Enzyme Immobilization on Polymer Membranes: A Quantum and Molecular Mechanics Study


1
Laboratory of Transport Phenomena and Biotechnology, Department of Computer Engineering, Modeling, Electronics and Systems (D.I.M.E.S.), University of Calabria, Ponte P. Bucci, Arcavacata, 87036 Rende (CS), Italy
2
Institute on Membrane Technology, ITM-CNR, Ponte P. Bucci, Arcavacata, 87036 Rende (CS), Cosenza, Italy
*
Author to whom correspondence should be addressed.
Computation 2019, 7(4), 56; https://0-doi-org.brum.beds.ac.uk/10.3390/computation7040056
Submission received: 26 August 2019
/
Revised: 18 September 2019
/
Accepted: 27 September 2019
/
Published: 28 September 2019
(This article belongs to the Special Issue Computational Design and Characterization of Membranes, Membrane Materials and Membrane Separation Processes)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Adsorption of the phosphotriesterase on a polysulfone membrane surface was investigated in this paper through a double-scale computational approach. Surface charges of the enzyme, as well as membrane, were calculated at sub and nanoscale while protein adsorption was simulated at larger scale. Adsorption energies were calculated as a function of the enzyme–surface distance, and for each distance, several protein rotations were tested to find the most stable orientations of the macromolecule. The results of this model were useful in obtaining information about the adhesion of the enzyme and to give indications on the orientations of its binding site. Adsorption energies agreed with the literature data. Furthermore, the binding site of the immobilized phosphotriesterase was less accessible with respect to native enzymes due to the steric hindrance of the polymer surface; thus, a reduction of its efficiency is expected. The proposed methodology made use of fundamental quantities, calculated without resorting to adjustable or empirical parameters, providing basic outputs useful for ascertaining enzymatic catalysis rate.
1. Introduction
Nowadays, significant growth in research activities in multiscale modeling is observed, with applications in many areas, including material sciences, fluid mechanics, chemistry, and biology. It is widely recognized that multiscale techniques will become an essential part of computational science and engineering. In this frame, however, the crucial point is represented by the reliable calculation of some key parameters that are associated with smaller scales [1,2].
In recent years, enzymatic productivity has been rapidly increasing through the improvement of genetic engineering, microbial cultivation technologies, and wild type strain screening technology, together with the understanding of enzymatic biosynthesis mechanisms [3]. This advance is providing different kinds of enzymes exhibiting improved activities, which is leading to a massive use of enzymes in industrial processes. Enzymes play key roles in numerous biotechnology products and processes that are commonly encountered in the production of food and beverages, cleaning supplies, clothing, paper products, pharmaceuticals, and monitoring devices [4]. At present, the most frequently used enzymes in biotechnology are hydrolases, which catalyze molecular breakdown. Enzymatic chiral selectivity has been exploited to prepare enantiomerically pure pharmaceuticals, agro-chemicals, chemical feedstocks, and food additives [5]. Most of the currently used industrial enzymes are hydrophilic, being used for the degradation of various natural substances. Proteases remain the dominant enzyme type for their extensive use in the detergent and dairy industries. In this frame, Phosphotriesterase (PTE) has also been used for the hydrolysis of common pesticides, such as glyphosate, due to the high efficiency of glyphosate abatement exhibited by this enzyme [6].
Immobilization of enzymes on the external or internal (pore) membrane surfaces is widely used in biotechnology industries [7,8], where immobilization is generally achieved by establishing non-covalent interactions between protein and polymeric surfaces without chemical modifications of membranes and enzymes [9]. As a result, non-covalent adsorption is an interesting research topic, and computational methodologies able to optimize and control this process on a larger scale [10,11] are highly desirable. However, due to some process limitations, several aspects need to be improved to increase the immobilized enzyme activity and re-usability. In this framework, it is essential to formulate advanced and reliable computational methodologies to optimize the enzyme immobilization without resorting to adjustable or empirical parameters.
Advanced modeling aimed at describing the enzyme adsorption on polymeric membranes surface was developed in this work. In particular, the immobilization of phosphotriesterase (PTE) on polysulfone (PSU) surface was studied at a fixed pH via a quantum and molecular mechanics approach (QM/MM) without resorting to adjustable parameters. This approach achieved fundamental results, such as adsorption energies and enzyme orientation on the polymer surface. The absorption energy is an important property to predict the enzyme adhesion on the membrane surface and hence, to predict possible releases of the macromolecule in solution. Moreover, the geometries of the immobilized protein, i.e., its orientations, can be used in mesoscale modeling to describe the diffusion of the substrate towards the catalytic site to check possible steric hindrance of the surface.
The proposed methodology was implemented to provide an innovative tool starting from the calculation of sub-nano quantities which can be used to develop advanced membrane bioreactors. In particular, this model was developed, in the frame of previous works [12,13], to provide basic outputs useful for ascertaining enzymatic catalysis rates for which both the molecular orientation and the adsorption energy are fundamental. In this way, a series of accurate simulations can be planned for achieving true optimization of PTE immobilization before performing experimental tests.
2. Computational Approach
2.1. Quantum Calculations
The adsorption of macromolecules on polymer surfaces is mainly controlled by electrostatic and hydrophobic interactions, as well as by the hydrogen bond at very short distances; thus, the surface charges of interacting systems are necessary to accurately evaluate short- and long-range electrostatic contributions. Surface charges of proteins largely depend on the nature of the external amino acids and conditions, such as pH, solvent, and ionic strength. In general, ab initio (quantum mechanics)-based methods are usually used for the calculation of partial charges considering the effect of specific conditions and avoiding the use of the classic force fields. However, the calculation of partial charges via quantum approaches is time-consuming, especially for proteins formed by thousands of atoms. For this reason, herein, a contact surface of the PTE was first determined through a home-made algorithm and using the crystallographic structure of the enzyme at pH = 7 [14]. The electrostatic charges on large fragments of external amino acids were calculated in the frame of density functional theory (DFT) using the Löwdin method as implemented in NWChem [15]. All quantum calculations were performed by using the B3LYP [16] hybrid functional and double-ζ basis set (6–31G*) for each atom of the enzyme and polysulfone surfaces. The thresholds for the energy convergence in the self-consistent field procedure and the root-mean-square of the electron density were set to 10−6 (au) and to 2 × 10−5 (au), respectively. The Löwdin method was chosen for its low dependence on the orbitals basis set, and it was also used to calculate the distributions of charges on the PSU model surface. It is important to emphasize that once the Cartesian coordinates of the atoms on the enzyme surface were determined, these were grouped in large fragments containing more than 500 atoms; then DFT calculations of the point charges were performed using these fragments and not on the individual amino acids. In this way, the protonated and deprotonated forms of the external amino acids can be evaluated more accurately via an ab initio approach. A dense polymeric surface was obtained through a molecular dynamics simulated annealing, as described in detail in [12], hence the atomic coordinates of the equilibrated surface model were used for the calculation of the partial charges at quantum mechanics level through the same computational approach used for the enzyme charges calculation. Nevertheless, in case of the surface charges of the polymer, the continuum conductor-like screening model (COSMO) [17] was used with a relative dielectric constant equal to 4 to consider the molecules of water in close contact with the polymer surface, i.e., confined. The equilibrated PSU unit cell with L = 7.2 nm, was translated in the X and Y directions obtaining 3 cells for each axis so that the QM/MM calculations did not suffer from the edges effect.
The pH was also considered in the calculations of PTE surface charges. Specifically, the protonated form of the external residues forming a fragment depends on the difference between pH and pKr, with Kr equal to the equilibrium constant of the amino acid’s functional group. For the deprotonated form is predominant, whereas protonated forms result in the most abundant for . However, for specific pH values, both forms had to be considered. Thus, the ratio between the deprotonated [D] and protonated [P] external amino acid was calculated at pH = 7 using the following relationship:
The external residues of the PTE were then protonated or deprotonated according to the ratios obtained from Relationship (1). It is worth noting that the pH value is closely related to the structure of the enzyme used to carry out the DFT calculations; thus, simulations at different pH would require the crystallographic structure of the protein corresponding to that value.
2.2. Molecular Mechanics Optimizations
Following the approach used in the previous work and by Mikael Lund and Bo Jönsson [12,18], the potential energy, characterizing the interaction between proteins and polymer surface, can be divided into three different parts.
The term takes into account the repulsion of electron clouds through hard-spheres [19]. The Coulomb interaction was considered in defined by the atomic surface charges, qi and qj, of the protein and polymer surface, respectively, and evaluated through the DFT calculations. εr and ε0 are the relative and vacuum dielectric permittivity, respectively; in agreement with the value used for the calculation of the partial charges on polysulfone, εr = 4.0 was used in Relationship (4) for very close interacting systems and εr = 78.2 for weakly interacting systems. Finally, to evaluate the hydrophobic (van der Waals) interaction, the Lennard–Jones 12–6 potential was used; σii and εii parameters were taken from [20,21].
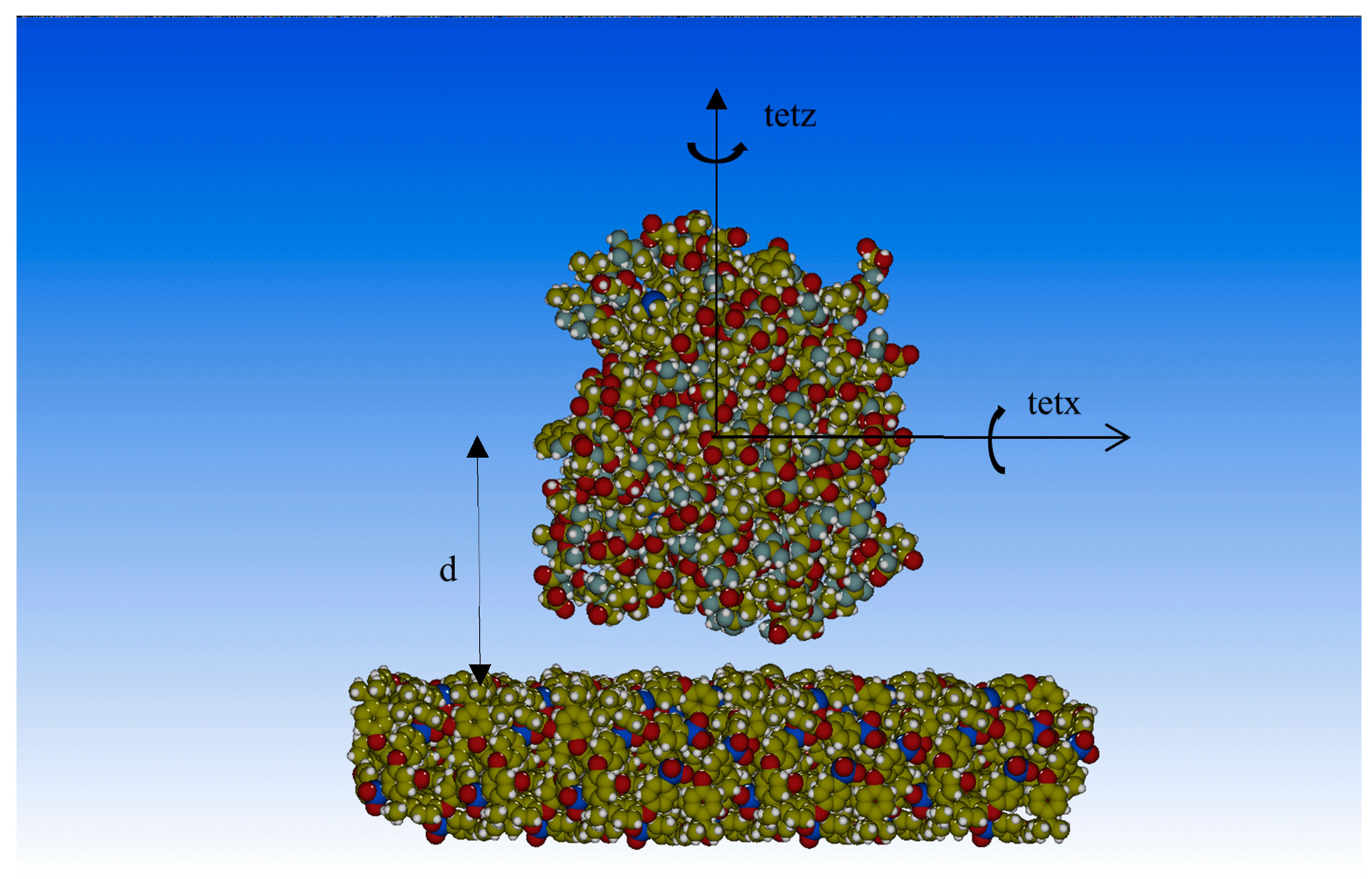
Bespoke MATLAB [22] functions were implemented to calculate the total interaction energy according to Equations (2)–(5) in which a 3D grid-type calculation was performed. The radial points refer to PTE-PSU distance, d, along the Z-axis perpendicular to the polymer surface, as shown in Figure 1, while the angular points are related to protein rotations with respect to Z and X (tetz, tetx) (Figure 1).
Potential energy was obtained as a function of the protein–surface distance (d), and global and local minima were characterized. The d values ranged between 24 Å and 60 Å, and a step of 1 Å was used, while, for each distance, 16 rotations of macromolecule were evaluated (Figure 1).
3. Results and Discussion
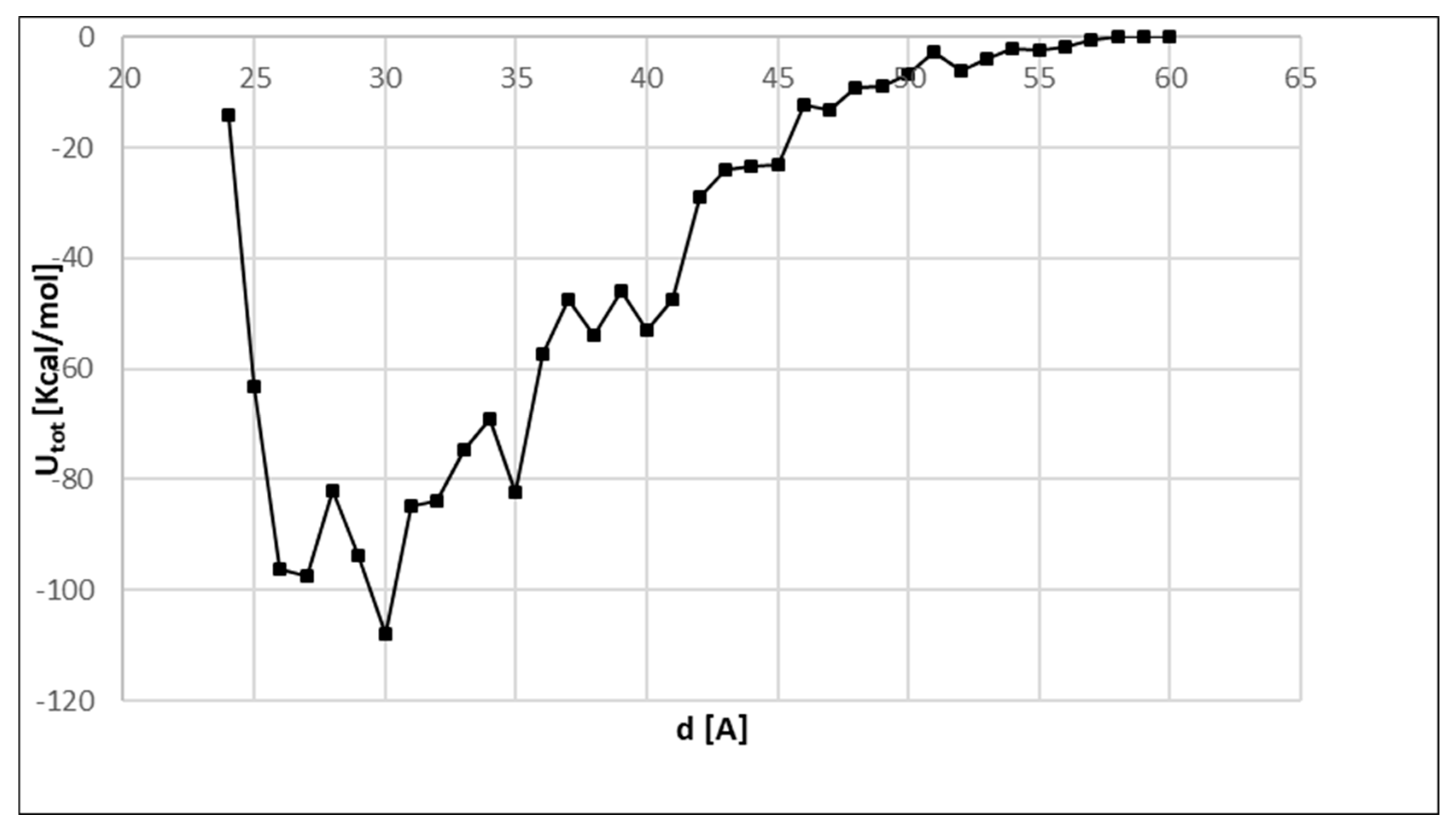
The interaction energy as a function of the enzyme–surface distance is shown in Figure 2, and as expected, various local minima and a global minimum were found. The potential energy profile showed a complete overview of the interaction energy from very close distances to weakly interacting systems or non-interacting systems, i.e., the enzyme in solution at 60 Å.
A global minimum was found at a distance of 30 Å from PSU surface with a total energy equal to −107.96 Kcal/mol. The nearest local minima were found at d = 27 Å and 35 Å with interactions energies of −97.57 and −82.44 Kcal/mol, respectively. Other local minima were found further from the surface with interaction energies considerably higher than −107.96 Kcal/mol. Thus, they were not considered in the analysis of the enzyme orientation since jumps for thermal vibrations in these minima are difficult, at room temperature. Instead, the energy differences between the global and the two nearest minima were 10 Kcal/mol and 25 Kcal/mol. Hence, considering a distribution of the macromolecules among these minima, an adsorption energy ranging between −82.44 Kcal/mol and −107.96 Kcal/mol is expected. Average solvation interaction energies and total average interaction energies were evaluated for five proteins on five polymer surfaces, considering 1633 different protein orientations and using log P of each amino acid residue belonging to the protein fragment and repetitive units of each polymeric surface [23]. The total average interaction energies ranged between −111.09 kcal/mol (for hydrophobic surface) and −39.53 kcal/mol (for hydrophilic surface) in good agreement with the values found herein. Moreover, the energies associated with the adsorption of bovine serum albumin on hydrophilic (polystyrene) and hydrophobic surfaces were evaluated through atomistic simulations [24]. The values were of the same order of magnitude of the global minimum; however, being the polystyrene more hydrophobic than polysulfone, larger energies were found [24]. Nevertheless, being able to reproduce the order of magnitude of the adsorption energy [23,24,25,26] through an ab initio quantum method, this can be considered a satisfactory result. Furthermore, this QM/MM model can be used in comparative analyses.
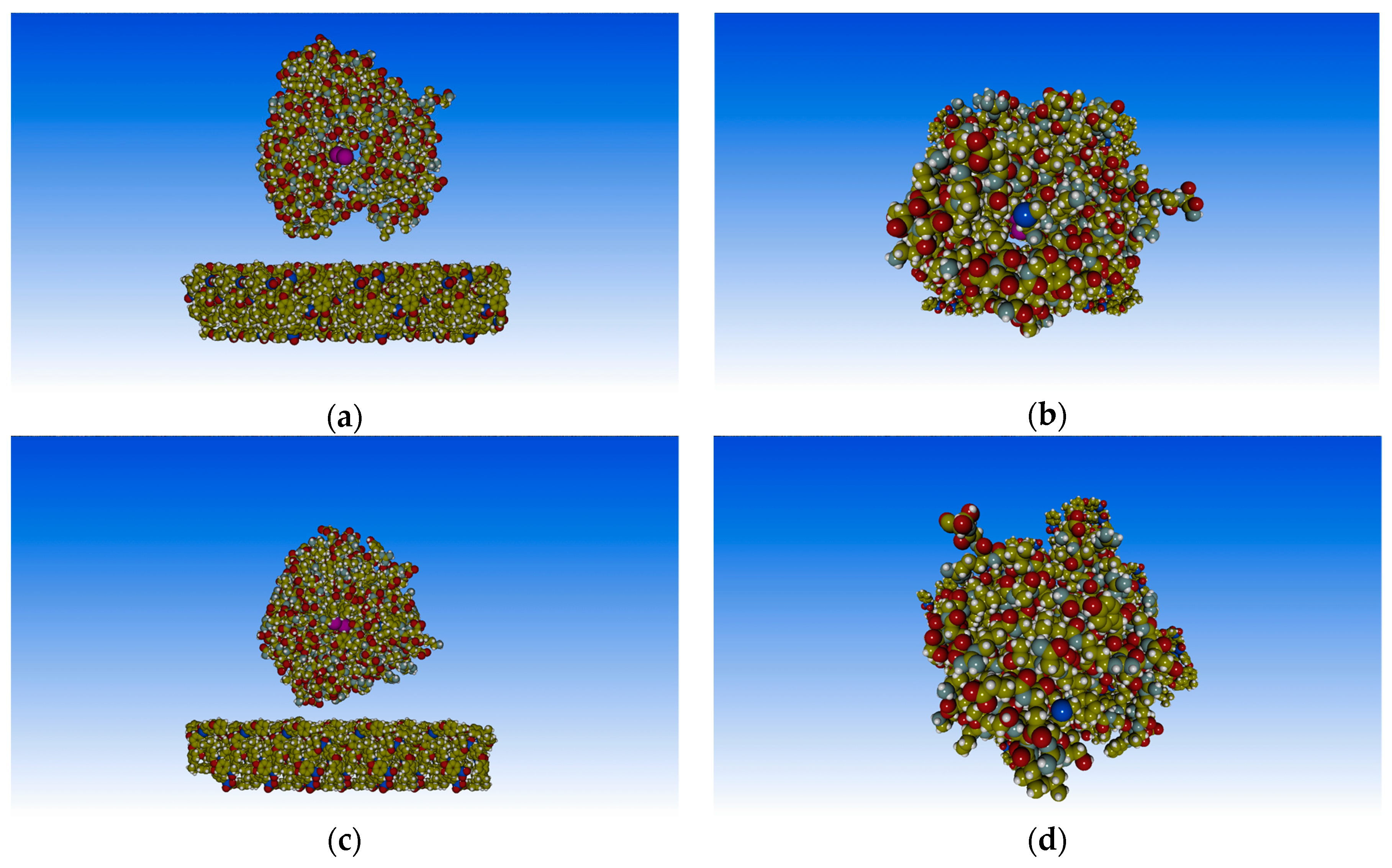
The orientation of the PTE on the PSU surface, corresponding to the global minimum, is shown in Figure 3. In particular, the orientation of the contact surface, used in the QM/MM calculations, is shown in Figure 3a and b, while the complete structure of the enzyme in the same orientation is reconstructed in Figure 3c and d. One of the goals of this work was to elucidate the stable orientations of the enzyme on the PSU membrane surfaces, then to provide supramolecular structures (enzyme–surface) for modeling the substrate diffusion at higher scale. Nevertheless, the supramolecular structures can be used to investigate the steric hindrance of the catalytic site due to the polymeric surface. This can be achieved by analyzing the position of the two zinc atoms present in the catalytic site of the PTE which are responsible for the glyphosate hydrolysis. In addition, the binding site, close to the enzyme surface and responsible for the selective adsorption of the substrate, can be monitored. Hence, by analyzing the position of these atoms with respect to the membrane surface, the steric hindrance can be predicted.
Comparing Figure 3a (side view) and 3b (top view), it is clear that the two zinc atoms are difficult to reach from the bulk (solution) side. On the contrary, these atoms were visible from the side of the supramolecular structure, which means that the substrate must arrive from this direction to reach the binding site. This result is clearer when the complete structure of the enzyme is shown in the same orientation, as in Figure 3c and d. In particular, the zinc atoms are still visible from the side view, Figure 3c, but when the supramolecular structure is rotated to show the top view (bulk side), these atoms are no longer visible (Figure 3d). In solution, the substrate molecules could easily reach the binding site without barriers because the enzyme was free. Instead, for adsorbed enzymes, the steric hindrance of the surface made this difficult.
Starting from the position corresponding to the global minimum, the enzyme was rotated by 90 degrees around the X-axis (tetx = 90°, Figure 1) to make the zinc atoms more accessible from the bulk side, as shown in Figure 4a. This figure shows that the two atoms of zinc were more visible with respect to Figure 3b, which refers to the enzyme at the same distance but not rotated. Unfortunately, the energy cost for this rotation is 83.09 Kcal/mol. This energy gap is too high to allow this spin at room temperature; thus, the enzyme fails to rotate and direct the binding site toward the bulk. As shown in Figure 4b (side view of the rotated enzyme), a rotation of the protein around the X-axis greater than 90 degrees (tetx > 90) would make the metal atoms more accessible for the substrate from the solution. However, in this case, the protruding amino acid residue would touch the surface of the polysulfone.
In regard to the orientations of the PTE associated with the local minima, the same conclusion can be drawn, as shown in Figure 5. For both minima, the two atoms of zinc were clearly visible from the side of the supramolecular structure, whereas they were difficult to reach for the substrate from the bulk side. Moreover, concerning the rotations of the enzyme around the X-axis to make metal atoms more accessible, i.e., tetx = 90 or tetx > 90, results similar to those found for the PTE rotations, adsorbed in the global minimum, were obtained.
In summary, this modeling showed that the binding site of the adsorbed PTE was less accessible with respect to the native enzyme due to the steric hindrance of the polymer surface. As a result, the adsorbed enzyme should reduce its activity; i.e., the enzyme’s efficiency should be reduced since the substrate should reach the binding site laterally and adjacent adsorbed enzymes could hinder its access. This depends on the enzyme–enzyme equilibrium distance and protein concentration.
The model still fails to incorporate various aspects, but being a first step towards developing a more comprehensive holistic approach, this work provides a good starting ground. Finally, the analysis of these aspects will be the subject of subsequent works. Nevertheless, the modeling showed that useful information about the adsorption of proteins on surfaces can be obtained from ab initio DFT-based calculations avoiding the use of empirical or adjustable parameters.
4. Conclusions
A model based on quantum and molecular mechanics calculations was proposed in this work to simulate the enzyme adsorption on the polysulfone membrane surface. The modeling aims to achieve the following objectives: the calculation of the enzyme–surface interaction energies without resorting to adjustable or empirical parameters and the orientation of the absorbed enzymes on the membrane surface to predict possible steric hindrance for the substrate. The surface charges of the PTE were calculated by an accurate the quantum mechanics approach at pH = 7, while a PSU surface model was defined by a molecular dynamics simulated annealing. Electrostatic charges of the polymer surface were also calculated using the same quantum approach. The interaction energy between enzyme and membrane surface was evaluated as a function of protein–surface distance considering several rotations of the PTE for each distance. The final result was the calculation of potential energy profiles as well as macromolecular structures that can be used as inputs in large scale model development. The computed interaction energies are in good agreement with adsorption free energies reported in the literature; this means that the considered enzyme can be effectively adsorbed on PSU. However, accurate analysis of the protein orientations suggested that the binding site of the immobilized enzyme was less accessible with respect to the pristine PTE due to the steric hindrance of the polymer surface; thus, a reduction of the enzyme efficiency was expected. The proposed methodology made use of fundamental quantities and it was designed to provide basic outputs useful for ascertaining enzymatic catalysis rates, i.e., kinetic proprieties from immobilization features, important for designing enzymatic bioreactors.
Author Contributions
Conceptualization, G.D.L.; methodology, G.D.L.; software, F.P.; validation, S.C. (S. Curcio); formal analysis, S.C. (S. Curcio) and G..D.L.; investigation, G..D.L., S.C. (S. Curcio), and F.P.; data curation, G.D.L., and F.P.; writing—original draft preparation, G.D.L., S.C. (S. Curcio); writing—review and editing, G..D.L. and S.C. (S. Curcio), and S.C. (Sudip Chakraborty); supervision, S.C. (S. Curcio) and S.C. (Sudip Chakraborty).
Funding
This research received no external funding.
Acknowledgments
The authors are grateful to CINECA for the use of High-Performance Computers.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Weinan, E.; Li, X.; Vanden-Eijnden, E. Some Recent Progress in Multiscale Modeling; Springer: Berlin/Heidelberg, Germany, 2004. [Google Scholar]
- Horstemeyer, M. Multiscale Modeling: A review. In Practical Aspects of Computational Chemistry; Springer: New York, NY, USA, 2009; pp. 87–135. [Google Scholar]
- Parekh, S.; Vinci, V.A.; Strobel, R.J. Improvement of microbial strains and fermentation processes. Appl. Microbiol. Biotechnol. 2000, 54, 287–301. [Google Scholar] [CrossRef] [PubMed]
- Gurung, N.; Ray, S.; Bose, S.; Rai, V. A broader view: Microbial enzymes and their relevance in industries, medicine, and beyond. BioMed Res. Int. 2013, 2013, 329121. [Google Scholar] [CrossRef] [PubMed]
- Underkofler, L.A.; Barton, R.R.; Rennert, S.S. Production of microbial enzymes and their applications. Appl. Microbiol. 1958, 6, 212–221. [Google Scholar] [PubMed]
- Raushel, F.M.; Holden, H.M. Phosphotriesterase: An enzyme in search of its natural substrate. Adv. Enzymol. Relat. Areas Mol. Biol. 2000, 74, 51–93. [Google Scholar] [PubMed]
- Nguyen, H.; Kim, M. An overview of techniques in enzyme immobilization. Appl. Sci. Converg. Technol. 2017, 26, 157–163. [Google Scholar]
- DiCosimo, R.; McAuliffe, J.; Poulose, A.J.; Bohlmann, G. Industrial use of immobilized enzymes. Chem. Soc. Rev. 2013, 42, 6437–6474. [Google Scholar] [CrossRef]
- Datta, S.; Christena, L.R.; Rajaram, Y.R.S. Enzyme immobilization: An overview on techniques and support materials. 3 Biotech 2013, 3, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kazemi, P.; Renka, R.J. A Levenberg–Marquardt method based on Sobolev gradients. Nonlinear Anal. 2012, 16, 6170–6179. [Google Scholar] [CrossRef]
- Han, L.; Neumann, M. Effect of dimensionality on the Nelder–Mead simplex method. Optim. Methods Softw. 2006, 21, 1–16. [Google Scholar] [CrossRef]
- Curcio, S.; Petrosino, F.; Morrone, M.; De Luca, G. Interactions between proteins and the membrane surface in multiscale modeling of organic fouling. J. Chem. Inf. Model. 2018, 58, 1815–1827. [Google Scholar] [CrossRef]
- De Luca, G.; Bisignano, F.; Paone, F.; Curcio, S. Multi-scale modeling of protein fouling in ultrafiltration process. J. Membr. Sci. 2014, 452, 400–414. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, H.; Liu, X.; Zhou, W.; Rao, Z. The crystal structure of the phosphotriesterase from M.tuberculosis, another member of phosphotriesterase-like lactonase family. Biochem. Biophys. Res. Commun. 2019, 510, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Valiev, M.; Bylaska, E.J.; Govind, N.; Kowalski, K.; Straatsma, T.P.; Van Dam, H.J.J.; Wang, D.; Nieplocha, J.; Apra, E.; Windus, T.L.; et al. NWChem: A comprehensive and scalable open-source solution for large scale molecular simulations. Comput. Phys. Commun. 2010, 181, 1477–1489. [Google Scholar] [CrossRef] [Green Version]
- Malki, Z.E.; Bouzzine, S.M.; Bejjit, L.; Haddad, M.; Hamidi, M.; Bouachrine, M. Density functional theory [B3LYP/6-311G(d,p)] study of a new copolymer based on carbazole and (3,4-Ethylenedioxythiophene) in their aromatic and polaronic states. J. Appl. Polym. Sci. 2011, 122, 3351–3360. [Google Scholar] [CrossRef]
- Klamt, A.; Schüürmann, G. COSMO: A new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc. Perkin Trans. 2 1993, 2, 799–805. [Google Scholar] [CrossRef]
- Lund, M.; Jönsson, B. A mesoscopic model for Protein-Protein interactions in solution. Biophys. J. 2003, 85, 2940–2947. [Google Scholar] [CrossRef]
- Zhou, A.Q.; O’Hern, C.S.; Regan, L. The power of hard-sphere models: Explaining side-chain dihedral angle distributions of thr and val. Biophys. J. 2012, 102, 2345–2352. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Huey, R.; Olson, A.J. Distributed automated docking of flexible ligands to proteins: Parallel applications of AutoDock 2.4. J. Comput.Aided Mol. Des. 1996, 10, 293–304. [Google Scholar] [CrossRef]
- Van der Waals Potential Energy. Available online: http://www.csb.yale.edu/userguides/datamanip/autodock/html/Using_AutoDock_305.a.html (accessed on 17 July 2019).
- Attaway, S. Matlab: A Practical Introduction to Programming and Problem Solving; Butterworth-Heinemann: Oxford, UK, 2009; ISBN 978-0-08-094325-1. [Google Scholar]
- Lu, D.R.; Lee, S.J.; Park, K. Calculation of solvation interaction energies for protein adsorption on polymer surfaces. J. Biomater. Sci. Polym. Ed. 1992, 3, 127–147. [Google Scholar] [CrossRef]
- Jeyachandran, Y.L.; Mielczarski, E.; Rai, B.; Mielczarski, J.A. Quantitative and qualitative evaluation of adsorption/desorption of bovine serum albumin on hydrophilic and hydrophobic surfaces. Langmuir 2009, 25, 11614–11620. [Google Scholar] [CrossRef]
- Salgın, S.; Takaç, S.; Özdamar, T.H. Adsorption of bovine serum albumin on polyether sulfone ultrafiltration membranes: Determination of interfacial interaction energy and effective diffusion coefficient. J. Membr. Sci. 2006, 278, 251–260. [Google Scholar] [CrossRef]
- May, S.; Harries, D.; Ben-Shaul, A. Lipid Demixing and Protein-Protein interactions in the adsorption of charged proteins on mixed membranes. Biophys. J. 2000, 79, 1747–1760. [Google Scholar] [CrossRef]
Figure 1.
Phosphotriesterase (PTE) on polysulfone surface, d = protein–surface distance, tetz and tetx protein rotations around the Z and X axis, respectively, used for the calculation of the interaction energy.
Figure 1.
Phosphotriesterase (PTE) on polysulfone surface, d = protein–surface distance, tetz and tetx protein rotations around the Z and X axis, respectively, used for the calculation of the interaction energy.

Figure 2.
Enzyme–polysulfone (PSU) interaction energy, Utot, versus enzyme–surface distance obtained by quantum and molecular mechanics (QM/MM) calculations.
Figure 2.
Enzyme–polysulfone (PSU) interaction energy, Utot, versus enzyme–surface distance obtained by quantum and molecular mechanics (QM/MM) calculations.

Figure 3.
Adsorbed PTE on the PSU model surface in the global minimum (−107.96 Kcal/mol at d = 30 Å). (a) side view and (b) top view of the enzyme surface, respectively; (c) side view and (d) top view of the full enzyme structure in the same orientation, respectively. Zinc atoms were shown in fuchsia.
Figure 3.
Adsorbed PTE on the PSU model surface in the global minimum (−107.96 Kcal/mol at d = 30 Å). (a) side view and (b) top view of the enzyme surface, respectively; (c) side view and (d) top view of the full enzyme structure in the same orientation, respectively. Zinc atoms were shown in fuchsia.

Figure 4.
Adsorbed PTE on the PSU surface at d = 30 Å but rotated by 90 degrees around the X-axis (Figure 1). (a) Top view and (b) side view of the enzyme surface. Zinc atoms were shown in fuchsia.
Figure 4.
Adsorbed PTE on the PSU surface at d = 30 Å but rotated by 90 degrees around the X-axis (Figure 1). (a) Top view and (b) side view of the enzyme surface. Zinc atoms were shown in fuchsia.

Figure 5.
Adsorbed PTE on the PSU model in the local minima. (a) Side view and (b) top view of the PTE-PSU structure corresponding to minimum with interaction energies of −97.57 Kcal/mol at d = 27 Å, respectively; (c) side view and (d) top view of the PTE-PSU structure corresponding to minimum with interaction energies of −82.44 Kcal/mol at d = 35 Å, respectively. Zinc atoms were shown in fuchsia.
Figure 5.
Adsorbed PTE on the PSU model in the local minima. (a) Side view and (b) top view of the PTE-PSU structure corresponding to minimum with interaction energies of −97.57 Kcal/mol at d = 27 Å, respectively; (c) side view and (d) top view of the PTE-PSU structure corresponding to minimum with interaction energies of −82.44 Kcal/mol at d = 35 Å, respectively. Zinc atoms were shown in fuchsia.

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Petrosino, F.; Curcio, S.; Chakraborty, S.; De Luca, G. Enzyme Immobilization on Polymer Membranes: A Quantum and Molecular Mechanics Study. Computation 2019, 7, 56. https://0-doi-org.brum.beds.ac.uk/10.3390/computation7040056
AMA Style
Petrosino F, Curcio S, Chakraborty S, De Luca G. Enzyme Immobilization on Polymer Membranes: A Quantum and Molecular Mechanics Study. Computation. 2019; 7(4):56. https://0-doi-org.brum.beds.ac.uk/10.3390/computation7040056
Chicago/Turabian StylePetrosino, Francesco, Stefano Curcio, Sudip Chakraborty, and Giorgio De Luca. 2019. "Enzyme Immobilization on Polymer Membranes: A Quantum and Molecular Mechanics Study" Computation 7, no. 4: 56. https://0-doi-org.brum.beds.ac.uk/10.3390/computation7040056
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.