CuSO4/[Cu(NH3)4]SO4-Composite Thermochemical Energy Storage Materials

 ,
,  ,
,
Abstract
:1. Introduction
- (1)
- Sensible heat storage [9,10,11]: Heat is stored by increasing the temperature of a liquid (oil, water, molten salts, …) or solid (brick, concrete, …) storage medium. To extract the stored heat, the process is reverted. Sensible heat storage is by far the simplest technologically implementable form of heat storage; thus, it is already operational on large scales in established processes. The two main drawbacks associated with this concept are the necessary heavy thermal insulation, preventing heat losses during storage through radiation, as well as the large volumes of storage medium.
- (2)
- Latent heat storage [12,13,14]: Heat is stored taking advantage of the energy demand/energy release, associated with a phase-transfer from solid to liquid, or vice-versa. Although the first commercial applications with latent heat storage are already available, the technological readiness is less than for sensible technologies. Associated drawbacks are the necessary thermal insulation, but also the potential ageing/degradation effects of the phase change material.
- (3)
- Thermochemical heat storage [15,16,17]. Thermochemical heat storage includes both heat stored via a reversible chemical reaction, as well as via sorption storage. Whereas sorption storage on, e.g., zeolites is a pure physical process, storage through a chemical reaction involves the reversible thermal decomposition of a (solid) storage material (A), liberating a reaction gas (B). The charged (solid) storage material (C) is stockpiled until, by reaction with the previously separated reaction gas, the initial discharged form of the storage material is reformedA(solid) + ΔH↔B(solid) + C(gas)Appealing advantages of this concept can be summarized as flexible operational temperature ranges, as for each application in principle a suitable storage reaction can be found; highest storage densities [18]; fast charging/discharging rates; and infinite storage, as in the absence of the reaction gas (C) no energy loss during storage can occur. Drawbacks are the notably higher technological efforts in operation, as well as the necessary development work for a custom-tailored process [5].
2. Materials and Methods
2.1. Materials
2.2. Preparation of CuSO4 on Support-Materials
2.3. Thermal Analysis
2.4. Scanning Electron Microscopy (SEM)
2.5. Nitrogen Physisorption Surface Area
2.6. X-ray Powder Diffraction (P-XRD)
2.7. Transient Hot Bridge (THB)
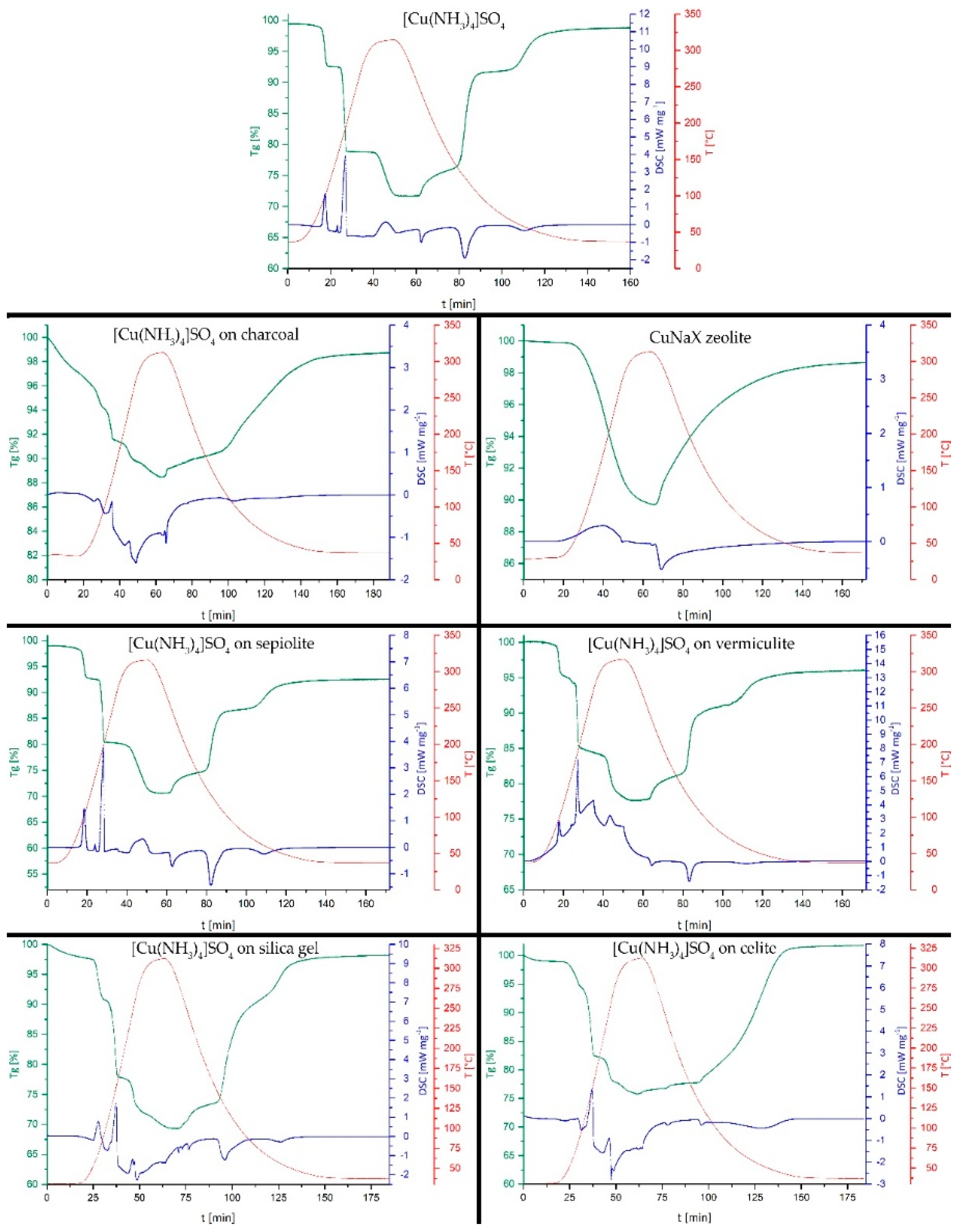
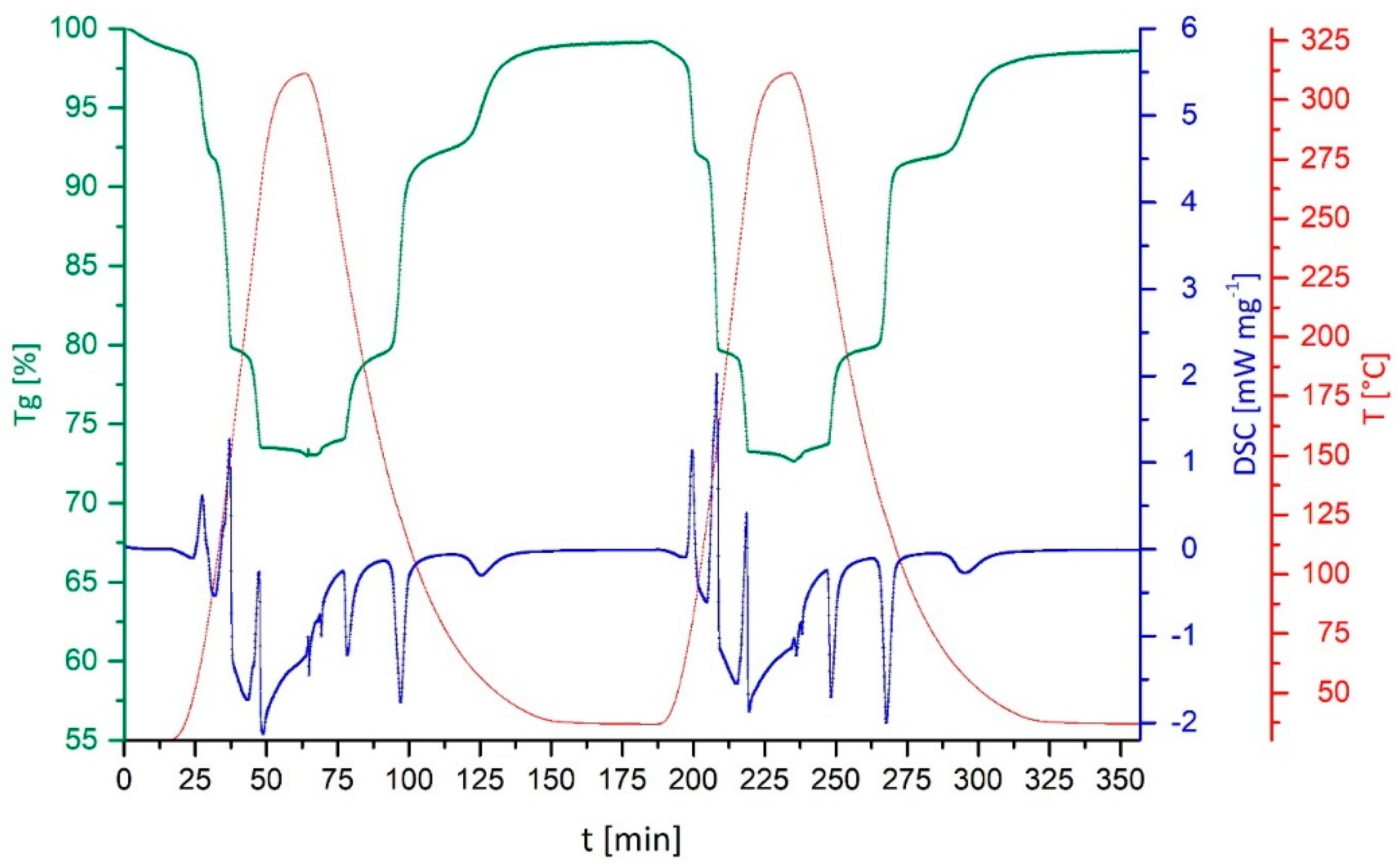
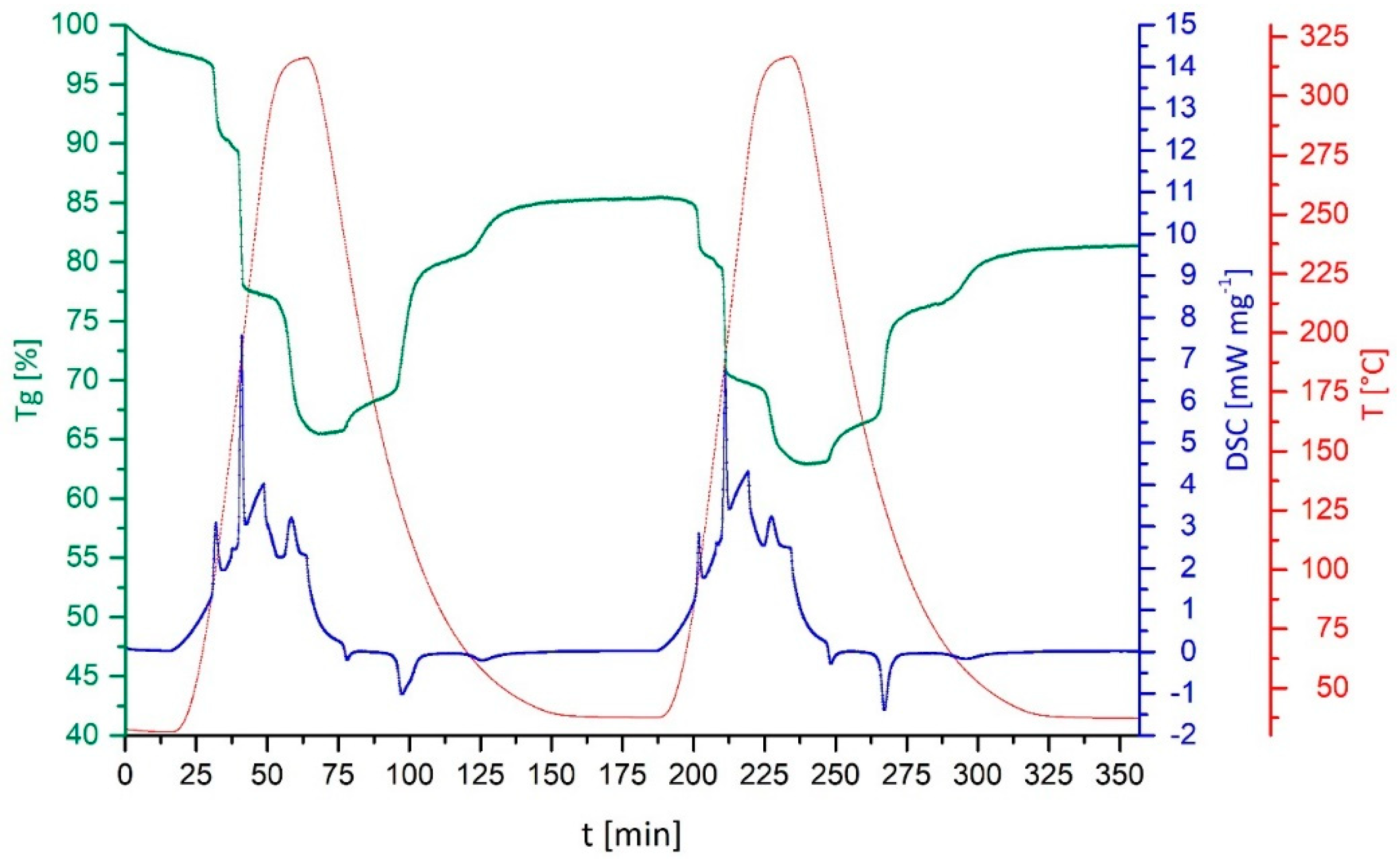
3. Discussion and Results
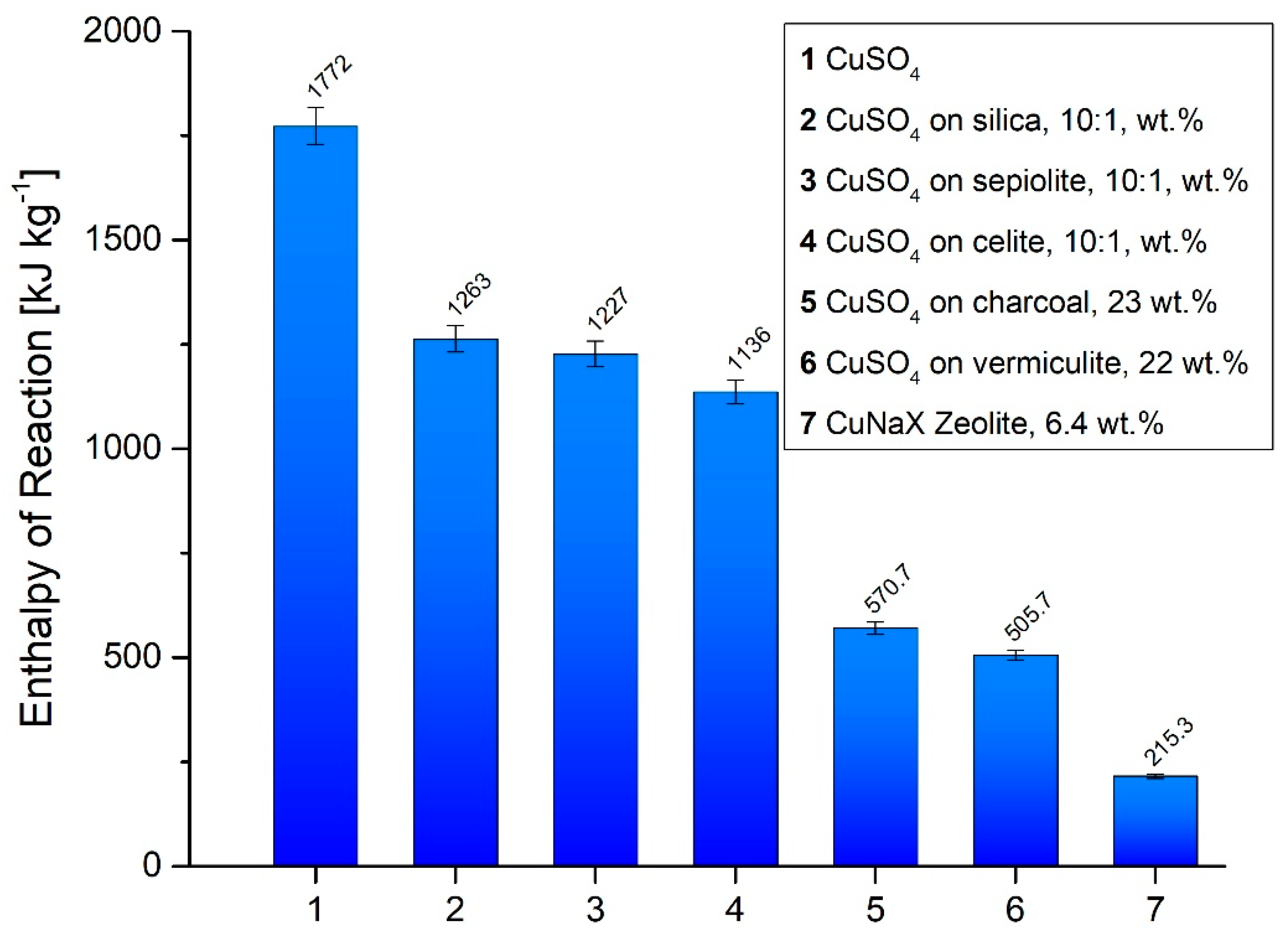
3.1. Reaction Enthalpy and Cycle Stability
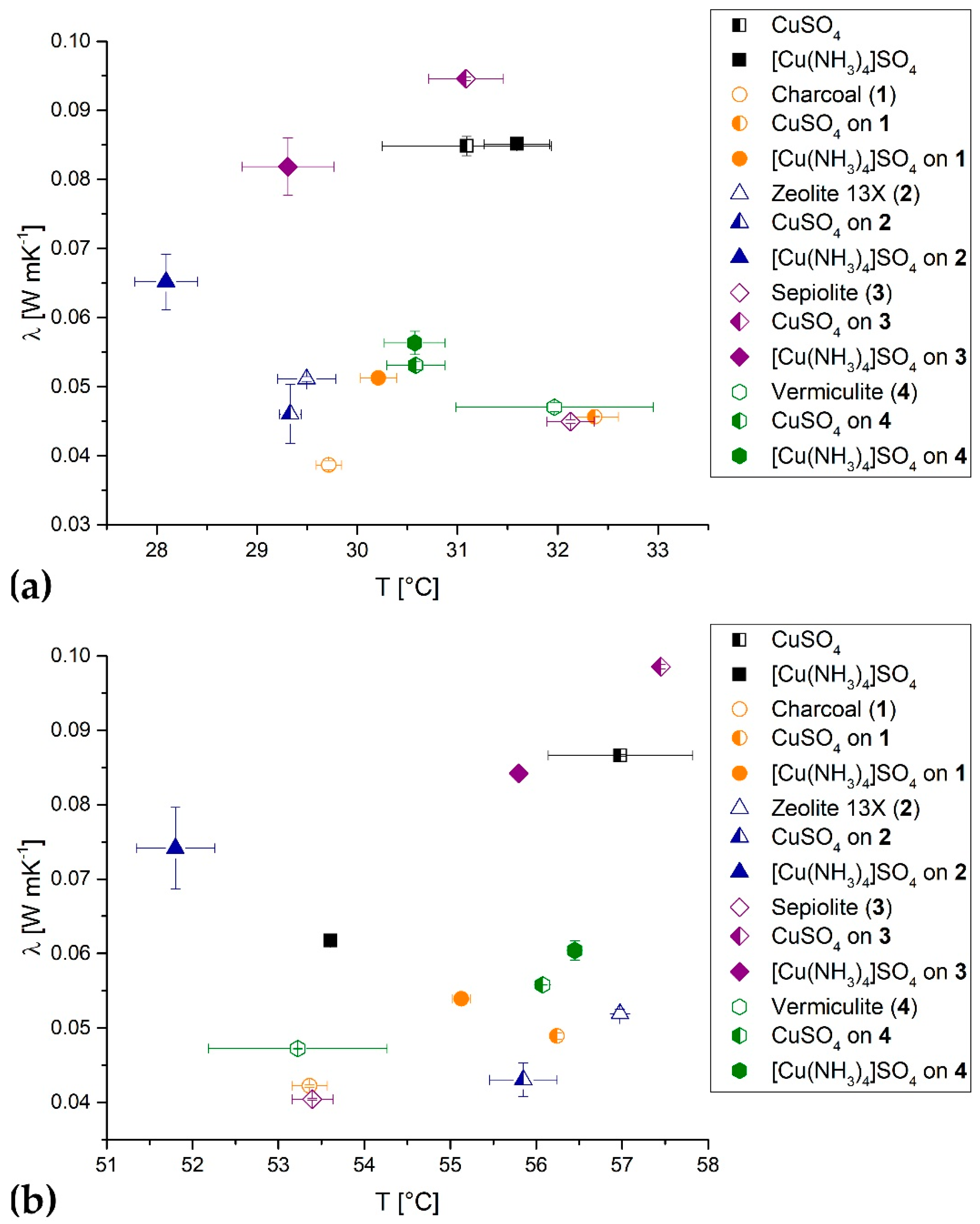
3.2. Thermal Conductivity
3.3. Scanning Electron Microscopy (SEM)
4. Conclusions
- (1)
- The formation of composites of CuSO4 with silica, sepiolite and celite (10:1 wt. %) results in energy storage densities between 1.14–1.26 MJ kg−1, around 30 % lower than for the pure salt. Adsorption and intercalation of CuSO4 on charcoal, in vermiculite and zeolite 13X substrate materials decreased the energy storage to 12.2 % (zeolite 13X) and 32.2 % (charcoal) of the initial value. For the zeolite, an ion exchange of Na+ to Cu2+ occurs, resulting a mixed CuNaX-zeolite, only capable of NH3− adsorption and desorption on the Cu-sites.
- (2)
- Only CuSO4 on sepiolite and on vermiculite retained the characteristic stepwise uptake and removal of the NH3-ligands. In terms of cycle stability, the composite with sepiolite provides full reversibility of the reaction, for the composite with vermiculite during the three investigated cycles, a certain mass-loss was observed as attributed to partial decomposition of the vermiculite substrate.
- (3)
- The effective thermal conductivities of the materials, determined at 30 °C and 55 °C, were not really improved compared to the pure CuSO4/[Cu(NH3)4]SO4. Only CuSO4 on sepiolite, especially at 55 °C, provides a higher effective thermal conductivity in the bulk.
- (4)
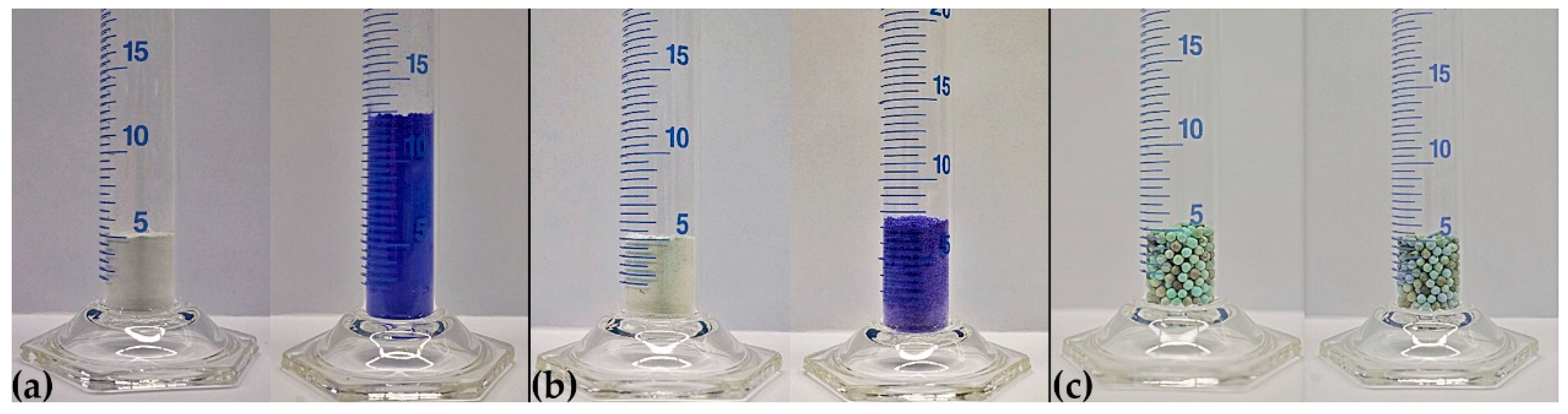
- Through the immobilization of the thermochemical energy storage material on the support/composite formation, a small reduction in energy content is traded for a drastically reduced volume change. The bulk volume expansion during reaction with NH3 is reduced for the sepiolite composite to a 1.3-fold expansion, as the composite material compensates for most of the molecular expansion during the reaction with NH3. Charcoal, vermiculite and CuNaX zeolite composites show no volume work during the reaction with NH3, the CuSO4 being incorporated in the matrix support.
- (5)
- Within further steps, a long-term cycling study of the sepiolite composite is suggested. Furthermore, other composite materials, presumably with better thermal conductivities (e.g., expanded natural graphite), should be investigated.
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Shine, K.P.; Fuglestvedt, J.S.; Hailemariam, K.; Stuber, N. Alternatives to the Global Warming Potential for Comparing Climate Impacts of Emissions of Greenhouse Gases. Clim. Chang. 2005, 68, 281–302. [Google Scholar] [CrossRef] [Green Version]
- IEA. Heating without Global Warming: Market Developments and Policy Considerations for Renewable Heat; IEA: Paris, France, 2014. [Google Scholar]
- UN Treatie. Paris Agreement, No. 54113; UN Treatie: Geneva, Switzerland, 2015. [Google Scholar]
- IEA. Co-Generation and Renewables. Solutions for a Low-Carbon Energy Future. 2011. Available online: https://www.iea.org/publications/freepublications/publication/co-generation-and-renewables-solutions-for-a-low-carbon-energy-future.html (accessed on 12 October 2020).
- Abedin, A.H. A Critical Review of Thermochemical Energy Storage Systems. Open Renew. Energy J. 2011, 4, 42–46. [Google Scholar] [CrossRef] [Green Version]
- Hasnain, S. Review on sustainable thermal energy storage technologies, Part I: Heat storage materials and techniques. Energy Convers. Manag. 1998, 39, 1127–1138. [Google Scholar] [CrossRef]
- Zhang, H.; Baeyens, J.; Cáceres, G.; Degrève, J.; Lv, Y. Thermal energy storage: Recent developments and practical aspects. Prog. Energy Combust. Sci. 2016, 53, 1–40. [Google Scholar] [CrossRef]
- Contribution of Working Groups I, II and III to the Fourth Assessment Report of the IntergoVernmental Panel on Climate Change. In Climate Change 2007: Synthesis Report; IPCC: Geneva, Switzerland, 2007.
- Bauer, T.; Steinmann, W.-D.; Laing, D.; Tamme, R. Thermal Energy Storage Materials and Systems. Annu. Rev. Heat Transf. 2012, 15, 131–177. [Google Scholar] [CrossRef]
- Xu, J.; Wang, R.; Li, Y. A review of available technologies for seasonal thermal energy storage. Sol. Energy 2014, 103, 610–638. [Google Scholar] [CrossRef]
- Bauer, T.; Pfleger, N.; Breidenbach, N.; Eck, M.; Laing, D.; Kaesche, S. Material aspects of Solar Salt for sensible heat storage. Appl. Energy 2013, 111, 1114–1119. [Google Scholar] [CrossRef]
- Cabeza, L.F.; Castell, A.; Barreneche, C.; De Gracia, A.; Fernández, A. Materials used as PCM in thermal energy storage in buildings: A review. Renew. Sustain. Energy Rev. 2011, 15, 1675–1695. [Google Scholar] [CrossRef]
- Tian, Y.; Zhao, C. A numerical investigation of heat transfer in phase change materials (PCMs) embedded in porous metals. Energy 2011, 36, 5539–5546. [Google Scholar] [CrossRef] [Green Version]
- Kuravi, S.; Goswami, Y.; Stefanakos, E.K.; Ram, M.; Jotshi, C.; Pendyala, S.; Trahan, J.; Sridharan, P.; Rahman, M.; Krakow, B. Thermal energy storage for concentrating solar power plants. Technol. Innov. 2012, 14, 81–91. [Google Scholar] [CrossRef]
- Cot-Gores, J.; Castell, A.; Cabeza, L.F. Thermochemical energy storage and conversion: A-state-of-the-art review of the experimental research under practical conditions. Renew. Sustain. Energy Rev. 2012, 16, 5207–5224. [Google Scholar] [CrossRef]
- Lele, A.F. State-of-Art of Thermochemical Heat Storage Systems. In A Thermochemical Heat Storage System for Households; Springer: Berlin/Heidelberg, Germany, 2016; pp. 15–58. [Google Scholar] [CrossRef]
- Prieto, C.; Cooper, P.; Fernández, A.I.; Cabeza, L.F. Review of technology: Thermochemical energy storage for concentrated solar power plants. Renew. Sustain. Energy Rev. 2016, 60, 909–929. [Google Scholar] [CrossRef] [Green Version]
- N’Tsoukpoe, K.E.; Liu, H.; Le Pierrès, N.; Luo, L. A review on long-term sorption solar energy storage. Renew. Sustain. Energy Rev. 2009, 13, 2385–2396. [Google Scholar] [CrossRef]
- Deutsch, M.; Müller, D.; Aumeyr, C.; Jordan, C.; Gierl-Mayer, C.; Weinberger, P.; Winter, F.; Werner, A. Systematic search algorithm for potential thermochemical energy storage systems. Appl. Energy 2016, 183, 113–120. [Google Scholar] [CrossRef]
- Aidoun, Z.; Ternan, M. Pseudo-stable transitions and instability in chemical heat pumps: The NH3–CoCl2 system. Appl. Therm. Eng. 2001, 21, 1019–1034. [Google Scholar] [CrossRef]
- Jiang, L.; Zhu, F.Q.; Wang, L.W.; Liu, C.Z.; Wang, R.Z. Experimental investigation on a MnCl2–CaCl2–NH3 thermal energy storage system. Renew. Energy 2016, 91, 130–136. [Google Scholar] [CrossRef] [Green Version]
- Trudel, J.; Hosatte, S.; Ternan, M. Solid–gas equilibrium in chemical heat pumps: The NH3–CoCl2 system. Appl. Therm. Eng. 1999, 19, 495–511. [Google Scholar] [CrossRef]
- Yamamoto, H.; Sanga, S.; Tokunaga, J.; Sakamoto, Y. Performance of thermal energy storage unit using CaCl2-NH3 system mixed with Ti. Can. J. Chem. Eng. 1990, 68, 948–951. [Google Scholar] [CrossRef]
- Müller, D.; Knoll, C.; Gravogl, G.; Jordan, C.; Eitenberger, E.; Friedbacher, G.; Artner, W.; Welch, J.M.; Werner, A.; Harasek, M.; et al. Medium-temperature thermochemical energy storage with transition metal ammoniates—A systematic material comparison. Appl. Energy 2020. submitted. [Google Scholar]
- Cammarata, A.; Verda, V.; Sciacovelli, A.; Ding, Y. Hybrid strontium bromide-natural graphite composites for low to medium temperature thermochemical energy storage: Formulation, fabrication and performance investigation. Energy Convers. Manag. 2018, 166, 233–240. [Google Scholar] [CrossRef] [Green Version]
- Zamengo, M.; Ryu, J.; Kato, Y. Thermochemical performance of magnesium hydroxide–expanded graphite pellets for chemical heat pump. Appl. Therm. Eng. 2014, 64, 339–347. [Google Scholar] [CrossRef]
- Zhao, Y.J.; Wang, R.Z.; Zhang, Y.N.; Yu, N. Development of SrBr 2 composite sorbents for a sorption thermal energy storage system to store low-temperature heat. Energy 2016, 115, 129–139. [Google Scholar] [CrossRef]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of Gases in Multimolecular Layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Degen, T.; Sadki, M.; Bron, E.; König, U.; Nénert, G. The HighScore suite. Powder Diffr. 2014, 29, S13–S18. [Google Scholar] [CrossRef] [Green Version]
- Available online: http://www.icdd.com (accessed on 12 October 2020).
- Hammerschmidt, U.; Meier, V. New Transient Hot-Bridge Sensor to Measure Thermal Conductivity, Thermal Diffusivity, and Volumetric Specific Heat. Int. J. Thermophys. 2006, 27, 840–865. [Google Scholar] [CrossRef]
- Rao, R. A note on the crystal structure of anhydrous copper sulphate. Acta Crystallogr. 1961, 14, 321–322. [Google Scholar] [CrossRef]
- Lutz, W. Zeolite Y: Synthesis, Modification, and Properties—A Case Revisited. Adv. Mater. Sci. Eng. 2014, 2014, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Valkov, M.; Simha, G. Vermiculite: Structural Properties and Examples of the Use. In Clay Minerals in Nature—Their Characterization, Modification and Application; Intech: London, UK, 2012. [Google Scholar] [CrossRef] [Green Version]
- Nonnen, T.; Beckert, S.; Gleichmann, K.; Brandt, A.; Unger, B.; Kerskes, H.; Mette, B.; Bonk, S.; Badenhop, T.; Salg, F.; et al. Erprobung eines thermochemischen Langzeitwärmespeichers auf Basis eines Zeolith/Salz-Komposits. Chem. Ing. Tech. 2016, 88, 363–371. [Google Scholar] [CrossRef]
- Wang, L.W.; Metcalf, S.J.; Critoph, R.E.; Thorpe, R.; Tamainot-Telto, Z. Thermal conductivity and permeability of consolidated expanded natural graphite treated with sulphuric acid. Carbon 2011, 49, 4812–4819. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Specific Surface Area [m2 g−1] | Pore Volume [cm3 g−1] | Average Pore Size [Å] | |
|---|---|---|---|
| Celite | 0.1570 ± 0.02 | n.d. | n.d. |
| Charcoal | 1694 ± 34 | 1.47 ± 0.03 | 34.775 ± 0.003 |
| Sepiolite | 709 ± 13 | 1.34 ± 0.09 | 76.046 ± 0.001 |
| Silica | 670 ± 2 | 0.95 ± 0.03 | 56.71 ± 0.012 |
| Vermiculite | 0.55 ± 0.19 | n.d. | n.d. |
| Zeolite 13X | 525 ± 16 | n.d. | n.d |
| bulk [Cu(NH3)4]SO4 | ||||||
| ↑ [°C] | ↓ [°C] | Δ [°C] | ||||
| 1st step | 79 | 66 | 13 | |||
| 2nd step | 168 | 138 | 30 | |||
| 3rd step | 307 | 248 | 59 | |||
| [Cu(NH3)4]SO4 + charcoal | CuNaX zeolite | |||||
| ↑ [°C] | ↓ [°C] | Δ [°C] | ↑ [°C] | ↓ [°C] | Δ [°C] | |
| 1st step | 65 | --- | --- | 46 | --- | --- |
| 2nd step | 119 | --- | --- | --- | --- | --- |
| 3rd step | 212 | 125 | 87 | --- | 288 | --- |
| [Cu(NH3)4]SO4 + sepiolite | [Cu(NH3)4]SO4 + vermiculite | |||||
| ↑ [°C] | ↓ [°C] | Δ [°C] | ↑ [°C] | ↓ [°C] | Δ [°C] | |
| 1st step | 77 | 74 | 3 | 85 | 96 | −11 |
| 2nd step | 170 | 138 | 32 | 171 | 129 | 42 |
| 3rd step | 302 | 265 | 37 | 305 | 236 | 69 |
| [Cu(NH3)4]SO4 + silica gel | [Cu(NH3)4]SO4 + celite | |||||
| ↑ [°C] | ↓ [°C] | Δ [°C] | ↑ [°C] | ↓ [°C] | Δ [°C] | |
| 1st step | 104 | 85 | 19 | 106 | --- | --- |
| 2nd step | 210 | 134 | 76 | 190 | --- | --- |
| 3rd step | 294 | 267 | 27 | 284 | 122 | 162 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Müller, D.; Knoll, C.; Gravogl, G.; Lager, D.; Welch, J.M.; Eitenberger, E.; Friedbacher, G.; Werner, A.; Artner, W.; Harasek, M.; et al. CuSO4/[Cu(NH3)4]SO4-Composite Thermochemical Energy Storage Materials. Nanomaterials 2020, 10, 2485. https://0-doi-org.brum.beds.ac.uk/10.3390/nano10122485
Müller D, Knoll C, Gravogl G, Lager D, Welch JM, Eitenberger E, Friedbacher G, Werner A, Artner W, Harasek M, et al. CuSO4/[Cu(NH3)4]SO4-Composite Thermochemical Energy Storage Materials. Nanomaterials. 2020; 10(12):2485. https://0-doi-org.brum.beds.ac.uk/10.3390/nano10122485
Chicago/Turabian StyleMüller, Danny, Christian Knoll, Georg Gravogl, Daniel Lager, Jan M. Welch, Elisabeth Eitenberger, Gernot Friedbacher, Andreas Werner, Werner Artner, Michael Harasek, and et al. 2020. "CuSO4/[Cu(NH3)4]SO4-Composite Thermochemical Energy Storage Materials" Nanomaterials 10, no. 12: 2485. https://0-doi-org.brum.beds.ac.uk/10.3390/nano10122485