A DFT Study on Structure and Electronic Properties of BN Nanostructures Adsorbed with Dopamine


1
Golestan Rheumatology Research Center, Golestan University of Medical Science, Gorgan 4934174515, Iran
2
Department of Chemistry, Azadshahr Branch, Islamic Azad University, Azadshahr, Golestan 4961789985, Iran
*
Author to whom correspondence should be addressed.
Computation 2019, 7(4), 61; https://0-doi-org.brum.beds.ac.uk/10.3390/computation7040061
Submission received: 31 August 2019
/
Revised: 22 October 2019
/
Accepted: 26 October 2019
/
Published: 1 November 2019
(This article belongs to the Special Issue Computational Studies of Adsorption on Nanoparticles and 2D-Materials)
Abstract
:Density functional theory calculations were carried out to investigate the adsorption behaviors of dopamine (DPM) on the BN nanostructures in gas and solvent phases. Our results revealed that the adsorption of DPM on BN nano-cages was stronger than other BN nanotubes. It was found that the adsorption of two DPM (−1.30 eV) upon B12N12 was weaker than those of a single DPM (−1.41 eV). The Ga-doped B12N12 had better conditions for the detection of DPM than that of the Al-doped B12N12 nano-cage. The solvation effects for the most stable systems were calculated which showed that it had positive impacts upon the adsorption behavior of the applied systems than those studied in gas phase. The available results are expected to provide a useful guidance for the adsorption of DPM and generation of the new hybrid compounds.
1. Introduction
Based on recent reports, nanotubes, nanowires, fullerenes, and nanoparticles are considered as bioconjugated nanostructure materials that have emerged as a new class of materials for biosensing and medical diagnostics implementation [1,2,3,4,5,6,7,8,9]. Carbon nanostructures (C24, C32, C60, and single-wall carbon nanotubes (SWCNT)) are used as nanovectors for drug delivery of anticancer and antitubercular compounds for tremendous medicinal applications [10,11,12]. Recently, many novel properties in several fields of life science including gas storage devices, biological applications, chemical sensors, and drug delivery systems have been reported [13,14,15]. Moreover, nanotubes containing atoms of groups III and V of the periodic table of elements are considered to be a suitable case as a replacement of CNTs. Boron nitride nanotubes (BNNTs) have been comprehensively investigated owing to their structural similarity with carbon nanotubes, holding promises for development in nano-bio systems. Subsequently, numerous experimental and theoretical investigations have been dedicated to considering the electronic properties and different structures of BNNTs. Besides, the BNNTs are considered to be a polar material due to the slight positive charges of boron (B) atoms and the slight negative charges of nitrogen (N) atoms; while there is no polarity in CNTs, this could be a reason to apply BNNTs in electronic and mechanical devices [16,17,18,19]. Dopamine (DPM) or 3,4-dihydroxyphenyl ethylamine exists in the mammalian central nervous system and is an important neurotransmitter, as it plays a pivotal role in communication with neurons [20,21]. Its electrochemical behavior has prominent impacts on its physiological functions which is an essential factor in the diagnosis of some diseases in clinical medicine [22]. For example, the adsorption of isoniazid drug molecule toward the electronic and structural properties of (5,5) and (10,0) BN nanotubes were studied by Deka et al. [23]. They reported that the interaction of drug molecules with (10,0) BNNT is thermodynamically notable in comparison with (5,5) BNNT. Anota and co-workers have shown the adsorption of the biopolymer chitosan upon (5,0) and (5,5) BN nanotubes [24]. It is found that the chitosan adsorption from its amine head attached to the B atom of the (5,0) BN nanotube is stronger than the (5,5) BN nanotube. Pandey and co-workers have an investigation focusing upon the interaction energies between tryptophan (Trp), aspartic acid (Asp), and arginine (Arg) with BNNTs. They inferred that the stabilization of the bioconjugated complexes are primarily indicated for electrostatic interactions. They also found that BNNTs have better sensitivity toward Asp and Arg in comparison to CNTs [25]. They have also performed another study concentrating upon the adsorption phenomena of adenine (A), guanine (G), cytosine (C), thymine (T), and uracil (U) on single-wall boron nitride nanotubes (SWBNNT), finding that the G-SWBNNT system has better adsorption energy than the other adsorption configurations owing to the hybridization of the molecular orbitals of G and the BNNT [26]. In the previous report, we explored the adsorption of adenine, uracil, and cytosine molecules over AlN and BN nano-cages using density functional theory (DFT) calculations [27]. Also, we studied the structure and electronic properties of B12N12 and Al12N12 nano-cages in reaction with caffeine and nicotine molecules, C6H5OH, SCN−, and OCN− by density functional theory [28,29]. The major goal of this study was to evaluate the most stable adsorption sites between the pure or metal-doped BN nanostructures and the DPM using DFT calculations. We also studied the structural, electronic properties, and vibration spectroscopy of the most stable configurations using the B3LYP method and 6-311G** standard basis set.
2. Materials and Methods
Full optimization, natural bonding orbital (NBO) analysis, density of states (DOS), quantum molecular descriptors (QMDs), and frontier molecular orbital (FMO) calculations were performed on the DPM/BN nanostructures systems applying DFT using the Gaussian 09 suite program [30]. The 6-311G** standard basis set was implemented for optimization through spin-unrestricted B3LYP method augmented with an empirical dispersion term (B3LYP-D) [31]. No symmetry constraint was imposed during the geometry relaxation. Basis set superposition error (BSSE) for the adsorption energies were corrected by using the counterpoise method. The B3LYP density functional has been previously shown to reproduce experimental proprieties and it has been commonly used for nanotube structures [32,33]. The effect of solvent (water) upon the adsorption of DPM interacting with B12N12 and B16N16 nano-cages was studied by using the B3LYP method with 6-311G** basis set. Solvation effects were included by using the polarized continuum model (PCM). A dielectric constant of 78.4 was used corresponding to that for water as the solvent [34]. By definition, a negative value of adsorption energy is referred to exothermic process. All frequency calculations were carried out using a hybrid DFT-B3LYP method with 6-311G** standard basis set. We introduced the adsorption energies (BE) of our systems as below:
where E cluster/Molecule is the total energy of the complex, E cluster is the energy of pristine clusters, and E Molecule is the total energy of a molecule. The quantum molecular descriptors [35,36] were computed by the following equations:
where I (−EHOMO) is the energy of the Fermi level and A (−ELUMO) is the first given value of the conduction band. Electronegativity (χ) is determined as the negative of chemical potential (µ), as follows: χ = −µ. In addition, the chemical hardness (η) can be determined using the Koopmans’ theorem. I (−EHOMO) is the ionization potential and A (−ELUMO) the electron affinity of the molecule.
BE = E cluster /Molecule − (E Molecule + E cluster) + EBSSE,
I = −E HOMO and A = −E LUMO,
η = (I − A)/2,
µ = −χ = −(I + A)/2,
S = 1/2η,
ω = (µ2/2 η),
3. Results and Discussion
3.1. Characteristics of DPM
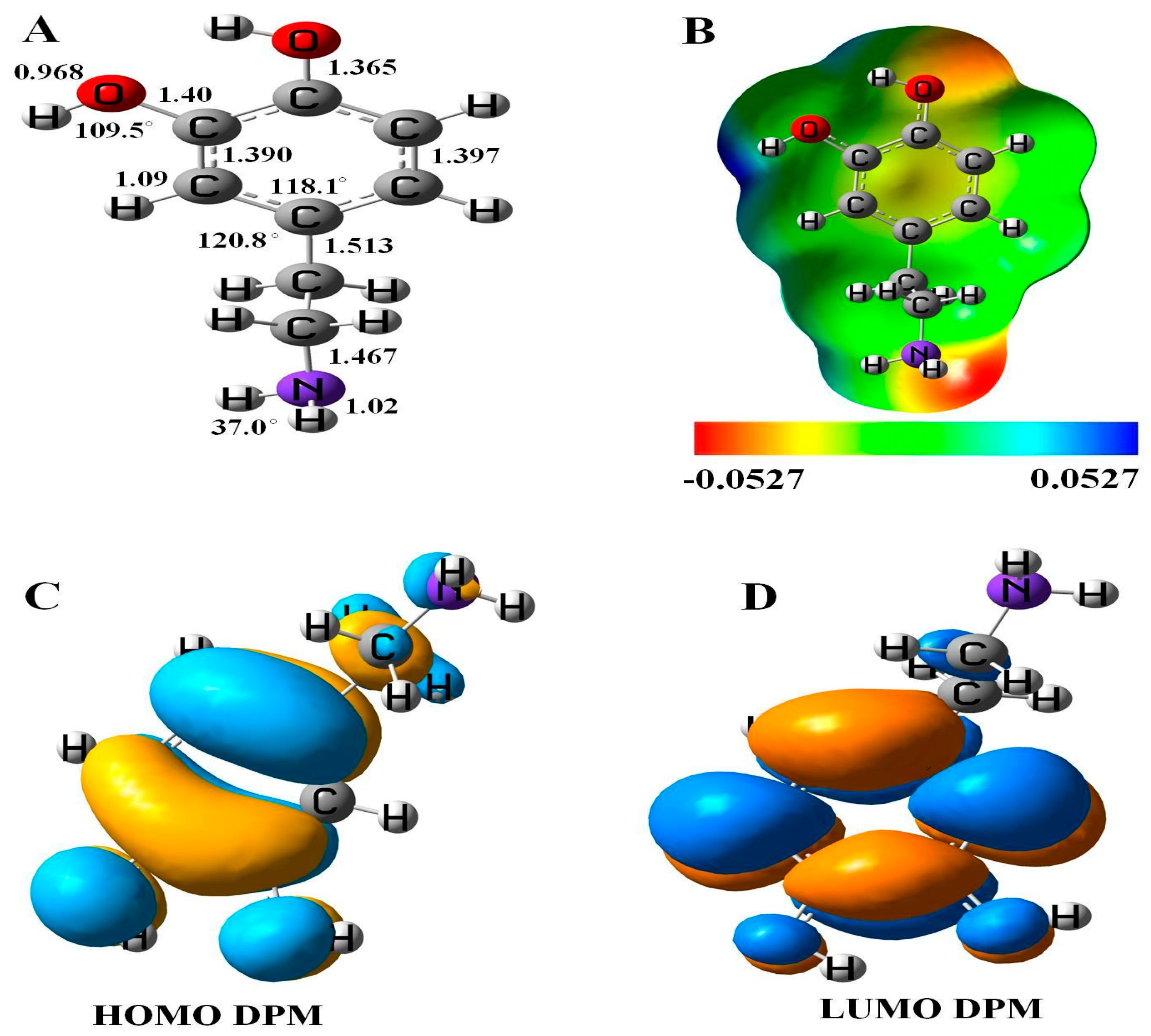
The relaxed geometry, molecular electrostatic potential surface (MEP), and frontier molecular orbital (FMO) of DPM was studied (see Figure 1). The bond lengths and angles between the constituent atoms of DPM calculated with DFT studies were consistent with previous reports [37]. The highest occupied molecular orbital (HOMO) of DPM was located upon the C–C atoms of ring and the electronegative nitrogen and oxygen atoms of a molecule with the energy of −5.49 eV, while the lowest unoccupied molecular orbital (LUMO) was located on the carbon atoms of the ring and slightly upon the oxygen atoms of DPM with the energy of 0.23 eV. These results were supported by quantum molecular descriptors calculations, as the DPM with high global hardness (2.63 eV) suggests it to be a highly stable drug having low reactivity. The calculated energy gap (Eg) and Fermi level energies (EF) of the pure molecule are 5.72 and −2.63 eV, respectively.
3.2. DPM Adsorption Phenomena upon the BN Nano-Cages
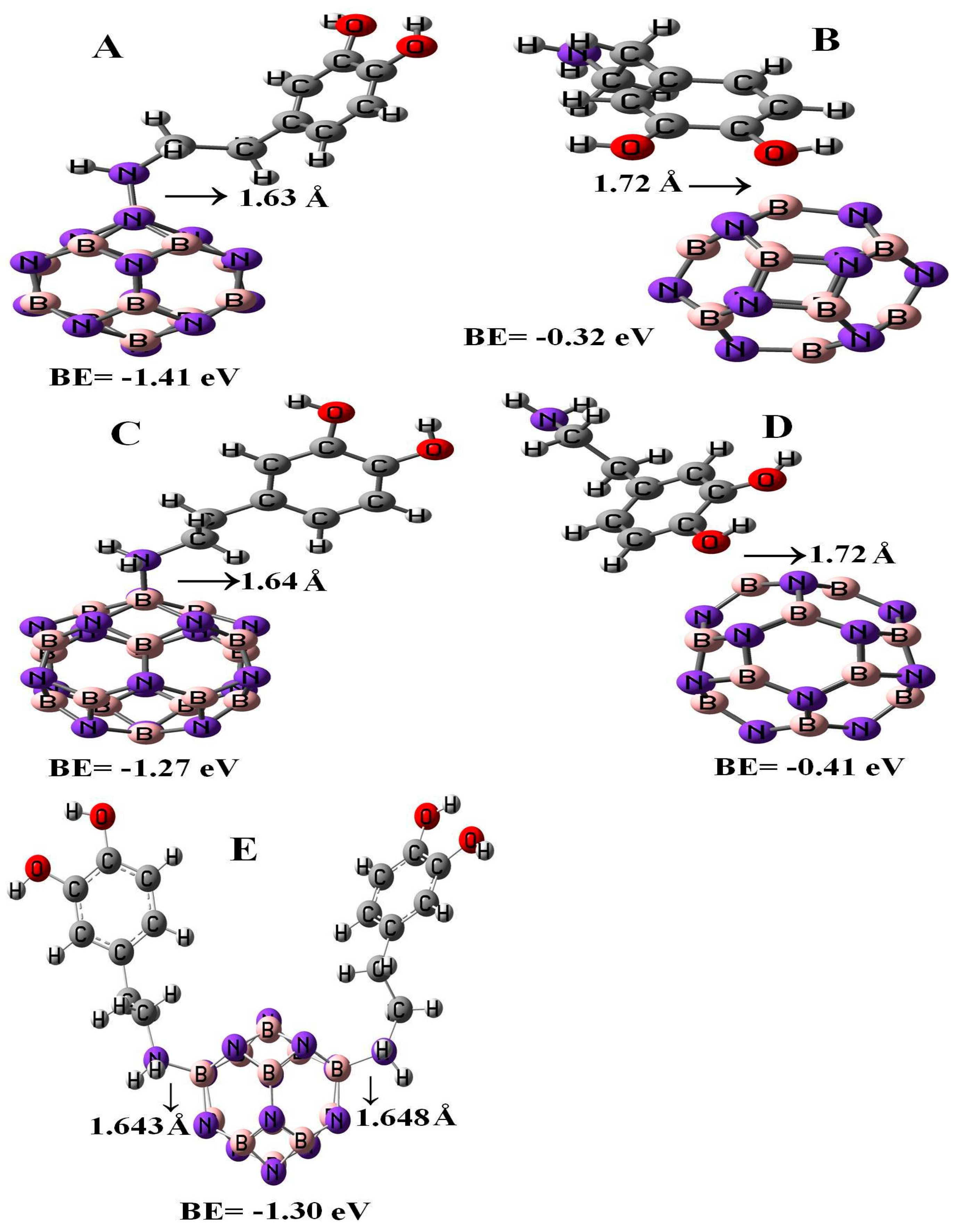
Figure 2 represents the configurations of the relaxed geometries of perfect B12N12 and B16N16 nano-cages at the side of functionalized BN nano-cages with DPM. As shown in Figure 2, the DPM was attached to the B atoms of B12N12 and B16N16 nano-cages from NH2 and OH groups. The chemisorption of DPM (from NH2 group) on the B atoms of B12N12 and B16N16 nano-cages were −1.41 and −1.27 eV with equilibrium distance of 1.63 and 1.64 Å, respectively. The calculated adsorption energies of DPM (from OH group) upon the top of the B atoms of B12N12 and B16N16 nano-cages were −0.32 and −0.41 eV which were less energetically notable than the NH2 group of DPM that interacted with the nano-cages. The binding energy of the adsorbed DPM (from the NH2 group) upon the B12N12 nano-cage was found to be larger than that on the surface of the B16N16 nano-cage. The boron–nitrogen bond lengths of the pristine B12N12 and B16N16 nano-cages were found to be 1.486 and 1.473 Å, respectively. Upon the adsorption of DPM, the boron–nitrogen bond lengths of B12N12 and B16N16 were increased to 1.515 and 1.557 Å, respectively, indicating the covalent interaction of DPM with B12N12 and B16N16 nano-cages that led to a change in hybridization from sp2 to sp3 at the adsorption sites. Our adsorption calculations indicated for a strong chemical interaction in nature.
The values of quantum molecular descriptors for the different configurations of DPM interacting with B12N12 and B16N16 nano-cages are presented in Table 1. We found that the global hardness was reduced from 3.42 eV in the perfect B12N12 to 2.86 eV in the DMP/B12N12 complex. Therefore, a reduction in global hardness led to a reduction in stability and the increment in the reactivity of the system. In addition, the adsorption of DMP by B12N12 dramatically declined the values of global hardness and electrophilicity index during the interaction process. The amount of charge transfer (∆N) between the drug molecule and the nano-cage was calculated. A negative value of ∆N suggests that the drug molecule acts as an electron donor while a positive value of ∆N suggests that the drug molecule acts as an electron acceptor. Upon the interaction of one DMP with B12N12, about 0.11 eV was transferred from the drug molecule to the perfect nano-cage.
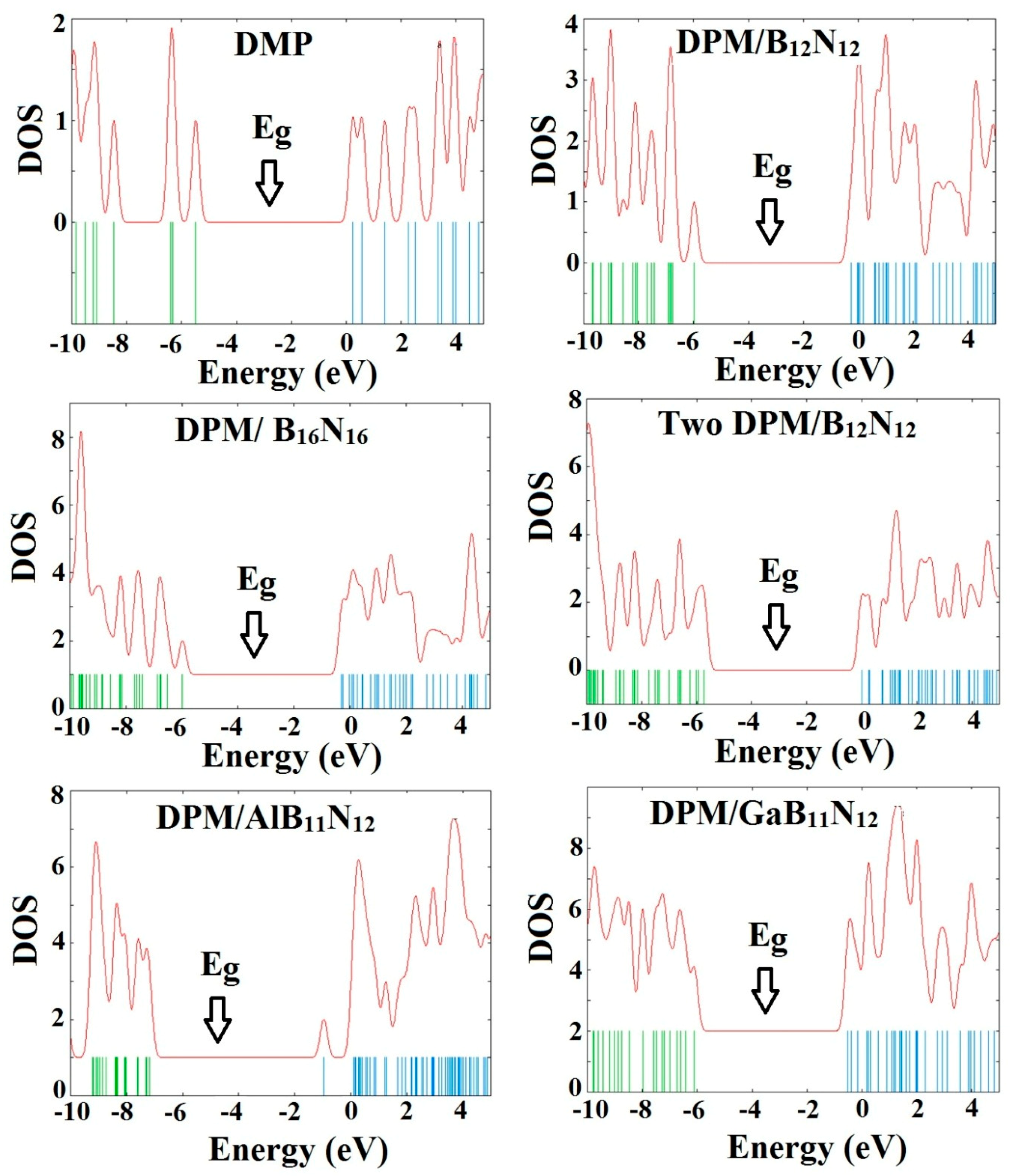
The calculated density of states (DOSs) of the investigated structures revealed that the DPM strongly interacted with B12N12 and B16N16 nano-cages, as shown in Figure 3. All the Fermi levels were at zero. Our calculations of these complexes indicated for strong coupling of the π-charge cloud of the BN nano-cage with that of the DPM. The analysis of DOS revealed dramatic changes owing to the drug adsorption upon B12N12 and B16N16 nano-cages in the gap regions of the total density of state (TDOS) spectrum. However, it could be inferred that the DOS near the HOMO level had the largest change compared with that of the BN nano-cages, so that some local energy levels became visible after the DPM interaction, resulting in the reduction of Eg. The energy gaps of the perfect B12N12 and B16N16 nano-cages were about 6.84 and 6.37 eV, respectively. The Fermi levels of the B12N12 and B16N16 nano-cages were significantly increased from −4.29 and −4.20 eV to −3.13 and −3.14 eV after the adsorption process, respectively, resulting in a charge transfer between the drug molecule and the nano-cage.
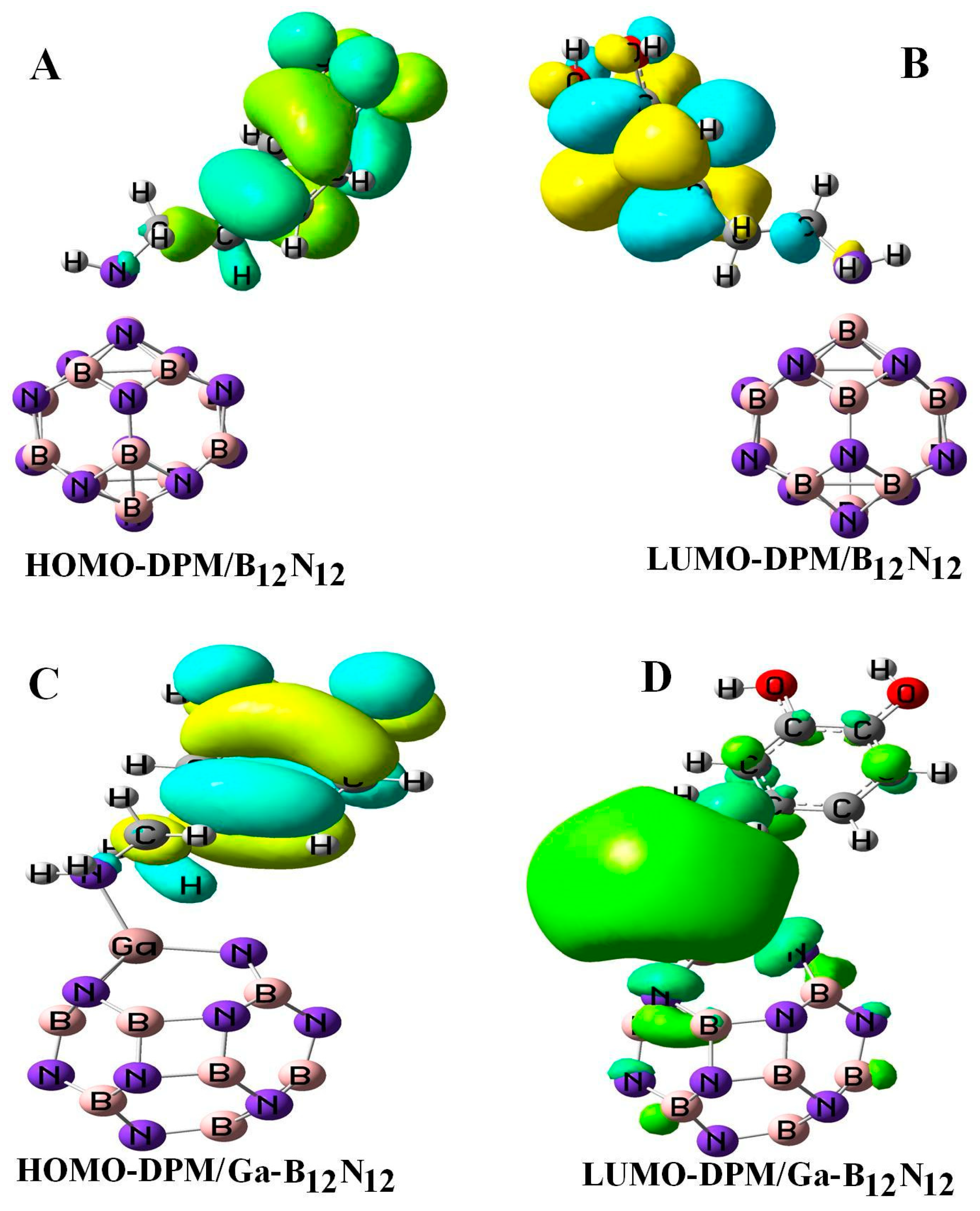
Figure 4 reveals the electron density distributions of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) for orbitals of DPM over the pure and Ga-doped B12N12 nano-cages. In the B12N12 nano-cage, the HOMO was mainly situated on the nitrogen atoms with the energy of −7.71 eV, while the LUMO was located upon the boron atoms of the nano-cage and was partly located on the B–N bonds with the energy of −0.87 eV, respectively. For DPM interacting with the B12N12 nano-cage, the HOMO and LUMO of this system were highly localized upon C–C bonds and the oxygen atoms with the energies of −5.98 and −0.27 eV, respectively (see Table 1). The theoretical studies of the infrared (IR) spectrum for DPM interacted with B12N12 were carried out in the range 4000–400 cm−1. The OH stretching vibrations for DPM/B12N12 system was observed at 3716 and 3773 cm−1, respectively. In the complex, the peak at 1659 cm−1 revealed a medium bond of the stretching vibration of N–H in DPM. The B–N bonds in DPM/B12N12 complex were observed at 857, 882, 902, 905, 910, 912, and 918 cm−1. Adsorption peaks at 1664 and 1676 cm−1 could be assigned to aromatic C–C stretching frequencies of DPM that interacted with the B12N12 nano-cage. The vibrations frequencies corresponding to the C–H bonds appeared at 3038, 3077, 3125, 3155, 3190, and 3220 cm−1 for DPM/B12N12 complex. The above theoretical results were in agreement with the experimental values [38,39]. Finally, the thermodynamic function changes were calculated. The calculations showed that both the enthalpy change (ΔH) and the entropic changes (ΔS) were negative (−30.49 kcal/mol and −48.67 cal/mol·K). The Gibbs free energy change (ΔG) was negative and equaled −15.975 kcal/mol, indicating that this molecule could be spontaneously adsorbed. However, the lower value of ΔG in comparison to that of ΔH was due to the entropic effect.
3.3. Adsorption of the Second DPM upon B12N12
Figure 2 represents the most stable relaxed configuration of the second DPM attached to the B12N12 nano-cage. The calculations revealed that the two DPM molecules (from NH2 group) were strongly loaded with the boron atoms of B12N12 with the interaction distances of 1.643 and 1.648 Å, suggesting strong adsorption. Geometrical parameters of the DPM in the strongly adsorbed system changed the B–N bond lengths to 1.567 Å. The H–N–H bond angles were diminished from 105.89¼ in the pure molecule to 106.4¼ in the two DPM/B12N12 complex while the length of N–H was increased from 1.019 Å in the pure molecule to 1.023 Å at the two DPM/B12N12 complex owing to a change in hybridization. The interaction result exhibited that the adsorption of DPM was comparatively strong. The negative adsorption energy for the second adsorbed DPM was calculated to be −1.30 eV, characterizing an exothermic process. We explored the influence of the second DPM adsorption upon the electronic property of the B12N12 nano-cage in the gas phase. The DOS analysis of this complex is shown in Figure 3. Eg and EF were reduced from 6.84 and −4.29 eV in the pure model to 5.72 and −2.87 eV in the two DPM/B12N12 complex. This result indicated that the energy gap of the second interacted DPM on B12N12 had a considerable change compared to the pure nano-cage. The DOS of this system introduced change owing to the second DPM adsorption in the gap region of the TDOS spectrum.
3.4. DPM Adsorption Phenomena upon the BN Nanotubes
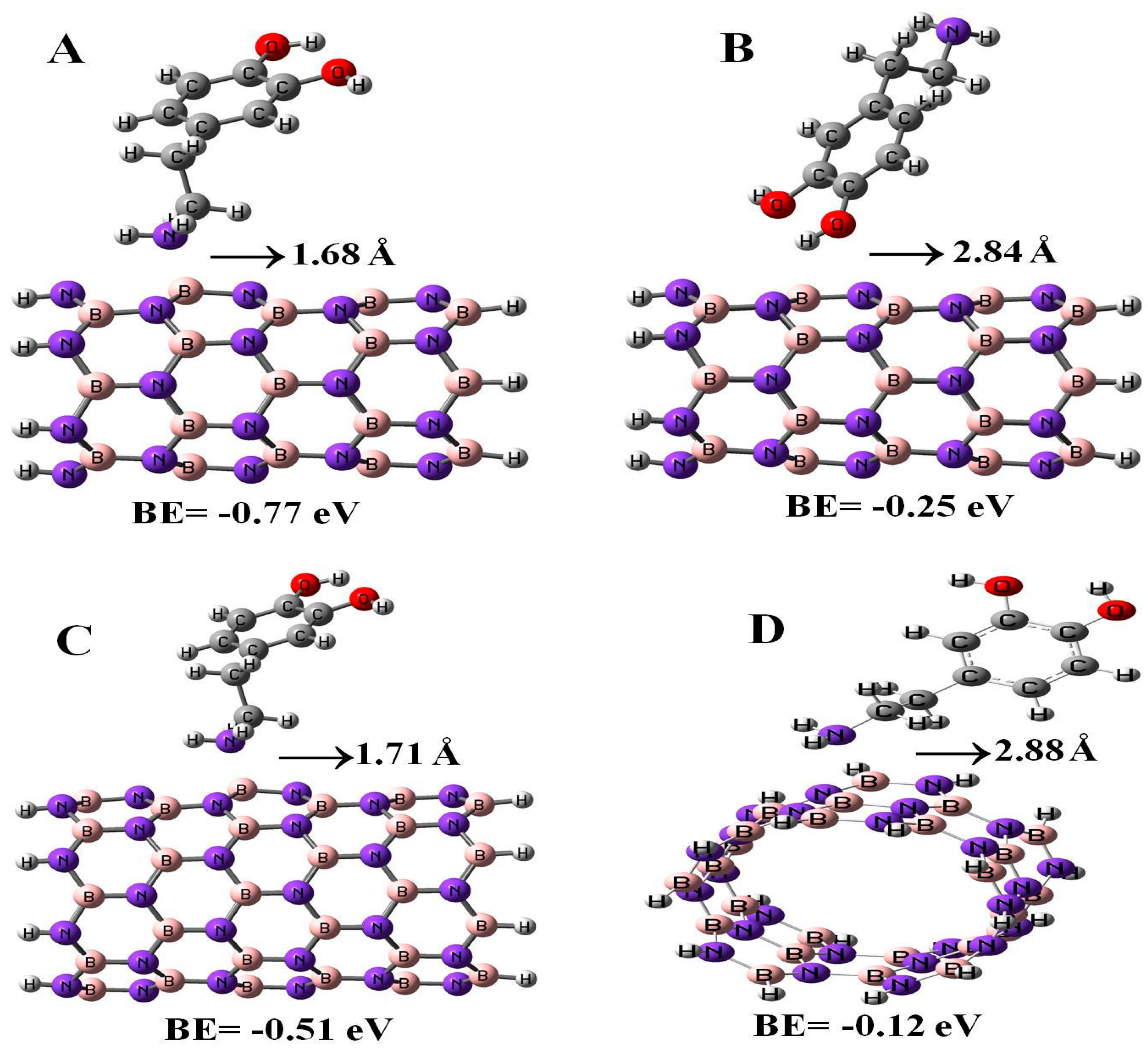
In the present study, we also calculated the adsorption properties of DPM interacting with (6,0), (8,0), and (5,5) BN nanotubes with the B3LYP method, see Figure 5. The binding energies of DPM (from NH2 group) on (6,0), (8,0), and (5,5) BN nanotubes were found to be −0.77, −0.51, and −0.12 eV with the relevant distances of 1.68, 1.71, and 2.88 Å, respectively. The energy and distance of DPM (from OH group) onto the surface of (6,0) BNNT were −0.25 eV and 2.84 Å, respectively. As it can be observed, the DPM adsorption on the BN nano-cages was more stable than those of the BN nanotubes. We found that when the BN nanotube diameter and length increased, the binding energy of the DPM (from NH2 group) at each configuration of the adsorption was different. Our results revealed that the type of BN nanotube plays an important role in determining the interaction between the two species. The influence of the DPM adsorption toward the electronic properties of the BN nanotubes was investigated by using DOS plots. As shown in Table 2, the energy gaps of (6,0), (8,0), and (5,5) BN nanotubes were reduced from 4.70, 5.39, and 6.31 eV in the pure tubes to 4.32, 5.09, and 5.51 eV in the complex configurations, respectively. As a result, the energy gap of the BN nanotube had no significant change in the adsorption of the DPM. Also, the DPM adsorption on the BN nano-cages was more stable than those of the BN nanotubes.
3.5. Adsorption of the DPM on the Al- and Ga-Doped B12N12
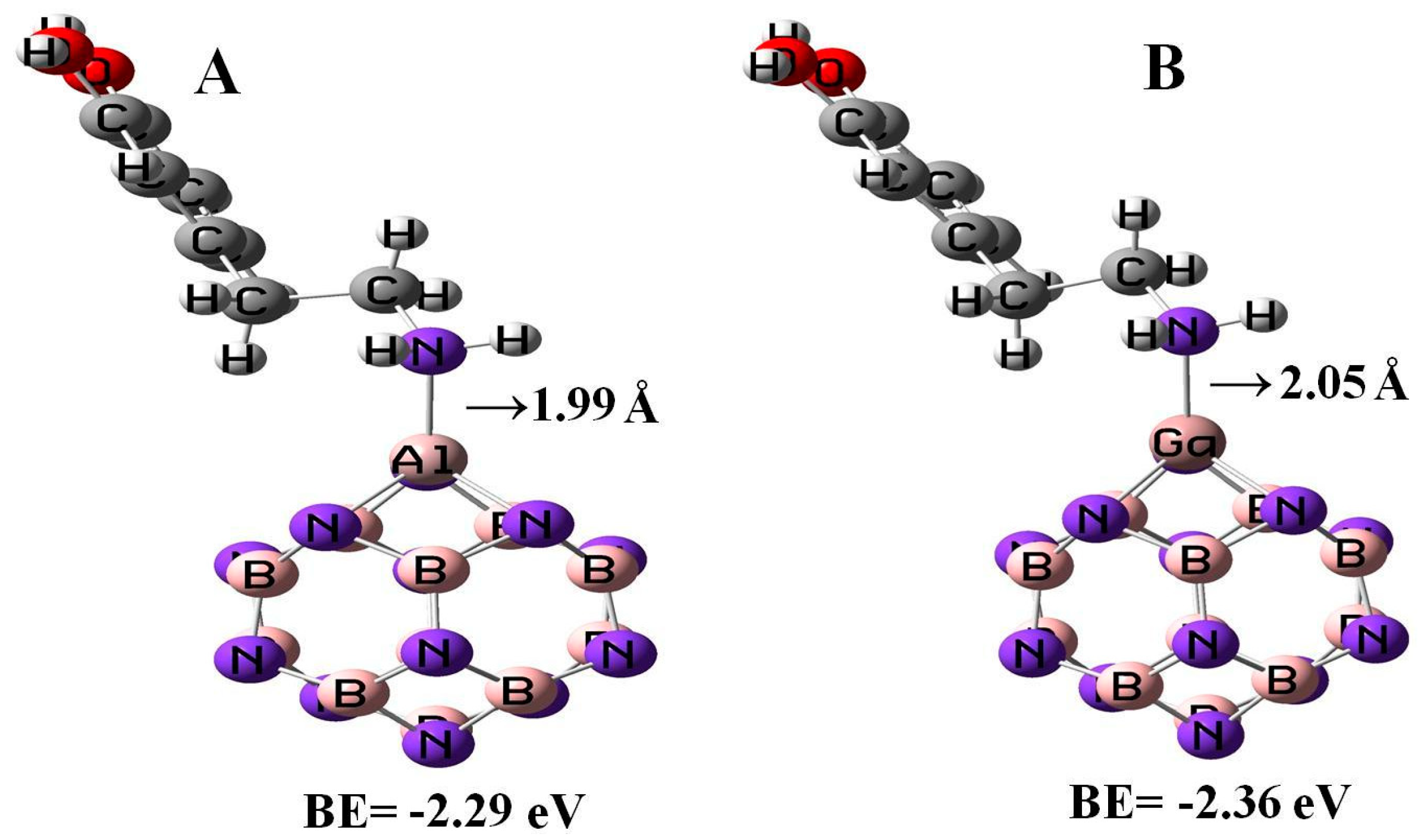
All the geometry relaxations of the DPM/Al- and Ga-doped B12N12 systems were performed (Figure 6). In Table 3, the bond length around the doping atom of Al–N and Ga–N of B12N12 expanded to 1.835 and 1.905 Å, respectively, while the bond length of B–N of B12N12 was 1.486 Å. We found that there were significant changes in the structures of Al- and Ga-doped B12N12 nano-cages after the adsorption process, the distances between the Al and Ga dopant atoms of B12N12 and the N atom of the molecule went to 1.99 and 2.05 Å with the adsorption energies of −2.29 and −2.36 eV, respectively. Therefore, the DPM/Ga–B11N12 bond was stronger than that of the DPM/Al–B11N12 system. Upon the adsorption processes, the Al–N and Ga–N bonds were increased to 1.864 and 1.923 Å while the N–Al–N and N–Ga–N bond angles were decreased from 83.2¼ and 79.8¼ in the pure models to 81.1¼ and 78.4¼ after the adsorption processes, respectively, which the NBO analysis showed that it can be attributed to a change in the hybridization of doped sites from sp2 to sp3. This above result reveals that the covalent interaction of DPM with Al- and Ga-doped B11N12 was strong in terms of binding energies. The charge analysis by the ∆N method revealed that in these forms about 0.08 electrons were transferred from the DPM to Al- and Ga-doped B12N12 nano-cages, respectively. The DOS plots of the pure and adsorbed DPM of Al- and Ga-doped B12N12 are shown in Figure 3. It could be found that for both Al- and Ga-doped B11N12 interacting with DPM, Eg was increased from 4.26 and 3.76 eV in the pure states to 5.67 and 5.58 eV in the complexes, respectively, while it did increase much in DPM/Ga–B11N12 in comparison with DPM/Al–B11N12 complex. Consequently, the electrical conductivity of the Al– and Ga–B11N12 significantly changed with the adsorption of DPM. The EF for the DPM added to Al– and Ga–B11N12 were significantly increased from −5.18 and −5.46 eV in clean systems to −3.30 and −3.31 eV after the adsorption processes. These results clearly indicate a charge transfer of DPM to Al– and Ga–B11N12 nano-cages. The electronic property between the DPM and Ga-B11N12 was delineated by the frontier orbital molecule (FMO) (see Table 3). The energy of HOMO for the pure Al- and Ga-doped B11N12 was about −7.31 and −7.34 eV and was calculated for LUMO of −3.05 and −3.58 eV, respectively. Upon the adsorption processes, the energy of HOMO for DPM interacting with Al- and Ga-doped B11N12 were slightly increased to −6.14 and −6.1 eV while the energy of LUMO for these complexes were significantly increased to −0.47 and −0.52 eV, respectively. The DOS plots of DPM attached to Al- and Ga-doped B11N12 revealed dramatic changes near the conduction band compared with that of the pure nano-cages. The HOMO of the DPM/Ga–B11N12 system was localized on the carbon atoms of the ring and from the oxygen and nitrogen atoms of a molecule but the LUMO was localized in the ring region with small distributions of the carbon atoms of a molecule and was localized upon the Ga, B, and N atoms of nano-cage (see Figure 4C,D). The above results showed that the DMP adsorption on the Al- and Ga-doped B12N12 systems were stronger than that of the pure nano-cages. Also, the DMP adsorption on the Ga–B11N12 system was stronger than that of the Al–B11N12 system.
3.6. The Solvent Effect on the Adsorption Phenomenon
We considered the adsorption of DPM from its NH2 group approaching to B12N12 and B16N16 nano-cages in a water solvent. Solvation energy (Esolv) for DPM interacted with B12N12 and B16N16 nano-cages in water were −1.63 and −1.43 eV, showing that they were more soluble in water than the pure nano-cages with energies of −1.41 and −1.27 eV, respectively. From the point of view that the electric dipole moment is one of the properties that are normally applied to discuss and elucidate the structure and reactivity of many chemical models [40,41], acquiring the electronic distribution in a molecule is of great importance. When a single DPM was added to the B atoms of B12N12 and B16N16 surfaces, the dipole moment of the complexes was significantly increased from 9.69 and 9.78 Debye in the gas phase to 12.06 and 13.08 Debye in the water phase. Solvation energy for the DPM attached to the Ga-doped B11N12 nano-cage was investigated. The Esolv for DPM adsorbed on the Ga–B11N12 surface in water phase was found to be −2.62 eV, which was more soluble than the gas phase with the value of −2.36 eV. The dipole moment value of the DPM/GaB11N12 complex in water phase was increased from 12.23 Debye in the gas phase to 16.73 Debye in the complex model. This result could be due to the polarization of the conducting electrons owing to the solvation.
4. Conclusions
The electronic and binding properties of DPM molecules upon BN nanostructures were investigated using DFT calculations and the following results were obtained:
- The DPM shows different binding characteristics on various BN nanostructures.
- The adsorption of DPM on BN nano-cages is stronger than other BN nanotubes.
- The most stable adsorption configuration relates to a single DPM–B12N12 system with adsorption energy of −1.41 eV.
- The doped BN nano-cages with Al and Ga atoms exhibit dramatic changes in adsorption and electronic properties with respect to their pristine counterparts.
- The adsorption of DPM on the Ga-doped B12N12 systems is stronger than that of the other studied systems.
- The adsorption of DPM on the studied systems in water phase is stronger than the gas phase.
We believe that our findings of the adsorption and electronic properties of DPM–BN nanostructures can help drug delivery scientists to perform novel drug delivery vehicles.
Author Contributions
M.T.B performed the conceptualization, methodology, formal analysis, investigation, software, supervision, project administration, and writing—review and editing. A.R.S was engaged in investigation, software, validation, resources, data curation, formal analysis, visualization, and writing—original draft preparation.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Chen, R.J.; Bangsaruntip, S.; Drouvalakis, K.A.; Kam, N.W.S.; Shim, M.; Li, Y.M.; Kim, W.; Utz, P.J.; Dai, H.J. Noncovalent functionalization of carbon nanotubes for highly specific electronic biosensors. Proc. Natl. Acad. Sci. USA 2003, 100, 4984. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.J.; Zhang, Y.G.; Wang, D.W.; Dai, H.J. Noncovalent Sidewall Functionalization of Single-Walled Carbon Nanotubes for Protein Immobilization. J. Am. Chem. Soc. 2001, 123, 3838. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Liu, Y.C.; Wang, Q.; Shen, J.W.; Wu, T.; Guan, W.J. On the spontaneous encapsulation of proteins in carbon nanotubes. Biomaterials 2009, 30, 2807. [Google Scholar] [CrossRef] [PubMed]
- Pantarotto, D.; Partidos, C.D.; Graff, R.; Hoebeke, J.; Briand, J.P.; Prato, M.; Bianco, A.J. Synthesis, structural characterization, and immunological properties of carbon nanotubes functionalized with peptides. Am. Chem. Soc 2003, 125, 6160. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.S.; Joselevich, E.; Woolley, A.T.; Cheung, C.L.; Lieber, C.M. Covalently functionalized nanotubes as nanometre- sized probes in chemistry and biology. Nature 1998, 394, 52. [Google Scholar] [CrossRef]
- Cui, Y.; Wei, Q.Q.; Park, H.K.; Lieber, C.M. Nanowire Nanosensors for Highly Sensitive and Selective Detection of Biological and Chemical Species. Science 2001, 293, 1289. [Google Scholar] [CrossRef]
- Benyamini, H.; Shulman-Peleg, A.; Wolfson, H.J.; Belgorodsky, B.; Fadeev, L.; Gozin, M. Interaction of C60-Fullerene and Carboxyfullerene with Proteins: Docking and Binding Site Alignment. Bioconjugate Chem 2006, 17, 378. [Google Scholar] [CrossRef]
- Hong, R.; Fischer, N.O.; Verma, A.; Goodman, C.M.; Emrick, T.; Rotello, V.M. Control of Protein Structure and Function through Surface Recognition by Tailored Nanoparticle Scaffolds. J. Am. Chem. Soc 2004, 126, 739. [Google Scholar] [CrossRef]
- You, C.C.; Agasti, S.S.; De, M.; Knapp, M.J.; Rotello, V.M. Modulation of the Catalytic Behavior of α-Chymotrypsin at Monolayer-Protected Nanoparticle Surfaces. J. Am. Chem. Soc 2006, 128, 14612. [Google Scholar] [CrossRef]
- Kia, M.; Golzar, M.; Mahjoub, K.; Soltani, A. A first-principles study of functionalized clusters and carbon nanotubes or fullerenes with 5-Aminolevulinic acid as vehicles for drug delivery. Superlattices Microstruct 2013, 62, 251–259. [Google Scholar] [CrossRef]
- Gallo, M.; Favila, A.; Glossman-Mitnik, D. DFT studies of functionalized carbon nanotubes and fullerenes as nanovectors for drug delivery of antitubercular compounds. Chem. Phys. Lett 2007, 447, 105–109. [Google Scholar] [CrossRef]
- Shukla, M.K.; Dubey, M.; Zakar, E.; Namburu, R.; Leszczynski, J. Density functional theory investigation of interaction of zigzag (7,0) single-walled carbon nanotube with Watson–Crick DNA base pairs. Chem. Phys. Lett 2010, 496, 128–132. [Google Scholar] [CrossRef]
- Wang, R.; Zhu, R.; Zhang, D. Adsorption of formaldehyde molecule on the pristine and silicon-doped boron nitride nanotubes. Chem. Phys. Lett 2008, 467, 131–135. [Google Scholar] [CrossRef]
- Choi, H.; Park, Y.C.; Kim, Y.-H.; Lee, Y.S. Ambient Carbon Dioxide Capture by Boron-Rich Boron Nitride Nanotube. J. Am. Chem. Soc 2011, 133, 2084–2087. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.-Y.; Yang, B.-S.; Wu, H.-S. Ab initio investigation of hydrogenation of (BN)16: A comparison with that of (BN)12. J. Mol. Struct. THEOCHEM 2010, 941, 144–149. [Google Scholar] [CrossRef]
- Mirzaei, M.; Yousefi, M. Boron nitride nanotubes with quadrangular cross sections: Density functional studies. Superlattices Microstruct 2012, 52, 648–652. [Google Scholar] [CrossRef]
- Soltani, A.; Ahmadian, N.; Amirazami, A.; Masoodi, A.; Lemeski, E.T.; Moradi, A.V. Theoretical investigation of OCN− adsorption onto boron nitride nanotubes. Appl. Surf. Sci 2012, 261, 262–267. [Google Scholar] [CrossRef]
- Soltani, A.; Ahmadian, N.; Kanani, Y.; Dehno khalaji, A.; Mighani, H. Ab initio investigation of the SCN− chemisorption of single-walled boron nitride nanotubes. Appl. Surf. Sci 2012, 258, 9536–9543. [Google Scholar] [CrossRef]
- Beheshtian, J.; Peyghan, A.A.; Bagheri, Z. Detection of phosgene by Sc-doped BN nanotubes: A DFT study. Sens. Actuators B Chem 2012, 171–172, 846–852. [Google Scholar] [CrossRef]
- Ribeiroa, F.A.D.; Tarley, C.R.T.; Borges, K.B.; Pereira, A.C. Development of a square wave voltammetric method for dopamine determination using a biosensor based on multiwall carbon nanotubes paste and crude extract of Cucurbita pepo L. Sens. Actuators B Chem 2013, 185, 743–754. [Google Scholar] [CrossRef]
- Njagi, J.; Chernov, M.M.; Leiter, J.C.; Silvana, A. Amperometric Detection of Dopamine in Vivo with an Enzyme Based Carbon Fiber Microbiosensor. Anal. Chem 2010, 82, 989–996. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Fei, J.; Hu, S. Simultaneous determination of dopamine and serotonin on a glassy carbon electrode coated with a film of carbon nanotubes. Anal. Biochem 2003, 318, 100–106. [Google Scholar] [CrossRef]
- Saikia, N.; Pati, S.K.; Deka, R.C. First principles calculation on the structure and electronic properties of BNNTs functionalized with isoniazid drug molecule. Appl. Nanosci 2012, 2, 389–400. [Google Scholar] [CrossRef] [Green Version]
- Juárez, A.R.; Anota, E.C.; Cocoletzi, H.H.; Riveros, A.F. Adsorption of chitosan on BN nanotubes: A DFT investigation. Appl. Surf. Sci 2013, 268, 259–264. [Google Scholar]
- Mukhopadhyay, S.; Scheicher, R.H.; Pandey, R.; Karna, S.P. Sensitivity of Boron Nitride Nanotubes toward Biomolecules of Different Polarities. J. Phys. Chem. Lett 2011, 2, 2442–2447. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Gowtham, S.; Scheicher, R.H.; Pandey, R.; Karna, S.P. Theoretical study of physisorption of nucleobases on boron nitride nanotubes: a new class of hybrid nano-biomaterials. Nanotechnology 2010, 21, 165703. [Google Scholar] [CrossRef] [Green Version]
- Baei, M.T.; Taghartapeh, M.R.; Lemeski, E.T.; Soltani, A. A computational study of adenine, uracil, and cytosine adsorption upon AlN and BN nano-cages. Physica B 2014, 444, 6–13. [Google Scholar] [CrossRef]
- Soltani, A.; Baei, M.T.; Lemeski, E.T.; Shahini, M. Sensitivity of BN nano-cages to caffeine and nicotine molecules. Superlattices Microstruct 2014, 76, 315–325. [Google Scholar] [CrossRef]
- Baei, M.T.; Taghartapeh, M.R.; Lemeski, E.T.; Soltani, A. Computational study of OCN− chemisorption over AlN nanostructures. Superlattices Microstruct 2014, 72, 370–382. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.02-SMP; Gaussian: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry, III. The role of exact exchange. J. Chem. Phys 1993, 98, 5648–5654. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anota, E.C.; Cocoletzi, G.H. GGA-based analysis of the metformin adsorption on BN nanotubes. Physica E 2014, 56, 134–140. [Google Scholar] [CrossRef]
- Parr, R.G.; Szentpaly, L.; Liu, S.J. Electrophilicity Index. Am. Chem. Soc 1999, 121, 1922. [Google Scholar] [CrossRef]
- Parr, R.G.; Pearson, R.G. Absolute hardness: companion parameter to absolute electronegativity. J. Am. Chem. Soc 1983, 105, 7512. [Google Scholar] [CrossRef]
- Mohammad-Shiri, H.; Ghaemi, M.; Riahi, S.; Akbari-Sehat, A. Computational and electrochemical studies on the redox reaction of dopamine in aqueous solution. Int. J. Electrochem. Sci 2011, 6, 317–336. [Google Scholar]
- Ge, B.; Tan, Y.; Xie, Q.; Ma, M.; Yao, S. Preparation of chitosan–dopamine-multiwalled carbon nanotubes nanocomposite for electrocatalytic oxidation and sensitive electroanalysis of NADH. Sens. Actuators B 2009, 137, 547–554. [Google Scholar] [CrossRef]
- Xuan, Y.; Jiang, G.; Li, Y.; Wang, J.; Geng, H. Inhibiting effect of dopamine adsorption and polymerization on hydrated swelling of montmorillonite. Colloids Surf. A Physicochem. Eng. Asp 2013, 422, 50–60. [Google Scholar] [CrossRef]
- Foresman, J.B.; Frisch, A.E. Exploring Chemistry with Electronic Structure Methods, 2nd ed.; Gaussian Inc.: Pittsburgh, PA, USA, 1996. [Google Scholar]
- Lewars, E. Computational Chemistry - Introduction to the Theory and Applications of Molecular and Quantum Mechanics; Kluwer Academic: Norwell, MA, USA, 2003. [Google Scholar]
Figure 1.
(A) Geometry, (B) molecular electrostatic potential surface (MEP) plot, (C) highest occupied molecular orbital (HOMO), and (D) lowest unoccupied molecular orbital (LUMO) of dopamine (DPM).
Figure 1.
(A) Geometry, (B) molecular electrostatic potential surface (MEP) plot, (C) highest occupied molecular orbital (HOMO), and (D) lowest unoccupied molecular orbital (LUMO) of dopamine (DPM).

Figure 2.
Single and double DPM adsorbed over the B12N12 and B16N16 nano-cages.

Figure 3.
Density of states plots of DPM over the pure, Al- and Ga-doped B12N12 and B16N16 nano-cages.
Figure 3.
Density of states plots of DPM over the pure, Al- and Ga-doped B12N12 and B16N16 nano-cages.

Figure 4.
HOMO and LUMO orbitals of DPM over the pure (A,B) and Ga-doped (C,D) B12N12 nano-cages.

Figure 5.
Adsorption of DPM over the (6,0) (A,B), (8,0) (C), and (5,5) (D) BN nanotubes

Figure 6.
Adsorption of a single DPM over the Al –doped (A) and Ga-doped (B) B12N12 nano-cages.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Calculated adsorption energy (Ead), charge (e), HOMO and LUMO energies (EHOMO and ELUMO), Fermi level energies (EF), energy gap (Eg), dipole moment (DM), and quantum molecular descriptors for optimized geometries of the DPM with B12N12 and B16N16 nano-cages in the gas phase.
Table 1.
Calculated adsorption energy (Ead), charge (e), HOMO and LUMO energies (EHOMO and ELUMO), Fermi level energies (EF), energy gap (Eg), dipole moment (DM), and quantum molecular descriptors for optimized geometries of the DPM with B12N12 and B16N16 nano-cages in the gas phase.
| System | B12N12 | DPM/B12N12 | B16N16 | DPM/ B16N16 | Two DPM/B12N12 |
|---|---|---|---|---|---|
| RB-N/Å | 1.486 | 1.569 | 1.473 | 1.557 | 1.567 |
| RB-N-B/¼ | 80.5 | 84.0 | 78.5 | 82.4 | 83.78 |
| RN-B-N/¼ | 98.2 | 91.7 | 99.3 | 92.9 | 92.0 |
| D/Å | - | 1.635 | - | 1.645 | 1.643 |
| Ead/eV | - | −1.41 | - | −1.27 | −1.30 |
| QB/e | 0.440 | 0.619 | 0.437 | 0.665 | 0.630 |
| QN/e | −0.440 | −0.512 | −0.437 | −0.514 | −0.511 |
| ΔN/e | - | −0.11 | - | −0.15 | −0.12 |
| EHOMO/eV | −7.71 | −5.98 | −7.38 | −5.98 | −5.73 |
| ELUMO/eV | −0.87 | −0.27 | −1.01 | −0.29 | −0.01 |
| Eg/eV | 6.84 | 5.71 | 6.37 | 5.69 | 5.72 |
| ∆Eg (%) | - | −19.8 | - | −10.7 | −16.37 |
| EFL/eV | −4.29 | −3.12 | −4.20 | −3.13 | −2.87 |
| μ/eV | −4.29 | −3.13 | −4.19 | −3.13 | −2.87 |
| η/eV | 3.42 | 2.86 | 3.19 | 2.84 | 2.86 |
| ω/eV | 2.69 | 1.71 | 2.76 | 1.73 | 1.44 |
| S/eV | 0.15 | 0.18 | 0.16 | 0.18 | 0.17 |
| DM/Debye | 0.0 | 9.69 | 0.0 | 9.78 | 7.58 |
Table 2.
Calculated adsorption energy (Ead), charge (e), HOMO and LUMO energies (EHOMO and ELUMO), Fermi level energies (EF), energy gap (Eg), dipole moment (DM), and quantum molecular descriptors for optimized geometries of the DPM with BN nanotubes in the gas phase.
Table 2.
Calculated adsorption energy (Ead), charge (e), HOMO and LUMO energies (EHOMO and ELUMO), Fermi level energies (EF), energy gap (Eg), dipole moment (DM), and quantum molecular descriptors for optimized geometries of the DPM with BN nanotubes in the gas phase.
| System | (6,0) BNNT | (5,5) BNNT | (8,0) BNNT | DPM/(6,0) BNNT | DPM/(5,5) BNNT | DPM/(8,0) BNNT |
|---|---|---|---|---|---|---|
| RB-N/Å | 1.488 | 1.450 | 1.449 | 1.511 | 1.463 | 1.510 |
| RB-N-B/¼ | 117.77 | 115.24 | 118.79 | 117.06 | 118.58 | 120.62 |
| RN-B-N/¼ | 119.85 | 119.62 | 119.97 | 114.26 | 119.23 | 114.54 |
| D/Å | - | - | - | 1.682 | 2.890 | 1.714 |
| Ead/eV | - | - | - | −0.77 | −0.12 | −0.51 |
| QB/e | 0.490 | 0.467 | 0.476 | 0.793 | 0.601 | 0.772 |
| QN/e | −0.490 | −0.467 | −0.476 | −0.564 | −0.459 | −0.553 |
| ΔN/e | - | - | - | −0.23 | −0.14 | −0.22 |
| EHOMO/eV | −6.62 | −6.40 | −6.47 | −5.94 | −5.51 | −5.94 |
| ELUMO/eV | −1.92 | −0.09 | −1.08 | −1.62 | 0.0 | −0.85 |
| Eg/eV | 4.70 | 6.31 | 5.39 | 4.32 | 5.51 | 5.09 |
| ∆Eg (%) | - | - | - | −8.08 | −12.68 | −5.56 |
| EFL/eV | −4.27 | −3.25 | −3.78 | −3.78 | −2.76 | −3.40 |
| μ/eV | −4.27 | −3.25 | −3.78 | −3.78 | −2.76 | −3.40 |
| η/eV | 2.35 | 3.20 | 2.70 | 2.16 | 2.76 | 2.54 |
| ω/eV | 3.88 | 1.67 | 2.64 | 3.31 | 1.38 | 2.26 |
| S/eV | 0.21 | 0.16 | 0.19 | 0.23 | 0.18 | 0.20 |
| DM/Debye | 7.96 | 0.00 | 11.87 | 10.06 | 3.98 | 12.89 |
Table 3.
Calculated adsorption energy (Ead), charge (e), HOMO and LUMO energies (EHOMO and ELUMO), Fermi level energies (EF), energy gap (Eg), dipole moment (DM), and quantum molecular descriptors for optimized geometries of the DPM with Al- and Ga-doped B12N12 nano-cages in the gas phase.
Table 3.
Calculated adsorption energy (Ead), charge (e), HOMO and LUMO energies (EHOMO and ELUMO), Fermi level energies (EF), energy gap (Eg), dipole moment (DM), and quantum molecular descriptors for optimized geometries of the DPM with Al- and Ga-doped B12N12 nano-cages in the gas phase.
| System | AlB11N12 | DPM/AlB11N12 | GaB11N12 | DPM/GaB11N12 |
|---|---|---|---|---|
| RAl-N/Å | 1.835 | 1.864 | - | - |
| RGa-N/Å | - | - | 1.905 | 1.923 |
| RAl-N-B/¼ | 83.6 | 84.9 | - | - |
| RN-Al-N/¼ | 83.2 | 81.1 | - | - |
| RGa-N-B/¼ | - | - | 84.9 | 85.9 |
| RN-Ga-N/¼ | - | - | 79.8 | 78.4 |
| D/Å | - | 1.998 | - | 2.05 |
| Ead/eV | - | –2.29 | - | –2.36 |
| QAl/e | 0.613 | 0.538 | - | - |
| QGa/e | - | - | 0.497 | 0.417 |
| QN/e | −0.548 | −0.575 | −0.613 | −0.592 |
| ΔN/e | - | −0.08 | - | −0.08 |
| EHOMO/eV | –7.31 | –6.14 | –7.34 | –6.10 |
| ELUMO/eV | –3.05 | −0.47 | –3.58 | −0.52 |
| Eg/eV | 4.26 | 5.67 | 3.76 | 5.58 |
| ∆Eg (%) | - | 33.1 | - | 48.4 |
| EFL/eV | –5.18 | –3.30 | –5.46 | –3.31 |
| μ/eV | –5.18 | –3.30 | –5.46 | –3.31 |
| η/eV | 2.13 | 2.83 | 1.88 | 2.79 |
| ω/eV | 6.30 | 1.92 | 7.93 | 1.96 |
| S/eV | 0.23 | 0.18 | 0.26 | 0.18 |
| DM/Debye | 3.24 | 12.49 | 2.76 | 12.23 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Soltani, A.R.; Baei, M.T. A DFT Study on Structure and Electronic Properties of BN Nanostructures Adsorbed with Dopamine. Computation 2019, 7, 61. https://0-doi-org.brum.beds.ac.uk/10.3390/computation7040061
AMA Style
Soltani AR, Baei MT. A DFT Study on Structure and Electronic Properties of BN Nanostructures Adsorbed with Dopamine. Computation. 2019; 7(4):61. https://0-doi-org.brum.beds.ac.uk/10.3390/computation7040061
Chicago/Turabian StyleSoltani, Ali Reza, and Mohammad T. Baei. 2019. "A DFT Study on Structure and Electronic Properties of BN Nanostructures Adsorbed with Dopamine" Computation 7, no. 4: 61. https://0-doi-org.brum.beds.ac.uk/10.3390/computation7040061
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.