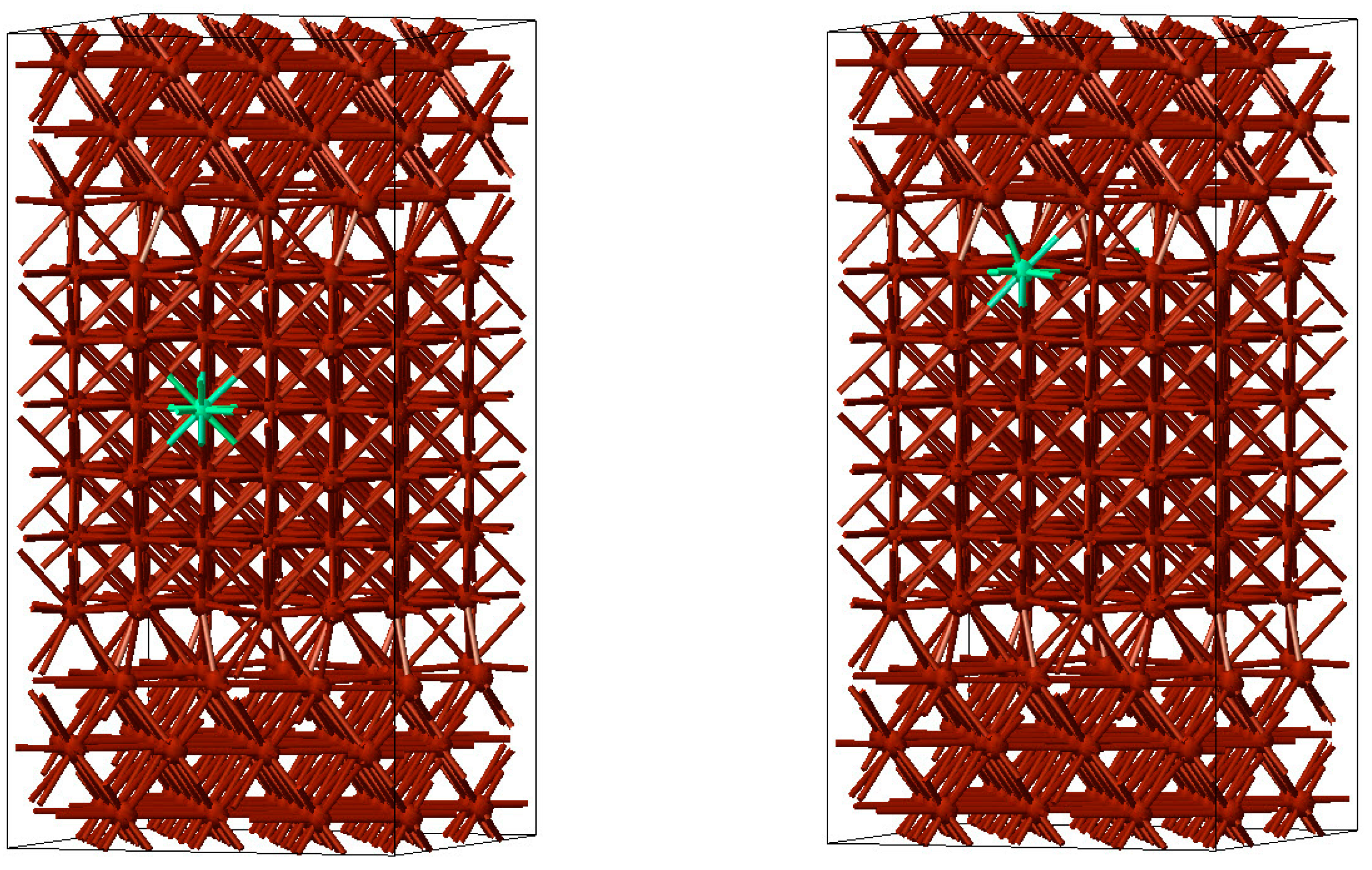
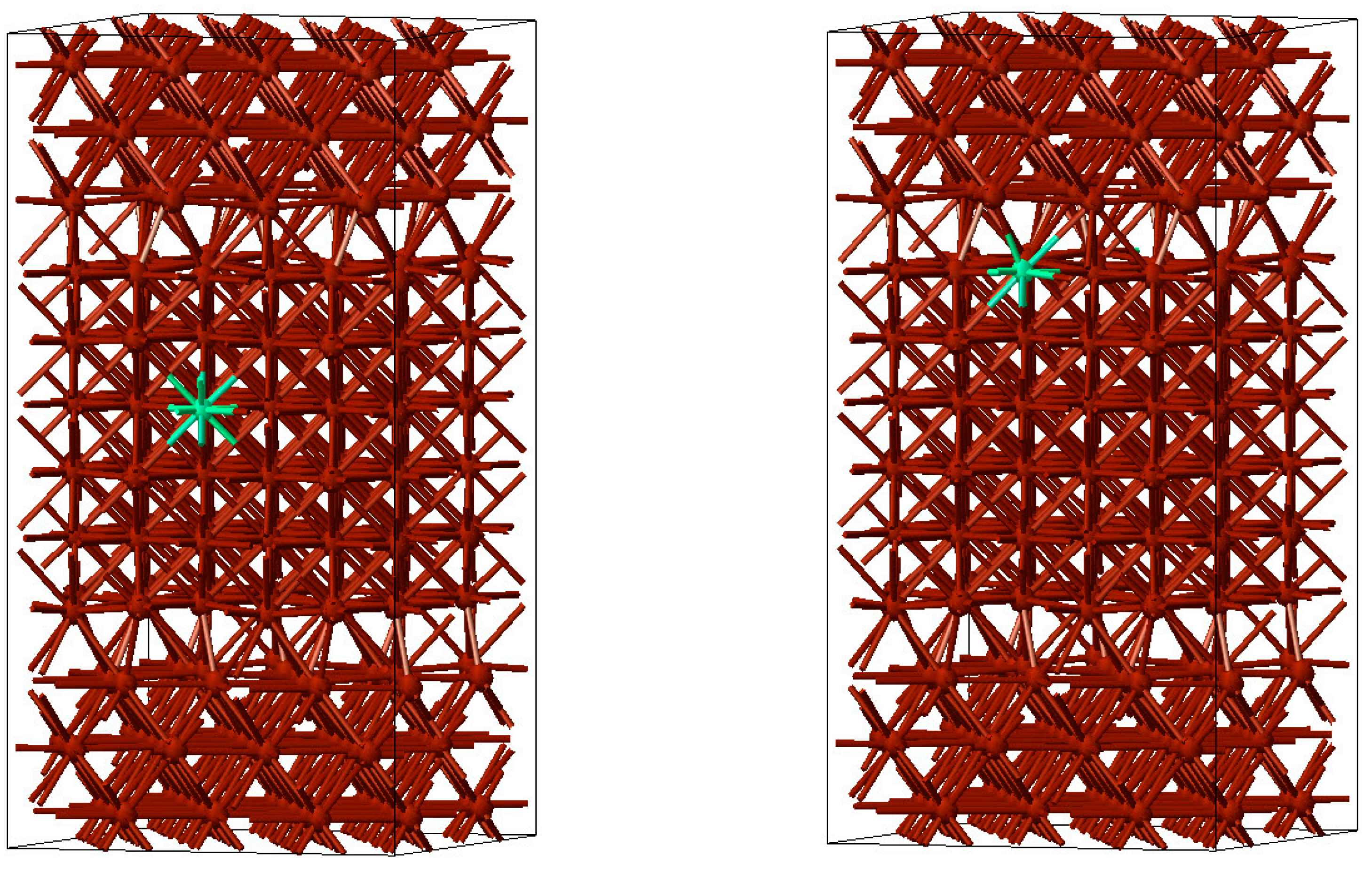
In a first step, slab models of face-centred cubic Cu were constructed with surfaces perpendicular to the [001] and [111] directions. These slabs were separated by a 10 Å wide vacuum region to simulate free surfaces. Subsequently, the slabs were combined to form Σ5(001) and Σ7(111) twist grain boundaries. As an example, a Σ5(001) twist grain boundary is displayed in
Figure 7.
In a second step, S and Cl atoms were inserted first at the centre of the slabs and then at their boundaries (free surfaces or grain boundaries) to estimate the segregation energies of these impurities from the differences in total energy. For a Cl atom at the centre of the grain and at a Σ5(001) grain boundary these situations are displayed in
Figure 8.
The calculated segregation energies are given in
Table 1 for both S and Cl impurities and for the segregation from the inside of a grain to both the free surfaces and the grain boundaries. Results are given for both the (001) and (111) surfaces and the Σ5(001) and Σ7(111) twist grain boundaries. In all cases negative segregation energies were obtained, that is, in all cases the impurities prefer a position at the surface and grain boundary, respectively. This trend is more pronounced for the free surfaces, where the impurity has more freedom to optimize its bonds with the neighbouring Cu atoms.
Finally, from a comparison of the total energies of the interface systems to those of the slabs with free surfaces the energies of separation were obtained, which gave a measure of the cleavage energy. These separation energies were evaluated with and without impurities to obtain a measure of the detrimental effect of the latter [
29]. The results are summarized in the
Table 2, which gives the energies of separation for both types of grain boundaries for pure Cu as well as for Cu microstructures with S and Cl impurities at the respective concentrations. Both S and Cl lead to a reduction of the energy of separation for both the Σ5(001) and Σ7(111) twist grain boundaries. While the reduction is of the order of 10% for sulphur, the effect is much more pronounced for chlorine, which induces a drastic weakening of these grain boundaries. The calculations thus explain the experimentally observed fracture of the Cu microstructures at the grain boundaries and assign the failure to chlorine impurities as the main cause.
The DFT simulations thus provide an explanation for the transition from intragranular plastic to intergranular brittle failure in electrodeposited Cu micro-tensile samples at elevated temperatures, which had been observed by electron backscatter diffraction experiments in a scanning electron microscope. While this embrittlement could be tentatively attributed to some additives in the sample, the exact mechanism as well as the role of particular elements was not understood. With the help of the simulations strong preference of the impurities to segregate to the grain boundaries and the free surfaces could be demonstrated, while the calculated cleavage energies revealed strong weakening of the grain boundaries by S and even more so by Cl.
For bulk Cu it had been shown that embrittlement at elevated temperatures could be reduced by addition of small amounts of Ti, V and Zr, whereas B, Nb, Fe and Ag were found to significantly strengthen Cu grain boundaries [
29]. More recent investigations have explored the mitigating influence of these additional dopant in detail providing similar quantitative explanations and insights into the origin of physical observations.
3.3. Structure and Bonding of Boron Carbide
Boron carbide, B
4C, is one of the hardest materials known, close to diamond and cubic boron nitride. Due to its mechanical properties, B
4C is used in many applications as an abrasive or shielding material. In nuclear reactors boron carbide is used to control the neutron flux due to the high neutron absorption of
10B and the radiation hardness and chemical stability of B
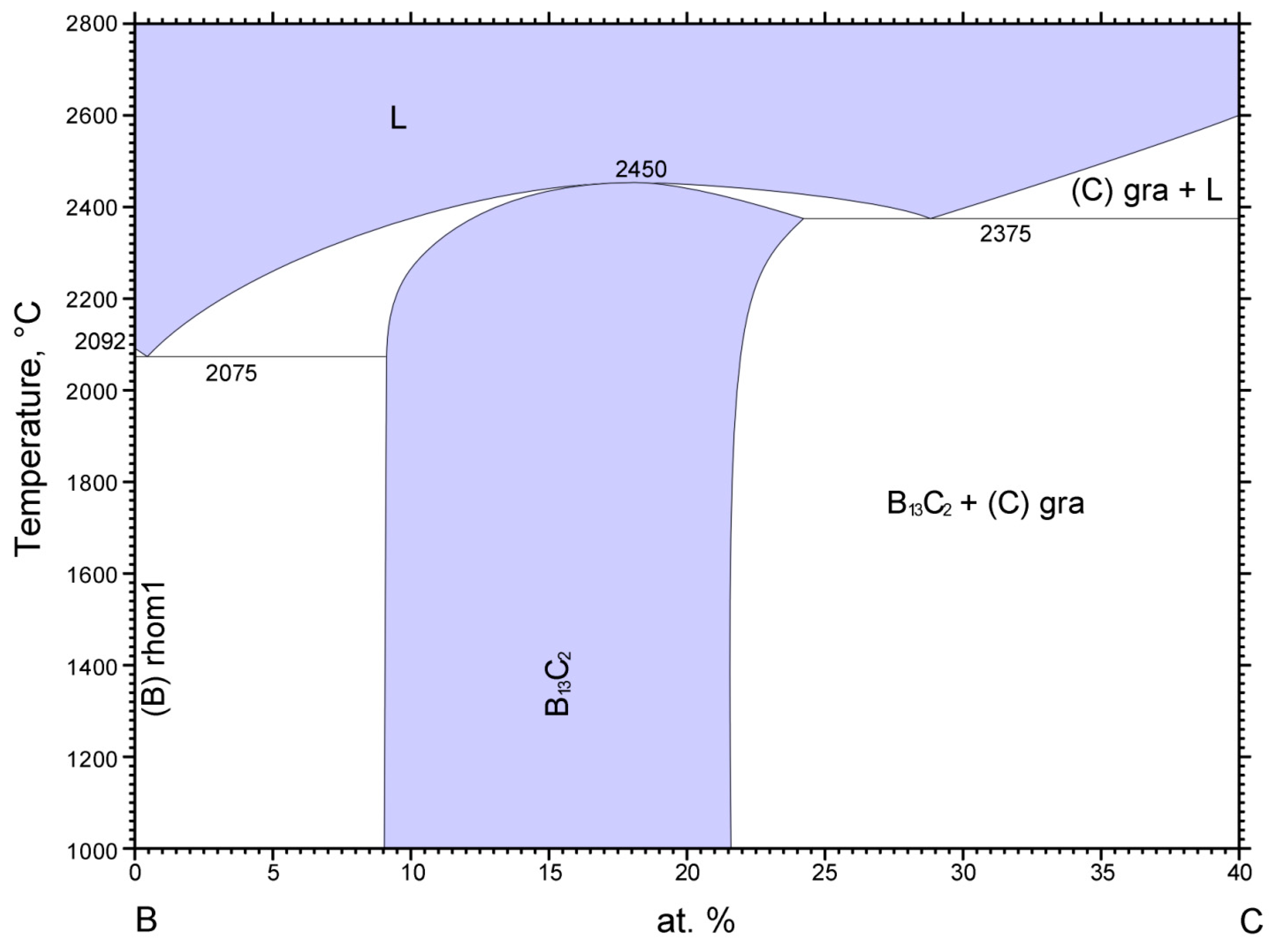
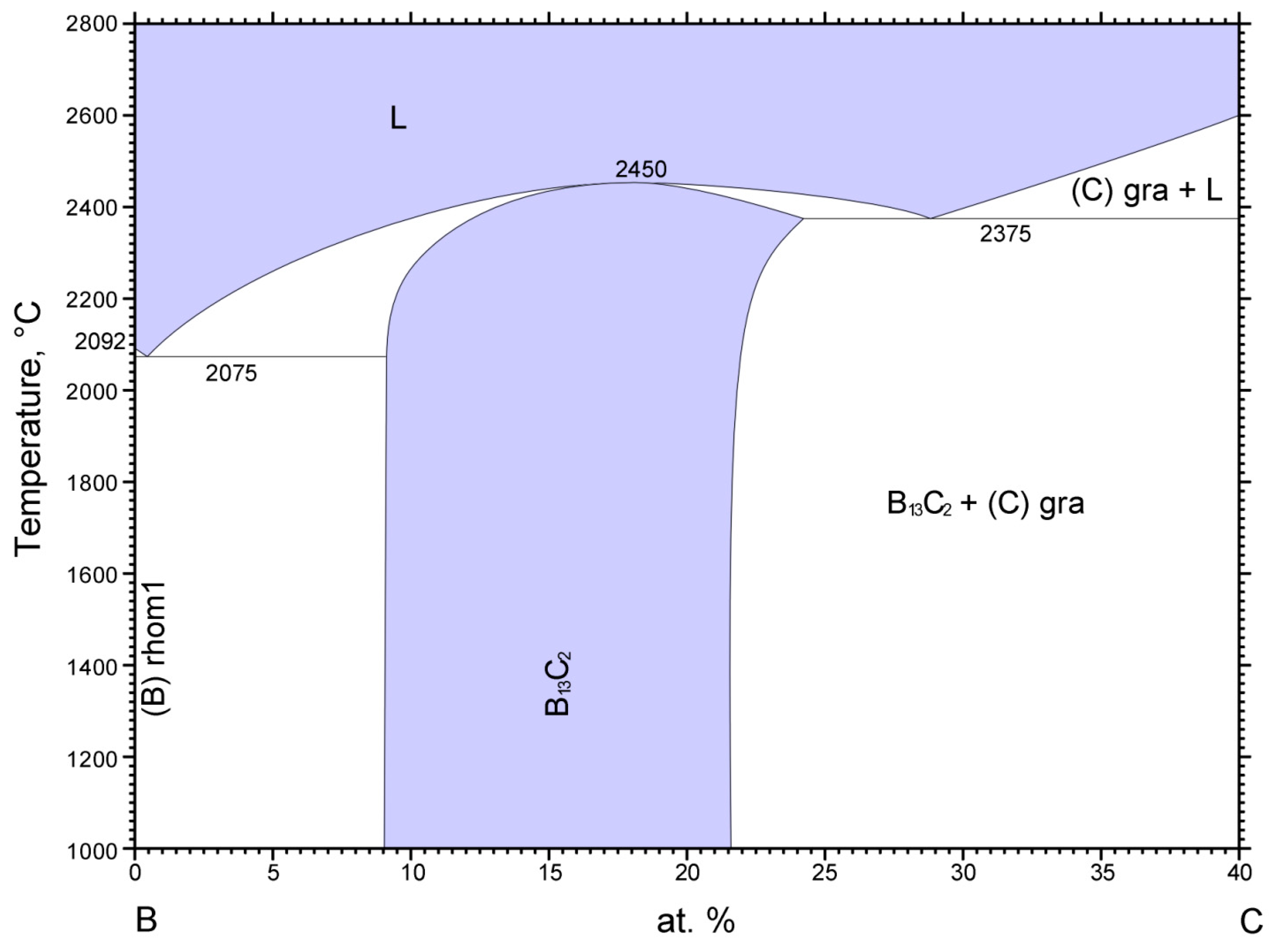
4C. According to the boron-carbon phase diagram displayed in
Figure 9, a boron carbide phase exists between approximately 9 at% to 22 at% C with a melting point reaching 2450 °C, which is complemented by carbide phases with excess boron and graphite, respectively, below and above this carbon concentration range [
31,
32].
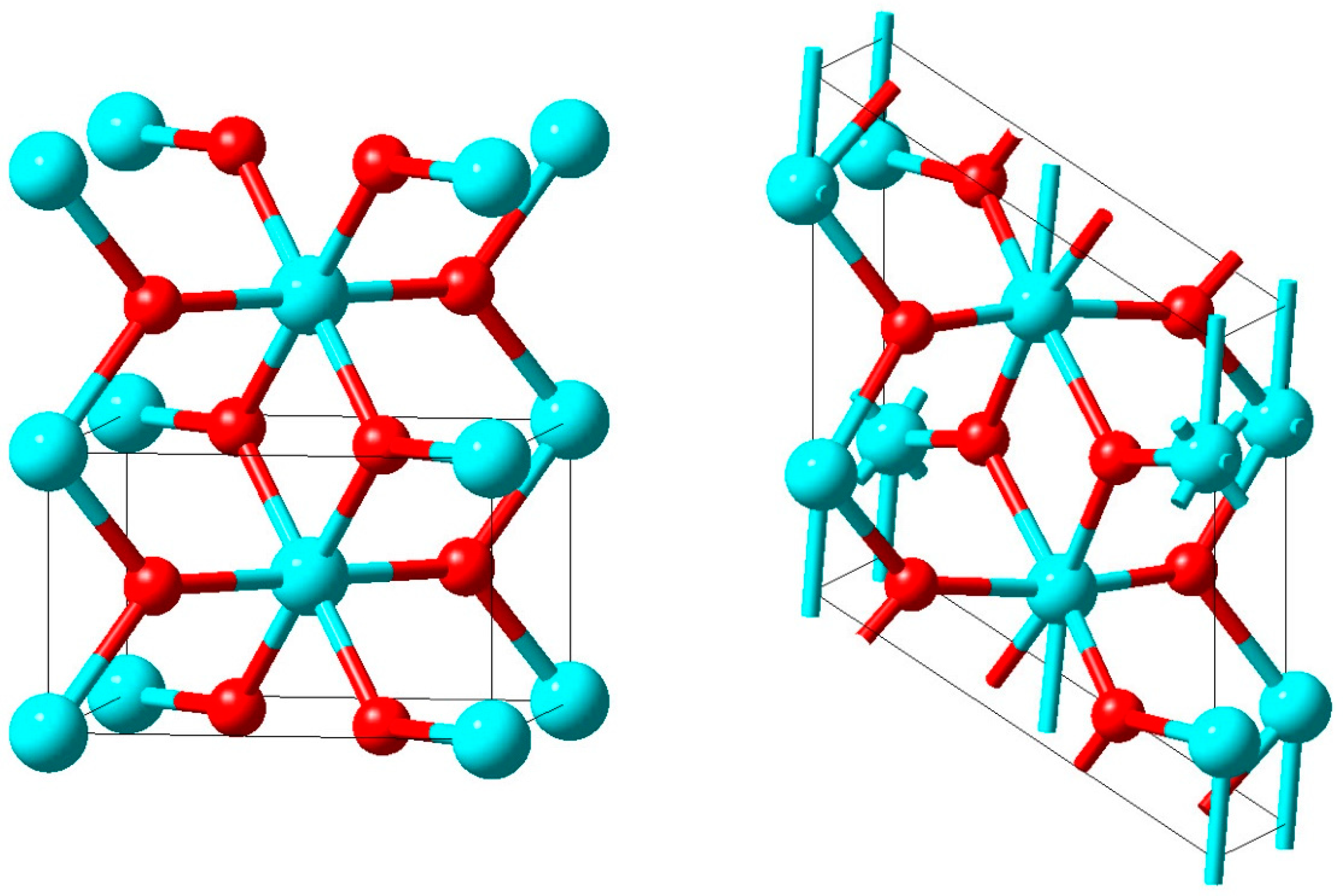
The crystal structure of boron carbide is characterized by a rhombohedral lattice with a single icosahedron at the corner of the unit cell and a three-atom linear rod parallel to the hexagonal c axis at the centre of the unit cell with lattice parameters of about a = 5.6 Å and c = 12.1 Å in hexagonal setting. However, the exact positions of the carbon atoms in this structure are far from being precisely known since B and C have very similar scattering cross sections for both neutron and X-rays. Larson obtained a C-B-C arrangement for both B
4C and boron-rich B
13C
2 within the rods from X-ray diffraction data and, hence, boron-pure icosahedra as shown on the left hand side of
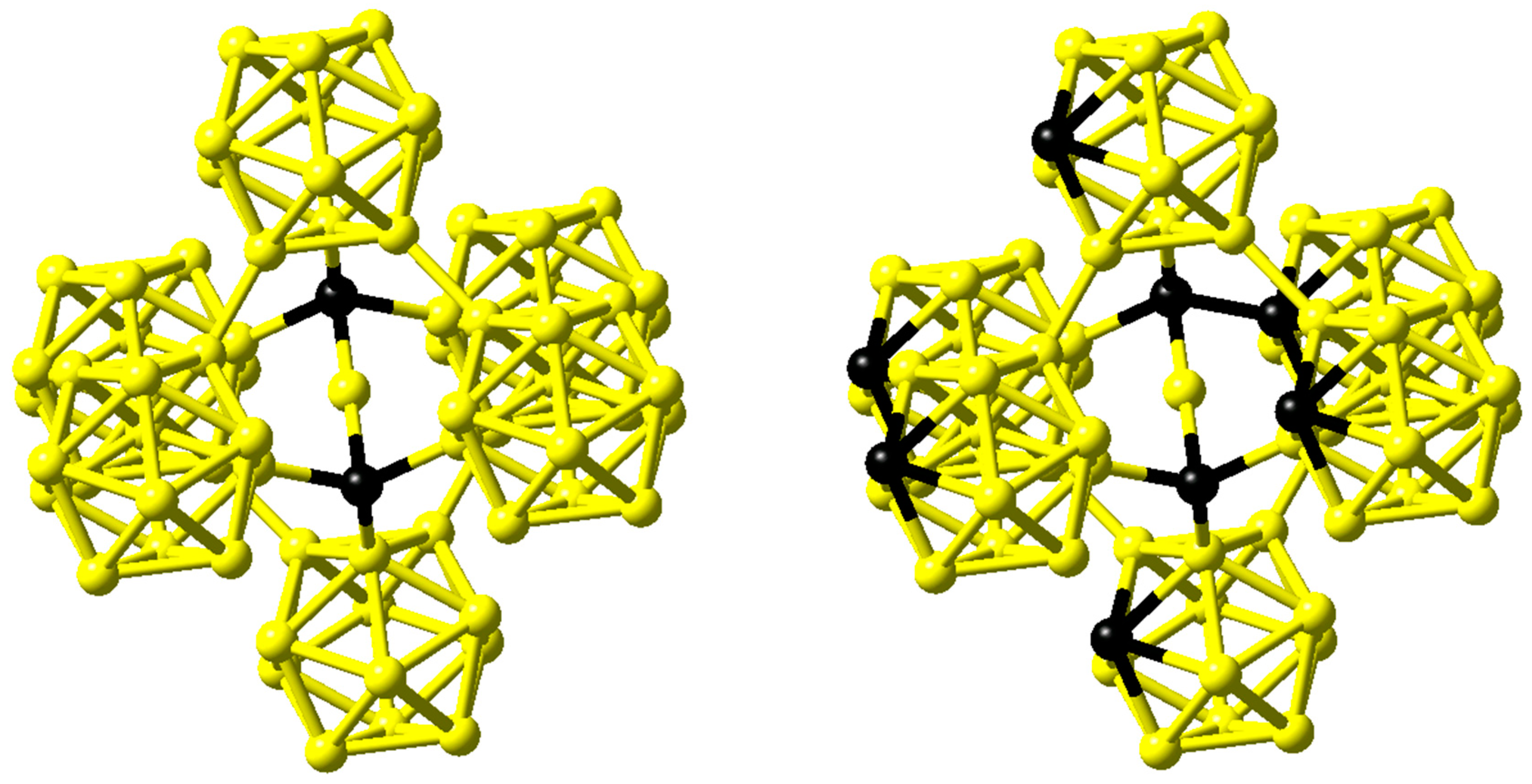
Figure 10 for the latter compound [
33]. Clark and Hoard gave a structure for B
4C with linear C-C-C rods and again icosahedra comprised of only B atoms [
34].
In contrast, comparing infrared absorption and Raman diffusion measurements to the predictions of ab initio calculations for B
4C, Lazzari et al. confirmed the finding of linear C-B-C rods reported by Larson but in addition were able to identify the carbon atom within the B
11C icosahedra at the polar sites, which form the top and bottom triangles [
35]. This structure is shown on the right hand side of
Figure 10.
The same result was obtained by Mauri et al., who compared the results of their ab initio calculations to
13C and
11B NMR chemical shifts [
36]. However, these groups considered only small sets of ordered structures.
In the present section, we describe a more comprehensive method going beyond all previous approaches, namely, the cluster expansion approach [
37] together with ab initio calculations to take into account disorder effects and unambiguously determine the distribution of the atoms within the structure. An implementation of this method is offered by the UNiversal CLuster Expansion (UNCLE) code, which has become state of the art in the description of disordered systems [
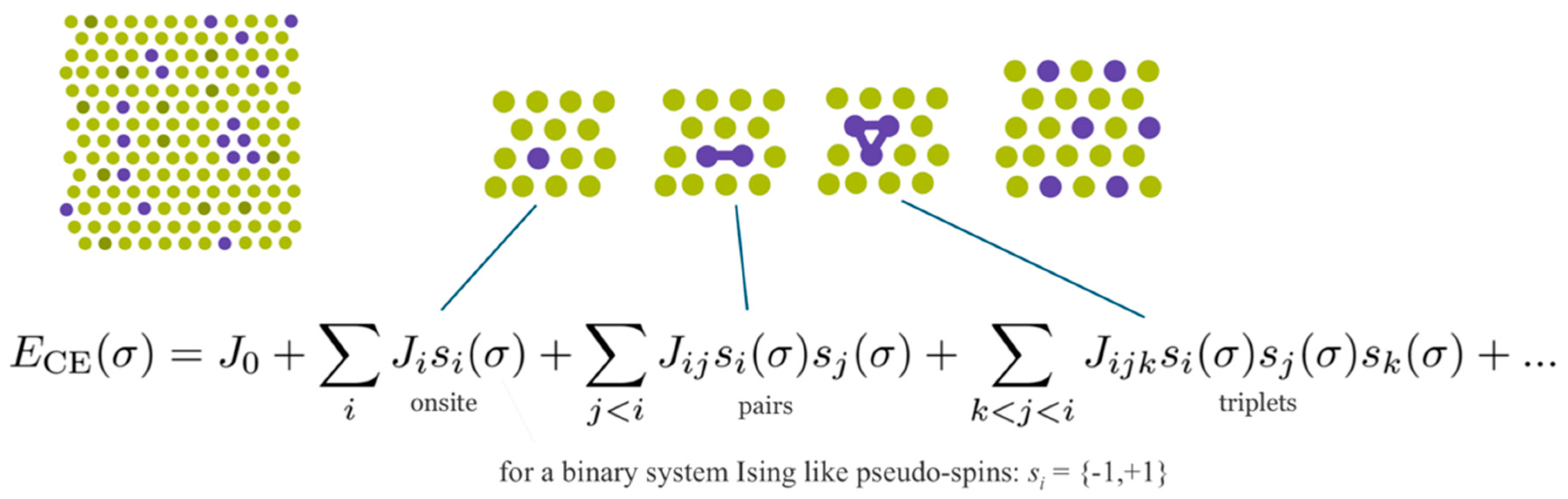
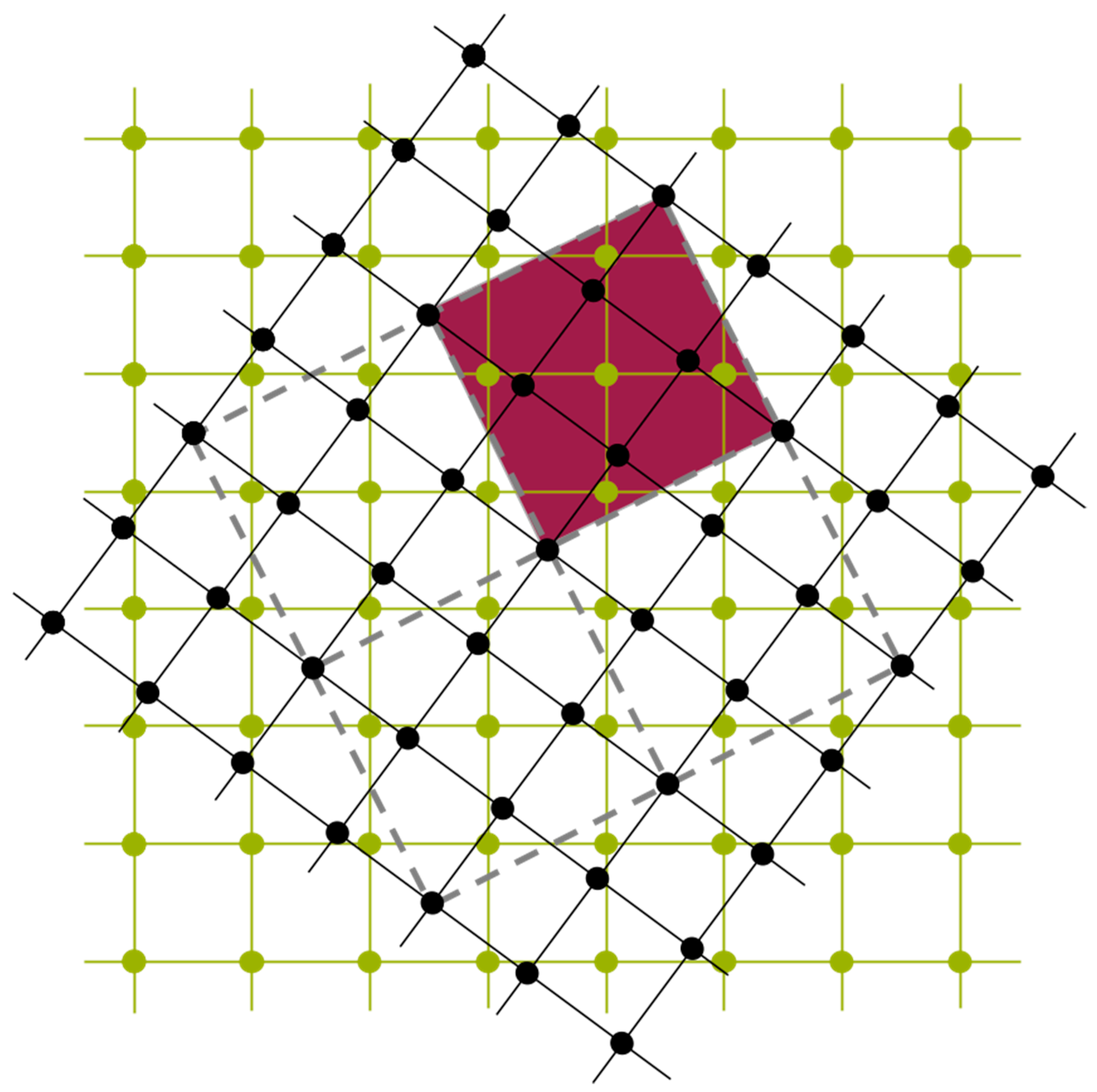
38]. In short, as indicated in
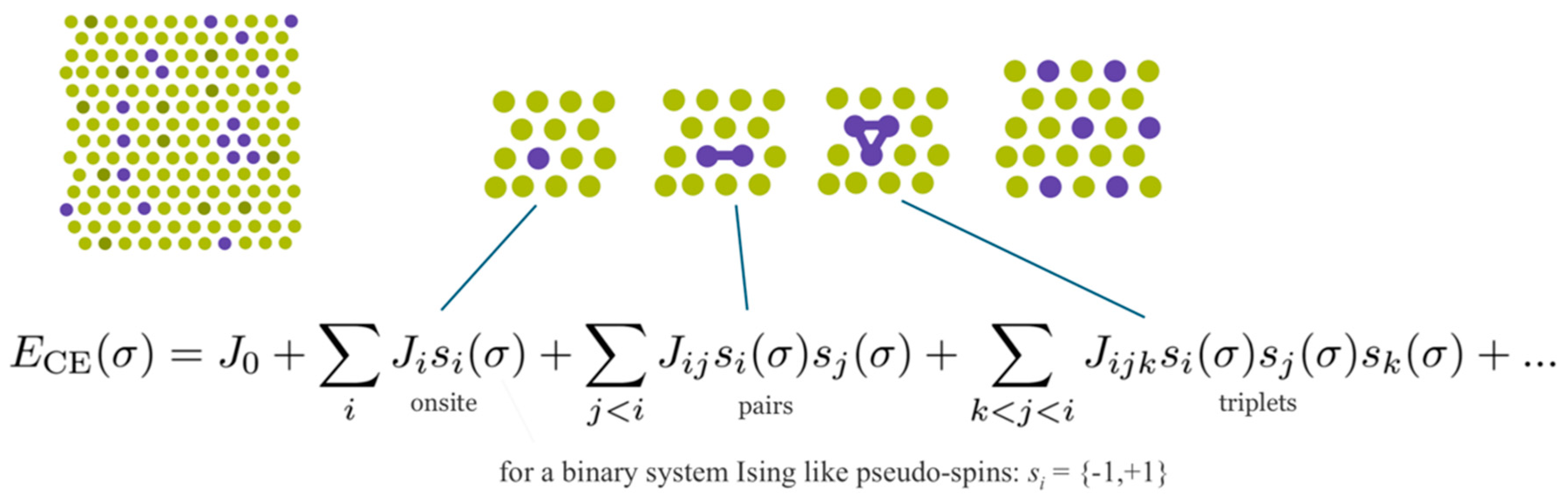
Figure 11, UNCLE expresses a desired quantity, for example the total energy, as an expansion in contributions from clusters including progressively larger numbers of vertices.
A crystal structure can be decomposed into an infinite set of clusters with different size and composed of different number of vertices, that is, two-body interactions, three-body interactions and so forth, as shown in
Figure 11. An effective interaction parameter is assigned to each of these clusters to reproduce the correct energy (or any other desired quantity). The core task is to truncate the cluster set at a reasonable size and to identify the effective interaction parameters.
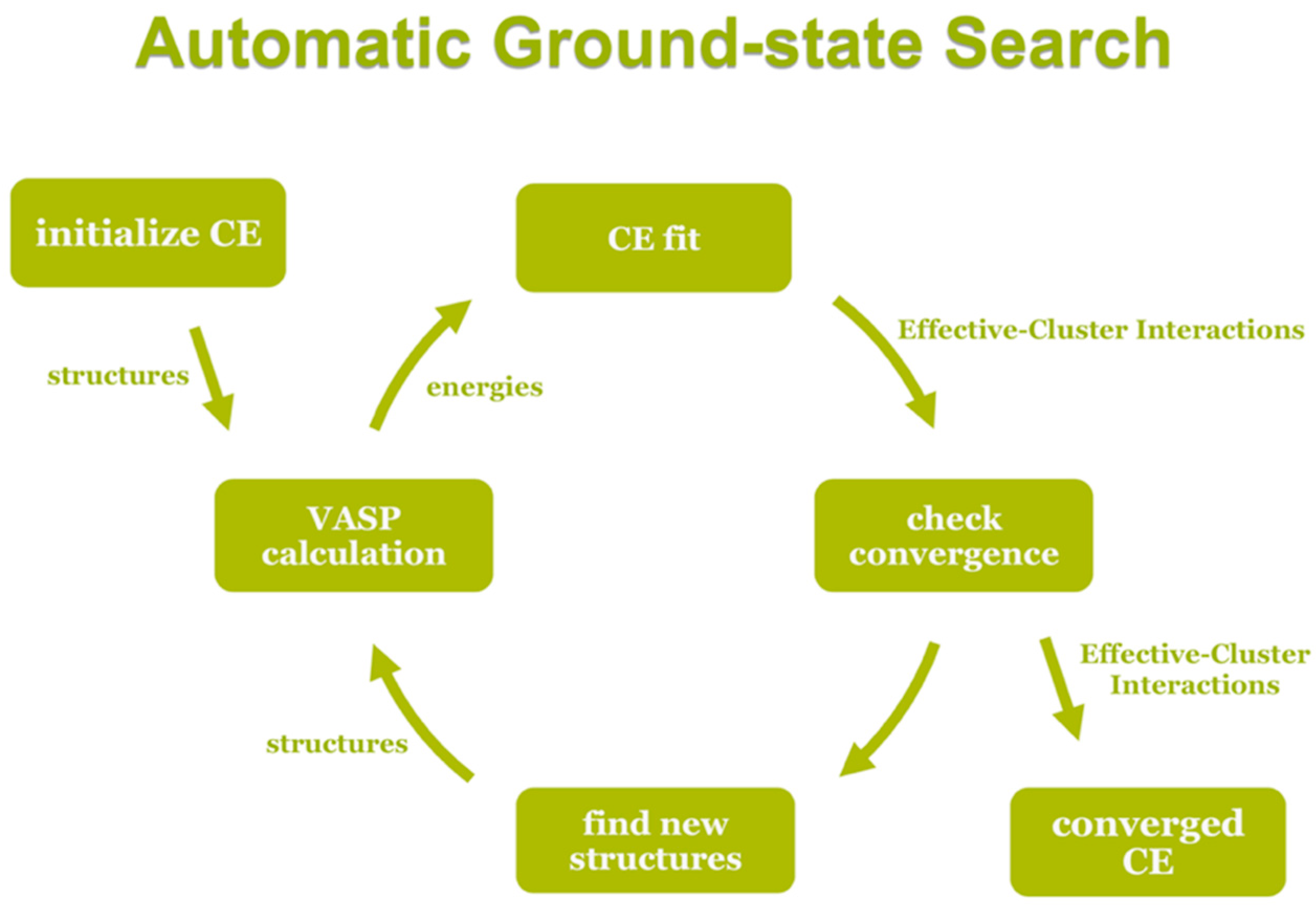
The UNCLE code uses a genetic algorithm to identify an optimum set of clusters with their corresponding interaction parameters obtained by a least-squares fit from the energies of structures contained in a training set. In turn these energies are calculated by using a DFT method. Structures are iteratively added to the training set in the procedure sketched in
Figure 12 until convergence is achieved.
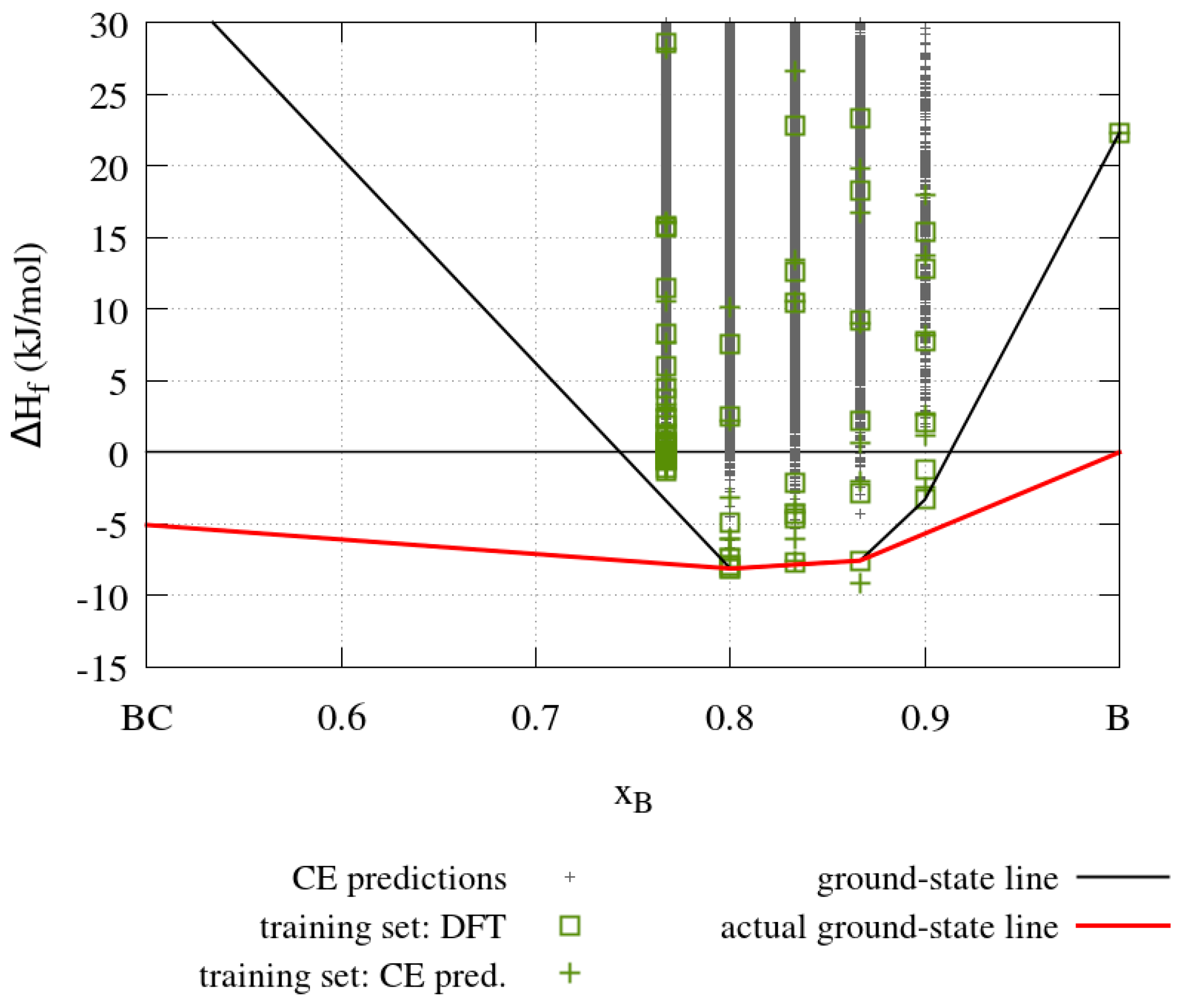
The results obtained from the cluster expansion calculations for the boron-carbide system are summarized in
Figure 13, which shows enthalpies of formation for boron carbides with five different boron concentrations. The solid black line in
Figure 13 indicates the convex hull obtained from the cluster expansion where the pure carbon and boron phases are based on the rhombohedral boron carbide crystal structure (cf.
Figure 10), in which all sites are occupied by carbon and boron atoms, respectively. The solid red line indicates the true ground-state line with graphite as the most stable carbon compound and α-boron as the most stable boron compound. The boron carbide crystal structure is thermodynamically stable where the cluster expansion ground state line coincides with the true ground state line, that is, for
in the range from 0.8 to 0.8667. The training set containing 63 structures with a maximum size of up to two unit cells is used to obtain the effective cluster interaction parameters. The energy of each structure in the training set is calculated with DFT. These effective cluster interaction parameters are then used to predict the energy of 838,511 structures with DFT accuracy.
The cluster expansion confirms the previous findings. However, since the cluster expansion takes 838,511 ordered structures into account with ab initio precision, the present results are general. Specifically, the ground-state structures obtained for B
13C
2 (x
B = 0.8667) and B
4C (x
B = 0.8) are identical to those proposed by Larson, Lazzari et al. and Mauri et al. shown in
Figure 10, which share the linear C-B-C motifs, whereas additional carbon atoms prefer the polar sites of the icosahedra.
Furthermore, with the ground-state structures for different boron concentrations at hand, the elastic and vibrational properties of these compounds are readily calculated. The elastic coefficients are computed using a fully automated, symmetry general approach [
39] as implemented in MedeA [
1]. The results as summarized in
Table 3 reveal considerable stiffening of the material with increasing C concentration between B
9C and B
4C as expected with the lowering of the enthalpy of formation within this sequence.
Finally, the computed phonon dispersions for B
4C confirm the thermodynamic stability of this compound and reveal an isolated high-frequency mode, which originates from bond-stretching vibrations of the B atoms in the C-B-C linear rods as illustrated in
Figure 14, which is consistent with the analysis of Lazzari et al. [
35].
In summary, using the UNiversal CLuster Expansion (UNCLE) code as well as calculations of the mechanical and vibrational properties we were able to identify the ground-state structures of boron carbides with boron concentrations varying between xB = 0.8 and xB = 0.9. For B4C and B13C2 the investigation confirmed the C-B-C sequence of the linear rods, whereas additional carbon atoms in B4C prefer the polar sites of the icosahedra. Computed elastic properties showed a stiffening of boron carbide with increasing carbon concentration, while analysis of vibrational spectra revealed an identifiable high-frequency mode connected to bond-stretching within the linear rods.
3.5. Accurate Band Gaps of Transition-Metal Oxides from Hybrid-Functional Calculations
The metal-insulator transition (MIT) of stoichiometric VO
2 at ambient pressure has been the subject of ongoing dispute since its discovery in the 1950s [
40,
41]. Occurring at the technologically interesting temperature of 340 K this metal-insulator transition is connected with a change in resistivity of several orders of magnitude leading to applications in surface coatings, sensors and imaging systems. Interestingly, this transition temperature is closely related to that of a structural transition from the high-temperature rutile structure to a low-temperature monoclinic M
1 structure, which deviates from the former by a pronounced V-V dimerization parallel to the rutile c-axis and a zigzag-like antiferroelectric displacement of the V atoms perpendicular to that axis.
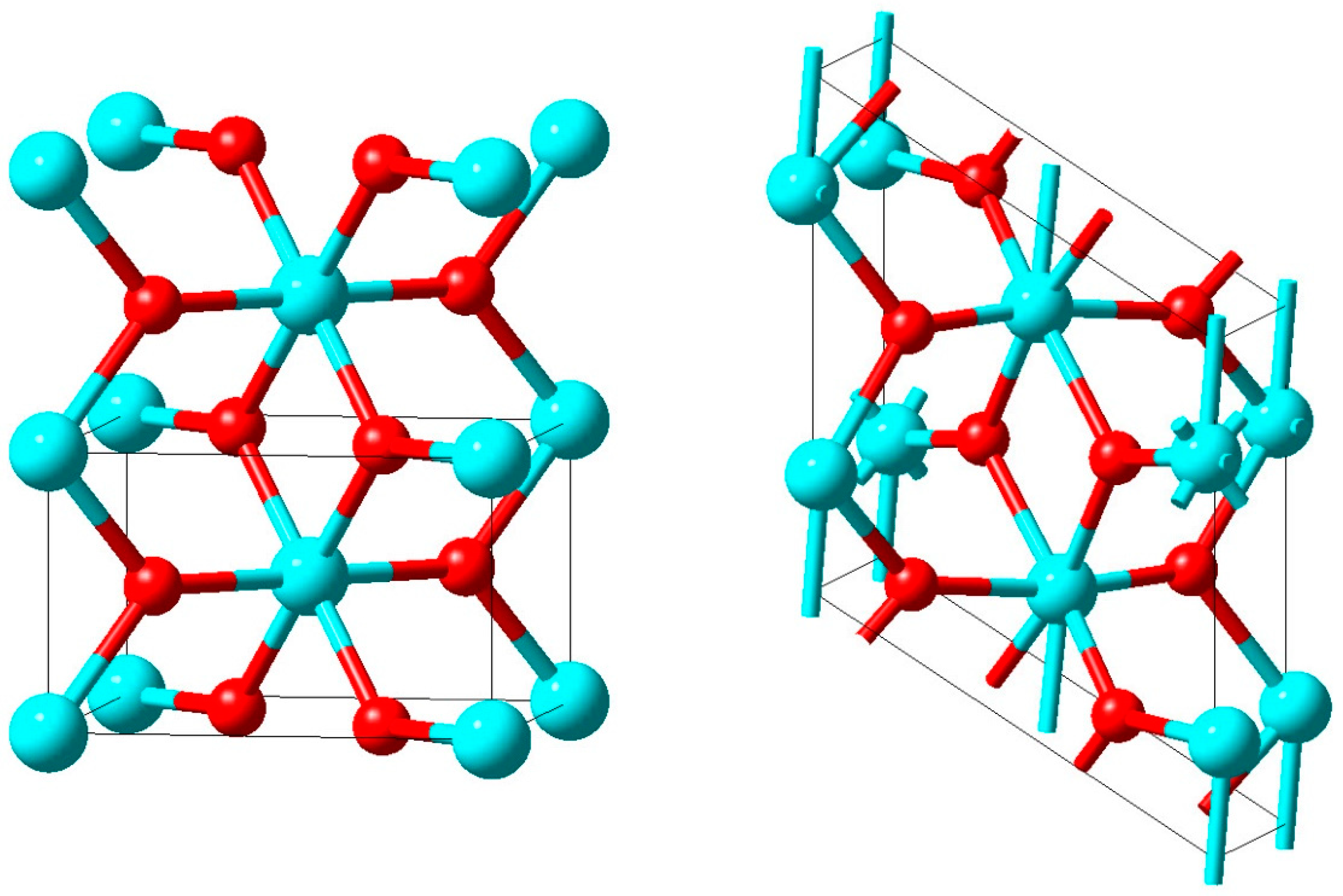
In
Figure 16, two unit cells of the rutile structure with the characteristic VO
6 octahedra and the vertical V-atom chains are displayed on the left hand side. In the monoclinic M
1 structure shown on the right hand side of that figure the unit cell is doubled and the vanadium atoms in the chains are dimerized as well as horizontally shifted off the octahedral centres.
As has been discussed by Goodenough, these atomic displacements can be nicely translated into energetic shifts and splittings of the relevant V 3
d and closely related O 2
p states. In doing so, Goodenough was indeed able to provide a qualitative understanding of the metal-insulator transition within the framework of an orbital or band picture [
42,
43]. However, this suggestion was soon questioned by Mott and co-workers, favouring the explanation that strong electron correlations play a dominant role for the metal-insulator transition and regarding the structural changes rather as an amendment [
44]. Indeed, first principles calculations as based on density functional theory and the local density or generalized-gradient approximation underrepresented the insulating behaviour and resulted in a slight semimetal-like overlap of the valence and conduction bands [
41,
45]. The situation became even more complicated by the discovery of the additional low-temperature insulating M
2 phase with a slightly different pattern of the structural distortions and long-range magnetic ordering [
46,
47]. Again, for this new phase, density functional theory together with a semi-local approximation failed to fully capture the insulating behaviour [
41].
While for many years these opposing perspectives could not be reconciled, modern hybrid functionals, which are known to accurately describe the electronic and magnetic properties of semiconducting phases of correlated electron materials [
18], offered the realistic chance to successfully account for the opening of a band gap in the semiconducting phases of VO
2 as well as the antiferromagnetic ordering of its M
2 phase and were thus expected to be able to also settle the long-standing dispute on the origin of the metal-insulator transitions of this cornerstone material [
48].
This perspective motivated hybrid-functional calculations for both the metallic high-temperature rutile phase as well as the insulating low-temperature monoclinic M
1 and M
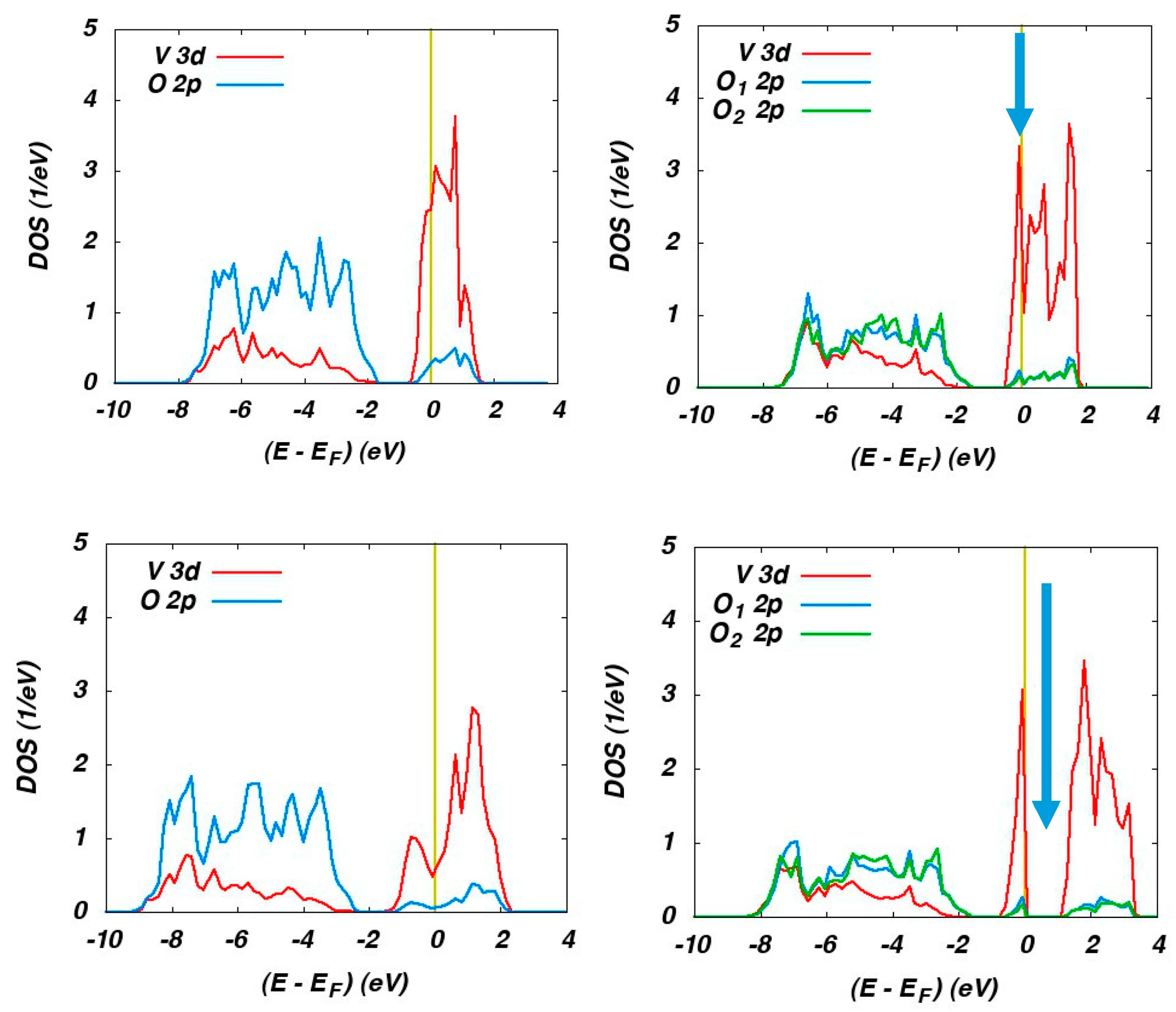
2 phases. The partial densities of states (DOS) calculated for the rutile phase using the semi-local GGA and HSE hybrid functional are displayed on the left-hand side of
Figure 17.
The O 2p and V 3d states (with small admixtures from the respective other type of orbital due to hybridization of these states) are easily recognized well below and at the Fermi energy, respectively. Both functionals result in metallic behaviour in accordance with the experimental findings. However, use of the hybrid functional leads to a much larger width of the V 3d bands in much better agreement with photoemission data.
The corresponding partial DOS for the monoclinic M
1 phase are displayed on the right-hand side of
Figure 17. Again, the O 2
p and V 3
d states are well separated for both functionals. However, GGA and HSE result in completely different behaviour. While the GGA leads to metallic behaviour as clearly indicated by the presence of electronic states at the Fermi energy, the HSE calculations reproduce the observed optical band gap as indicated by the arrow.
Finally, spin-polarized antiferromagnetic calculations were performed for the insulating M2 phase. For this phase, GGA calculations likewise fail to reproduce the insulating behaviour, whereas HSE calculations yield an optical band gap and well localized magnetic moments at the vanadium sites, thus impressively underlining the striking progress offered by hybrid functionals.
Overall, the calculations thus demonstrated for the first time that first-principles calculations using hybrid functionals are entirely capable of describing the electronic and magnetic properties of the insulating phases of VO
2, one of the key representatives of the broad class of electronically correlated materials. Furthermore, the calculations allowed for a deeper understanding of the origin of the metal-insulator transition, which was shown to be intimately connected to the structural changes occurring at the phase transition as suggested by Goodenough and co-workers. Nevertheless, follow-up studies indicated that methods based on density functional theory have difficulties capturing the correct magnetic states of the rutile and M
1 phases. A lot of work has been devoted to this issue as well as to investigations regarding the simultaneous occurrence of the electronic and structural transitions leading from the rutile phase to the monoclinic M
1 phase [
49].

 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}