Improved Sampling in Ab Initio Free Energy Calculations of Biomolecules at Solid–Liquid Interfaces: Tight-Binding Assessment of Charged Amino Acids on TiO2 Anatase (101)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. System Preparations
2.2. Metadynamics Setup
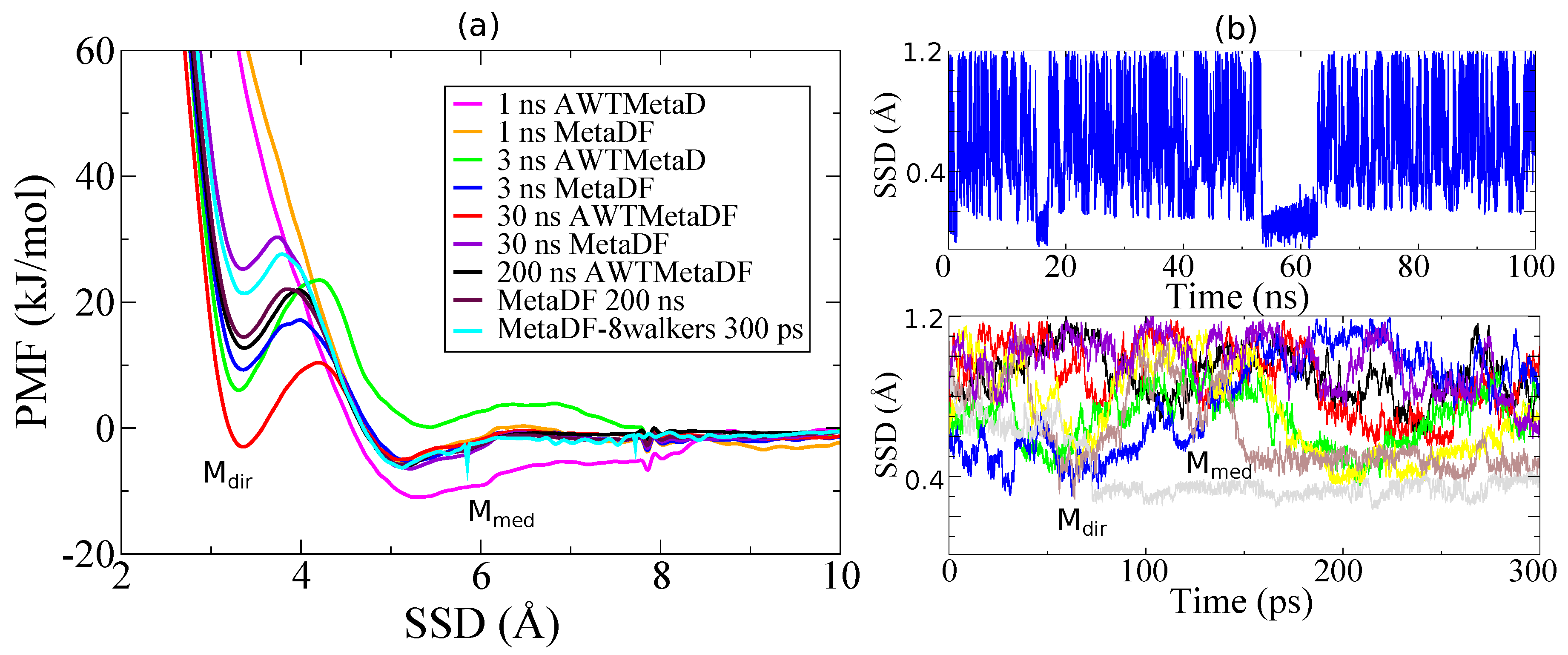
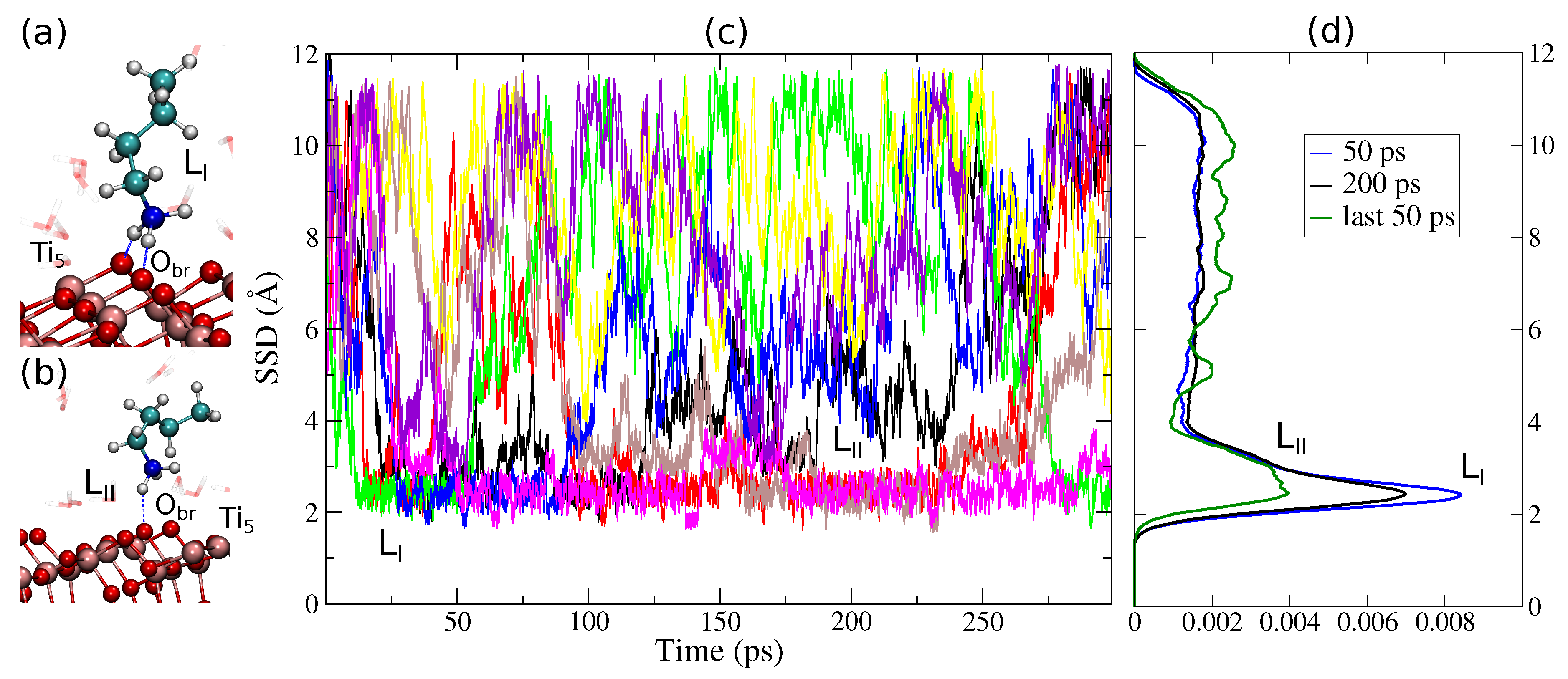
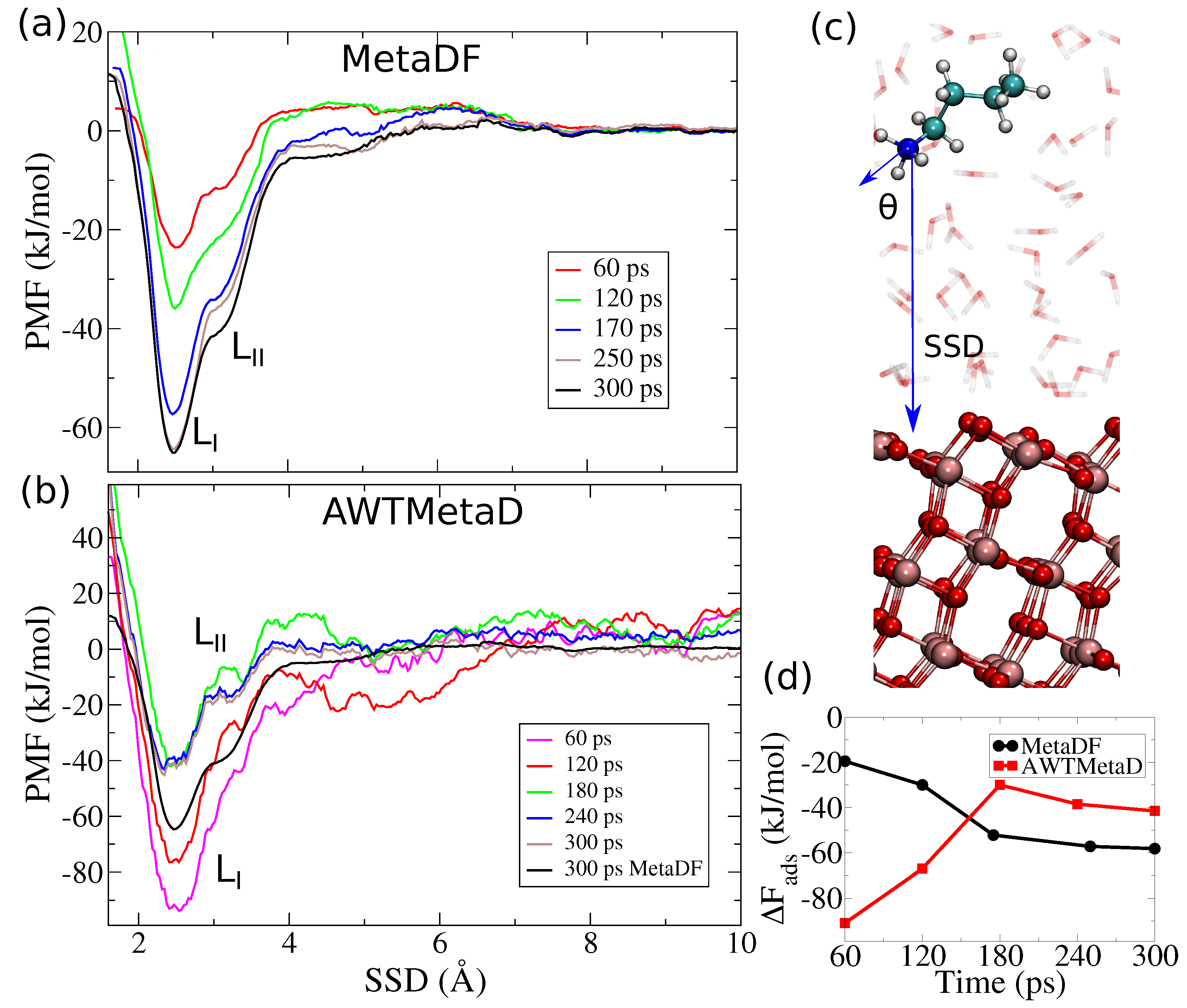
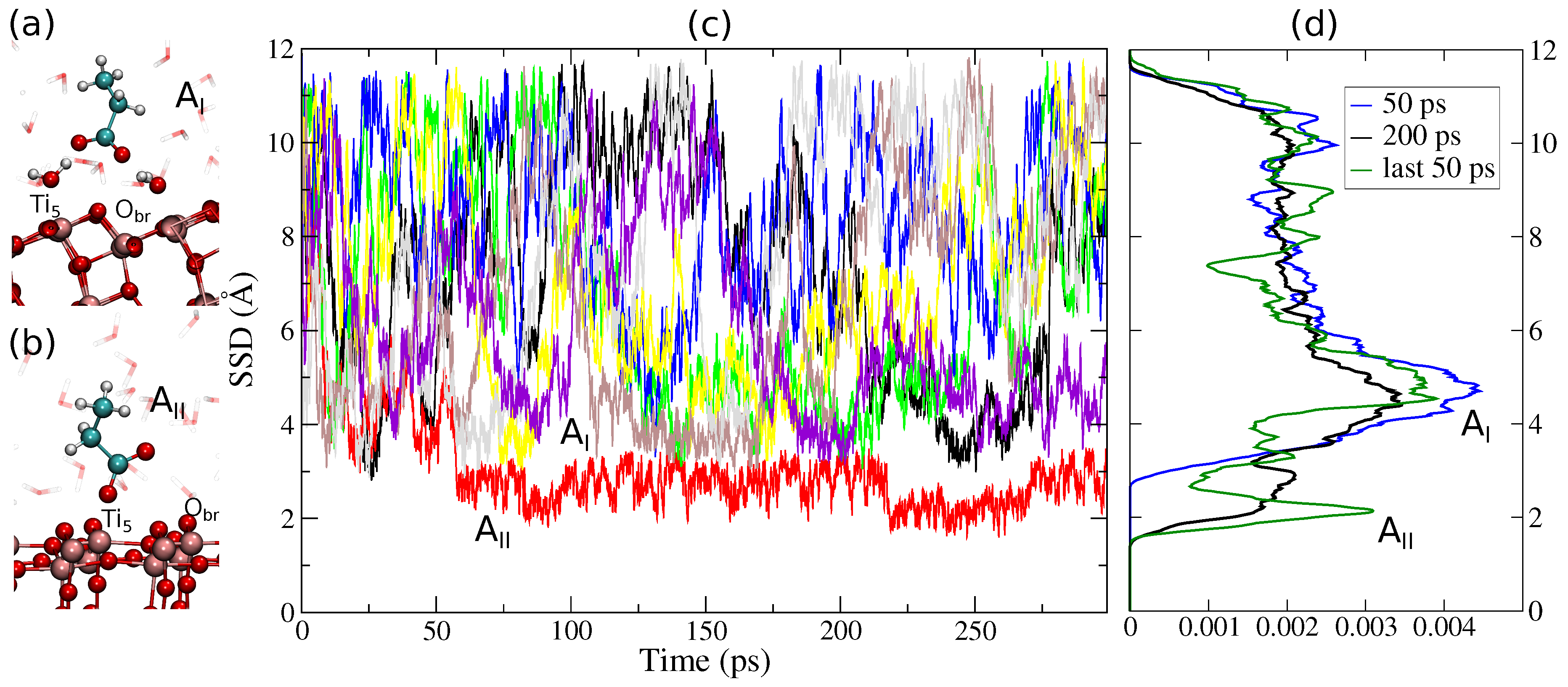
3. Results and Discussion
3.1. Method Validation
3.2. Tight-Binding Results
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shiba, K. Exploitation of Peptide Motif Sequences and Their Use in Nanobiotechnology. Curr. Opin. Biotechnol. 2010, 21, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Hartgerink, J.D.; Beniash, E.; Stupp, S.I. Self-Assembly and Mineralization of Peptide-Amphiphile Nanofibers. Science 2001, 294, 1684–1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowak, A.P.; Breedveld, V.; Pakstis, L.; Ozbas, B.; Pine, D.J.; Pochan, D.; Deming, T.J. Rapidly Recovering Hydrogel Scaffolds From Self-Assembling Diblock Copolypeptide Amphiphiles. Nature 2002, 417, 424–428. [Google Scholar] [CrossRef] [PubMed]
- Sarikaya, M.; Tamerler, C.; Jen, A.K.Y.; Schulten, K.; Baneyx, F. Molecular Biomimetics: Nanotechnology Through Biology. Nat. Mater. 2003, 2, 577–585. [Google Scholar] [CrossRef]
- Ellis-Behnke, R.G.; Liang, Y.X.; You, S.W.; Tay, D.K.C.; Zhang, S.; So, K.F.; Schneider, G.E. Nano Neuro Knitting: Peptide Nanofiber Scaffold for Brain Repair and Axon Regeneration with Functional Return of Vision. Proc. Natl. Acad. Sci. USA 2006, 103, 5054–5059. [Google Scholar] [CrossRef] [Green Version]
- Khatayevich, D.; Page, T.; Gresswell, C.; Hayamizu, Y.; Grady, W.; Sarikaya, M. Selective Detection of Target Proteins By Peptide-Enabled Graphene Biosensor. Small 2014, 10, 1505–1513. [Google Scholar] [CrossRef]
- Yemini, M.; Reches, M.; Rishpon, J.; Gazit, E. Novel Electrochemical Biosensing Platform Using Self-Assembled Peptide Nanotubes. Nano Lett. 2005, 5, 183–186. [Google Scholar] [CrossRef]
- Skorb, E.V.; Andreeva, D.V. Surface Nanoarchitecture for Bio-Applications: Self-Regulating Intelligent Interfaces. Adv. Funct. Mater. 2013, 23, 4483–4506. [Google Scholar] [CrossRef]
- Byrne, H.; Ahluwalia, A.; Boraschi, D.; Fadeel, B.; Gehr, P. The Bio-Nano-Interface in Predicting Nanoparticle Fate and Behaviour in Living Organisms: Towards Grouping and Categorising Nanomaterials and Ensuring Nanosafety by Design. BioNanoMaterials 2013, 14, 195–216. [Google Scholar] [CrossRef] [Green Version]
- Lynch, I.; Feitshans, I.L.; Kendall, M. Bio-Nano Interactions: New Tools, Insights and Impacts: Summary of the Royal Society Discussion Meeting. Phil. Trans. R. Soc. B Biol. Sci. 2015, 370, 20140162. [Google Scholar] [CrossRef] [Green Version]
- Lynch, I.; Salvati, A.; Dawson, K.A. Protein-Nanoparticle Interactions: What Does the Cell See? Nat. Nanotechnol. 2009, 4, 546–547. [Google Scholar] [CrossRef]
- Geetha, M.; Singh, A.K.; Asokamani, R.; Gogia, A.K. Ti Based Biomaterials, the Ultimate Choice for Orthopaedic Implants—A Review. Prog. Mater. Sci. 2009, 54, 397–425. [Google Scholar] [CrossRef]
- Song, D.P.; Chen, M.J.; Liang, Y.C.; Bai, Q.S.; Chen, J.X.; Zheng, X.F. Adsorption of Tripeptide Rgd on Rutile TIO2 Nanotopography Surface in Aqueous Solution. Acta Biomater. 2010, 6, 684–694. [Google Scholar] [CrossRef] [PubMed]
- Rajh, T.; Dimitrijevic, N.M.; Bissonnette, M.; Koritarov, T.; Konda, V. Titanium Dioxide in the Service of the Biomedical Revolution. Chem. Rev. 2014, 114, 10177–10216. [Google Scholar] [CrossRef] [PubMed]
- Fujishima, A.; Honda, K. Electrochemical Photolysis of Water at a Semiconductor Electrode. Nature 1972, 238, 37–38. [Google Scholar] [CrossRef]
- Brandt, E.G.; Agosta, L.; Lyubartsev, A.P. Reactive Wetting Properties of TiO2 Nanoparticles Predicted by Ab Initio Molecular Dynamics Simulations. Nanoscale 2016, 8, 13385–13398. [Google Scholar] [CrossRef]
- Gala, F.; Agosta, L.; Zollo, G. Water Kinetics and Clustering on the (101) TiO2 Anatase Surface. J. Phys. Chem. C 2016, 120, 450–456. [Google Scholar] [CrossRef]
- Roddick-Lanzilotta, A.D.; McQuillan, A.J. An in Situ Infrared Spectroscopic Study of Glutamic Acid and of Aspartic Acid Adsorbed on TIO2: Implications for the Biocompatibility of Titanium. J. Coll. Interface Sci. 2000, 227, 48–54. [Google Scholar] [CrossRef]
- Roddick-Lanzilotta, A.D.; Connor, P.A.; McQuillan, A.J. An in Situ Infrared Spectroscopic Study of the Adsorption of Lysine to TIO2 from an Aqueous Solution. Langmuir 1998, 14, 6479–6484. [Google Scholar] [CrossRef]
- Vitale, E.; Zollo, G.; Agosta, L.; Gala, F.; Brandt, E.G.; Lyubartsev, A. Stress Relief and Reactivity Loss of Hydrated Anatase (001) Surface. J. Phys. Chem. C 2018, 122, 22407–22417. [Google Scholar] [CrossRef]
- Schneider, J.; Ciacchi, L.C. Specific Material Recognition by Small Peptides Mediated by the Interfacial Solvent Structure. J. Am. Chem. Soc. 2012, 134, 2407–2413. [Google Scholar] [CrossRef]
- Skelton, A.A.; Liang, T.; Walsh, T.R. Interplay of Sequence, Conformation, and Binding at the Peptide–titania Interface as Mediated by Water. ACS Appl. Mater. Interfaces 2009, 1, 1482–1491. [Google Scholar] [CrossRef]
- Agosta, L.; Zollo, G.; Arcangeli, C.; Buonocore, F.; Gala, F.; Celino, M. Water Driven Adsorption of Amino Acids on the (101) Anatase TiO2 Surface: An Ab Initio Study. Phys. Chem. Chem. Phys. 2015, 17, 1556–1561. [Google Scholar] [CrossRef] [PubMed]
- Geada, I.L.; Ramezani-Dakhel, H.; Jamil, T.; Sulpizi, M.; Heinz, H. Insight into Induced Charges at Metal Surfaces and Biointerfaces Using a Polarizable Lennard-Jones Potentials. Nat. Commun. 2018, 9, 716. [Google Scholar] [CrossRef] [PubMed]
- Shchelokov, A.; Palko, N.; Potemkin, V.; Grishina, M.; Morozov, R.; Korina, E.; Uchaev, D.; Krivtsov, I.; Bol’shakov, O. Adsorption of Native Amino Acids on Nanocrystalline TIO2: Physical Chemistry, Qspr, and Theoretical Modeling. Langmuir 2019, 35, 538–550. [Google Scholar] [CrossRef]
- Yazdan-Yar, A.; Aschauer, U.; Bowen, P. Adsorption Free Energy of Single Amino Acids at the Rutile (110)/Water Interface Studied by Well-Tempered Metadynamics. J. Phys. Chem. C 2018, 122, 11355–11363. [Google Scholar] [CrossRef]
- Sultan, A.M.; Hughes, Z.E.; Walsh, T.R. Binding Affinities of Amino Acid Analogues at the Charged Aqueous Titania Interface: Implications for Titania-Binding Peptides. Langmuir 2014, 30, 13321–13329. [Google Scholar] [CrossRef] [PubMed]
- Monti, S.; Walsh, T.R. Free Energy Calculations of the Adsorption of Amino Acid Analogues at the Aqueous Titania Interface. J. Phys. Chem. C 2010, 114, 22197–22206. [Google Scholar] [CrossRef]
- Brandt, E.G.; Lyubartsev, A.P. Molecular Dynamics Simulations of Adsorption of Amino Acid Side Chain Analogues and a Titanium Binding Peptide on the TiO2 (100) Surface. J. Phys. Chem. C 2015, 119, 18126–18139. [Google Scholar] [CrossRef]
- Liu, S.; Meng, X.-Y.; Perez-Aguilar, J.M.; Zhou, R. An in Silico Study of TiO2 Nanoparticles Interaction with Twenty Standard Amino Acids in Aqueous Solution. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- Ozboyaci, M.; Kokh, D.B.; Corni, S.; Wade, R.C. Modeling and Simulation of Protein-Surface Interactions: Achievements and Challenges. Q. Rev. Biophys. 2016, 49, e4. [Google Scholar] [CrossRef] [Green Version]
- Agosta, L.; Brandt, E.G.; Lyubartsev, A.P. Diffusion and Reaction Pathways of Water Near Fully Hydrated TiO2 Surfaces from Ab Initio Molecular Dynamics. J. Chem. Phys. 2017, 147, 024704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Kotsis, K.; Manzhos, S. Comparative Density Functional Theory and Density Functional Tight Binding Study of Arginine and Arginine-Rich Cell Penetrating Peptide Tat Adsorption on Anatase TiO2. Phys. Chem. Chem. Phys. 2016, 18, 19902–19917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luschtinetz, R.; Frenzel, J.; Milek, T.; Seifert, G. Adsorption of Phosphonic Acid at the TIO2 Anatase (101) and Rutile (110) Surfaces. J. Phys. Chem. C 2009, 113, 5730–5740. [Google Scholar] [CrossRef]
- Dolgonos, G.; Aradi, B.; Moreira, N.H.; Frauenheim, T. An Improved Self-Consistent-Charge Density-Functional Tight-Binding (SCC-DFTB) Set of Parameters for Simulation of Bulk and Molecular Systems Involving Titanium. JCTC 2010, 6, 266–278. [Google Scholar] [CrossRef]
- Raiteri, P.; Laio, A.; Gervasio, F.L.; Micheletti, C.; Parrinello, M. Efficient Reconstruction of Complex Free Energy Landscapes by Multiple Walkers Metadynamics. J. Phys. Chem. C 2006, 110, 3533–3539. [Google Scholar] [CrossRef]
- Cuendet, M.A.; Tuckerman, M.E. Free Energy Reconstruction From Metadynamics or Adiabatic Free Energy Dynamics Simulations. J. Chem. Theory Comput. 2014, 10, 2975–2986. [Google Scholar] [CrossRef]
- Pantaleone, S.; Rimola, A.; Sodupe, M. Canonical, Deprotonated, or Zwitterionic? A Computational Study on Amino Acid Interaction With the TiO2 (101) Anatase Surface. J. Phys. Chem. C 2017, 121, 14156–14165. [Google Scholar] [CrossRef]
- Elstner, M.; Porezag, D.; Jungnickel, G.; Elsner, J.; Haugk, M.; Frauenheim, T.; Suhai, S.; Seifert, G. Self-consistent-charge density-functional tight-binding method for simulations of complex materials properties. Phys. Rev. B 1998, 58, 7260–7268. [Google Scholar] [CrossRef]
- Hutter, J.; Iannuzzi, M.; Schiffmann, F.; VandeVondele, J. CP2K: Atomistic Simulations of Condensed Matter Systems. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 15–25. [Google Scholar] [CrossRef] [Green Version]
- Martínez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. Packmol: A Package for Building Initial Configurations for Molecular Dynamics Simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S. Semiempirical GGA-Type Density Functional Constructed with a Long-Range Dispersion Correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Goedecker, S.; Teter, M.; Hutter, J. Separable Dual-Space Gaussian Pseudopotentials. Phys. Rev. B 1996, 54, 1703–1710. [Google Scholar] [CrossRef] [Green Version]
- Krack, M. Pseudopotentials for H To Kr Optimized for Gradient-Corrected Exchange-Correlation Functionals. Theor. Chem. Acc. 2005, 114, 145–152. [Google Scholar] [CrossRef] [Green Version]
- VandeVondele, J.; Hutter, J. Gaussian Basis Sets for Accurate Calculations on Molecular Systems in Gas and Condensed Phases. J. Chem. Phys. 2007, 127, 114105. [Google Scholar] [CrossRef] [Green Version]
- Tribello, G.A.; Bonomi, M.; Branduardi, D.; Camilloni, C.; Bussi, G. Plumed2: New Feathers for an Old Bird. Comp. Phys. Comm. 2014, 185. [Google Scholar] [CrossRef] [Green Version]
- Barducci, A.; Bussi, G.; Parrinello, M. Well-Tempered Metadynamics: A Smoothly Converging and Tunable Free-Energy Method. Phys. Rev. Lett. 2008, 100, 020603. [Google Scholar] [CrossRef] [Green Version]
- Valsson, O.; Tiwary, P.; Parrinello, M. Enhancing Important Fluctuations: Rare Events and Metadynamics From a Conceptual Viewpoint. Annu. Rev. Phys. Chem. 2016, 67, 159–184. [Google Scholar] [CrossRef]
- Branduardi, D.; Bussi, G.; Parrinello, M. Metadynamics with Adaptive Gaussians. J. Chem. Theory Comput. 2012, 8, 2247–2254. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Piccinin, S.; Fabris, S. Reaction Mechanisms of Water Splitting and H2 Evolution by a Ru(II)-Pincer Complex Identified with Ab Initio Metadynamics Simulations. ACS Catal. 2012, 2, 1500–1506. [Google Scholar] [CrossRef]
- Jämbeck, J.P.M.; Lyubartsev, A.P. Exploring the Free Energy Landscape of Solutes Embedded in Lipid Bilayers. J. Phys. Chem. Lett. 2013, 4, 1781–1787. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Sano, K.I.; Shiba, K.; Kumashiro, Y.; Iwahori, K.; Yamashita, I.; Hara, M. Mechanism Underlying Specificity of Proteins Targeting Inorganic Materials. Nano Lett. 2006, 6, 515–519. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Shindo, H.; Asakura, T. Structure and Dynamic Properties of a Ti-Binding Peptide Bound to TiO2 Nanoparticles As Accessed by 1H Nmr Spectroscopy. J. Phys. Chem. B 2016, 120, 4600–4607. [Google Scholar] [CrossRef] [PubMed]
- Deighan, M.; Pfaendtner, J. Exhaustively Sampling Peptide Adsorption With Metadynamics. Langmuir 2013, 29, 7999–8009. [Google Scholar] [CrossRef] [PubMed]
- Kästner, J.; Thiel, W. Analysis of the Statistical Error in Umbrella Sampling Simulations by Umbrella Integration. J. Chem. Phys. 2006, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monti, S.; van Duin, A.C.T.; Kim, S.Y.; Barone, V. Exploration of the Conformational and Reactive Dynamics of Glycine and Diglycine on TIO2: Computational Investigations in the Gas Phase and in Solution. J. Phys. Chem. C 2012, 116, 5141–5150. [Google Scholar] [CrossRef]
- Sultan, A.M.; Westcott, Z.C.; Hughes, Z.E.; Palafox-Hernandez, J.P.; Giesa, T.; Puddu, V.; Buehler, M.J.; Perry, C.C.; Walsh, T.R. Aqueous Peptide-TIO2 Interfaces: Isoenergetic Binding Via Either Entropically or Enthalpically Driven Mechanisms. ACS Appl. Mater. Interfaces 2016, 8, 18620–18630. [Google Scholar] [CrossRef] [Green Version]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agosta, L.; Brandt, E.G.; Lyubartsev, A. Improved Sampling in Ab Initio Free Energy Calculations of Biomolecules at Solid–Liquid Interfaces: Tight-Binding Assessment of Charged Amino Acids on TiO2 Anatase (101). Computation 2020, 8, 12. https://0-doi-org.brum.beds.ac.uk/10.3390/computation8010012
Agosta L, Brandt EG, Lyubartsev A. Improved Sampling in Ab Initio Free Energy Calculations of Biomolecules at Solid–Liquid Interfaces: Tight-Binding Assessment of Charged Amino Acids on TiO2 Anatase (101). Computation. 2020; 8(1):12. https://0-doi-org.brum.beds.ac.uk/10.3390/computation8010012
Chicago/Turabian StyleAgosta, Lorenzo, Erik G. Brandt, and Alexander Lyubartsev. 2020. "Improved Sampling in Ab Initio Free Energy Calculations of Biomolecules at Solid–Liquid Interfaces: Tight-Binding Assessment of Charged Amino Acids on TiO2 Anatase (101)" Computation 8, no. 1: 12. https://0-doi-org.brum.beds.ac.uk/10.3390/computation8010012