Nanomedicines to Deliver mRNA: State of the Art and Future Perspectives

 , , , , and
, , , , and
Abstract
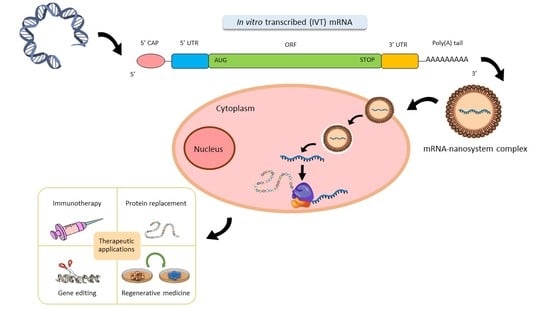
:
1. Introduction
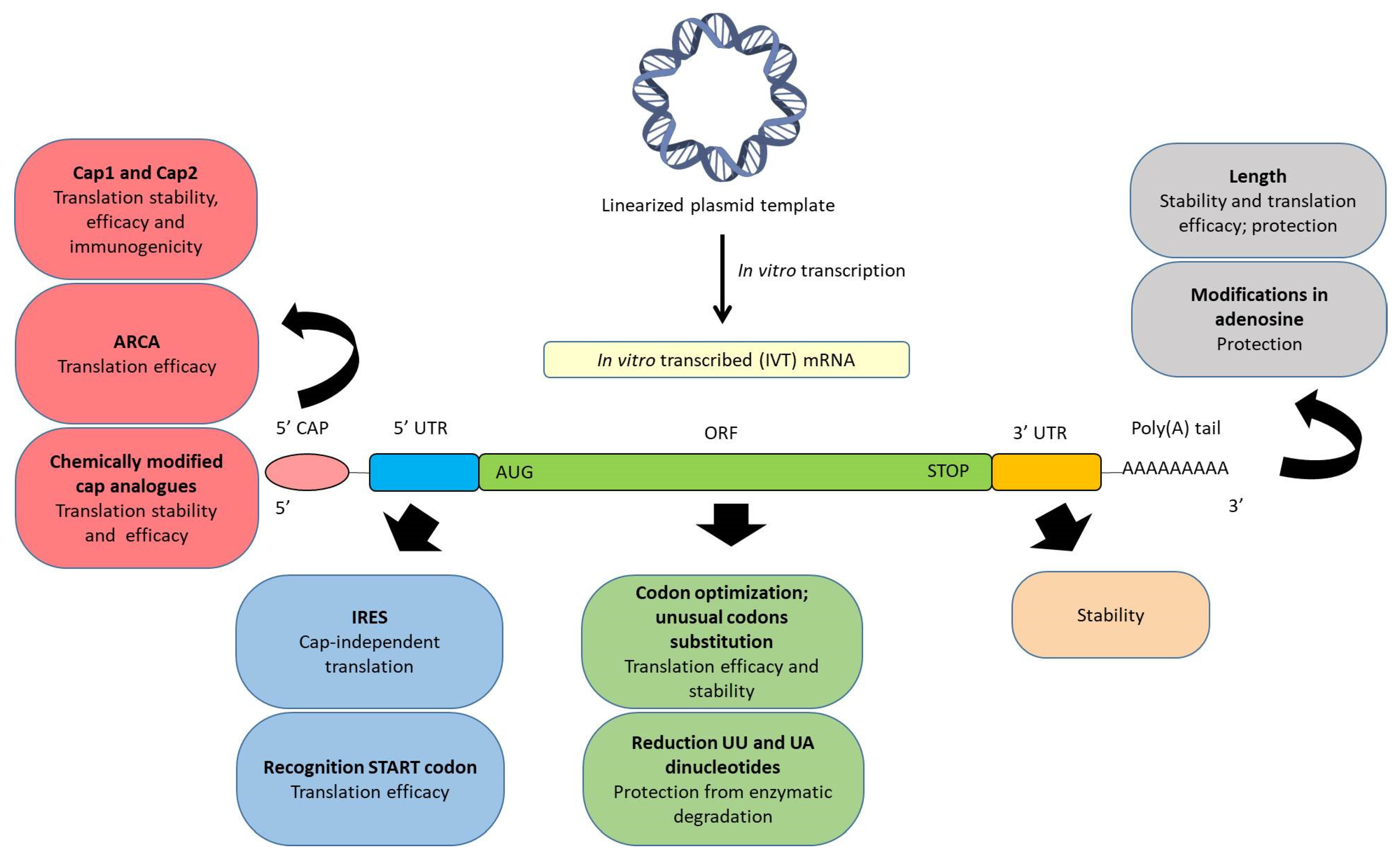
2. Structure of Synthetic IVT mRNA and Chemical Modifications
2.1. 5’ Cap
2.2. ORF
2.3. Poly(A) Tail
2.4. UTRs
2.5. Modified Nucleosides
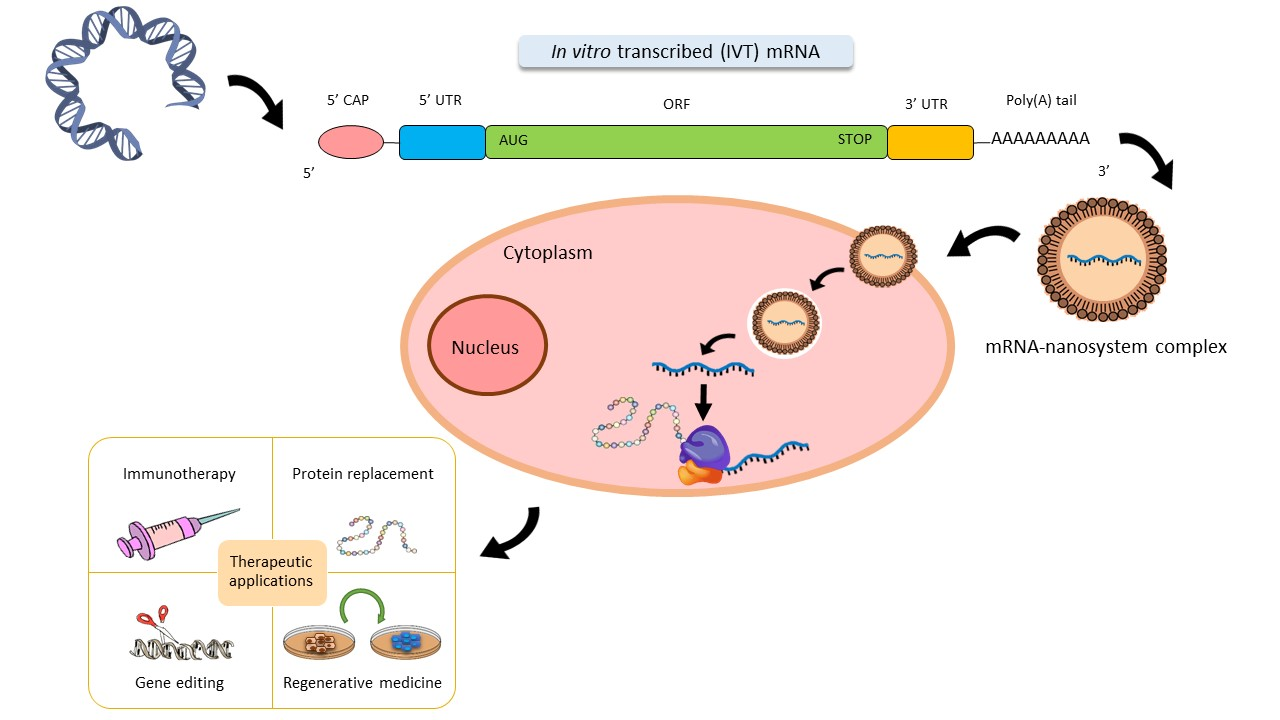
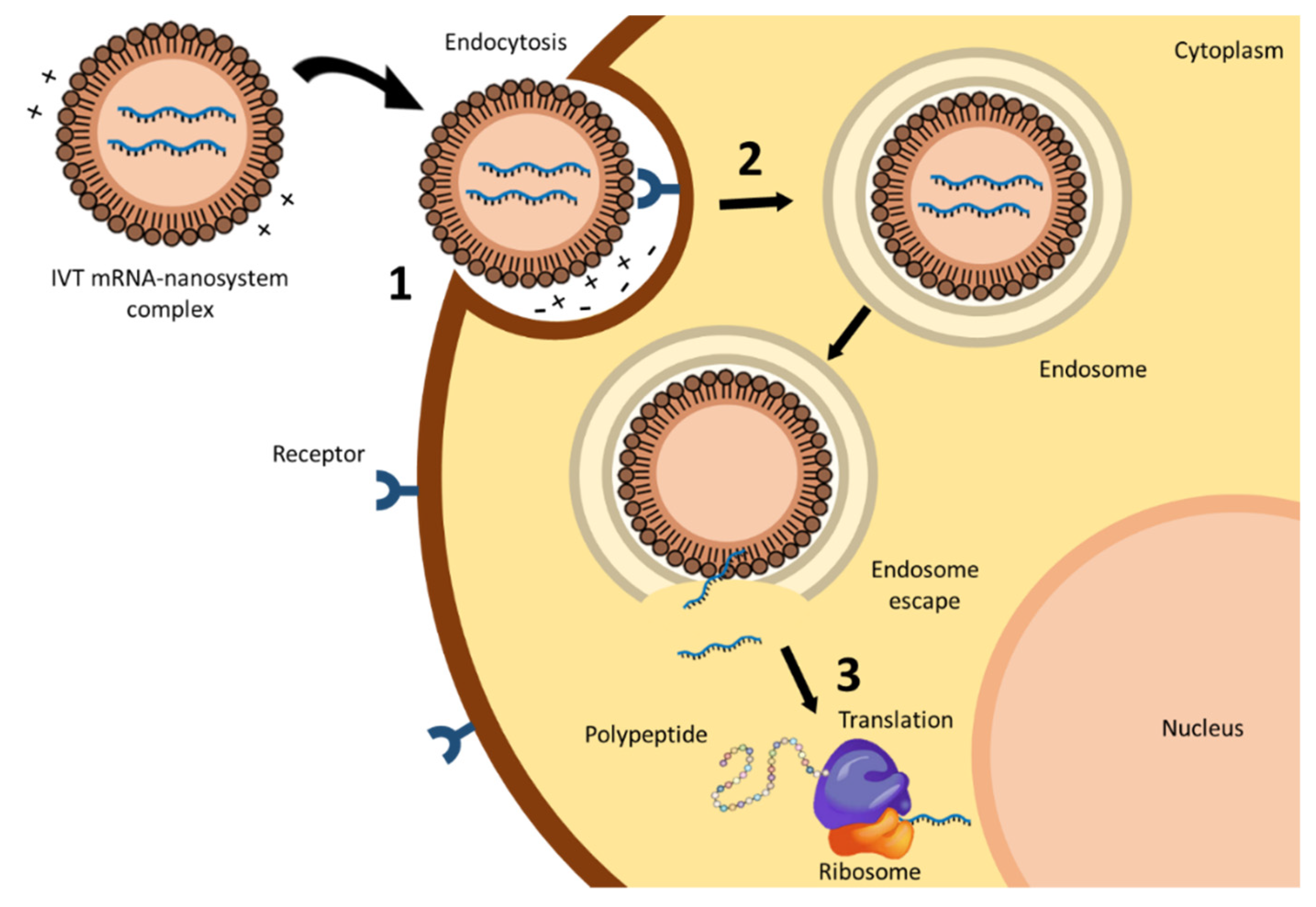
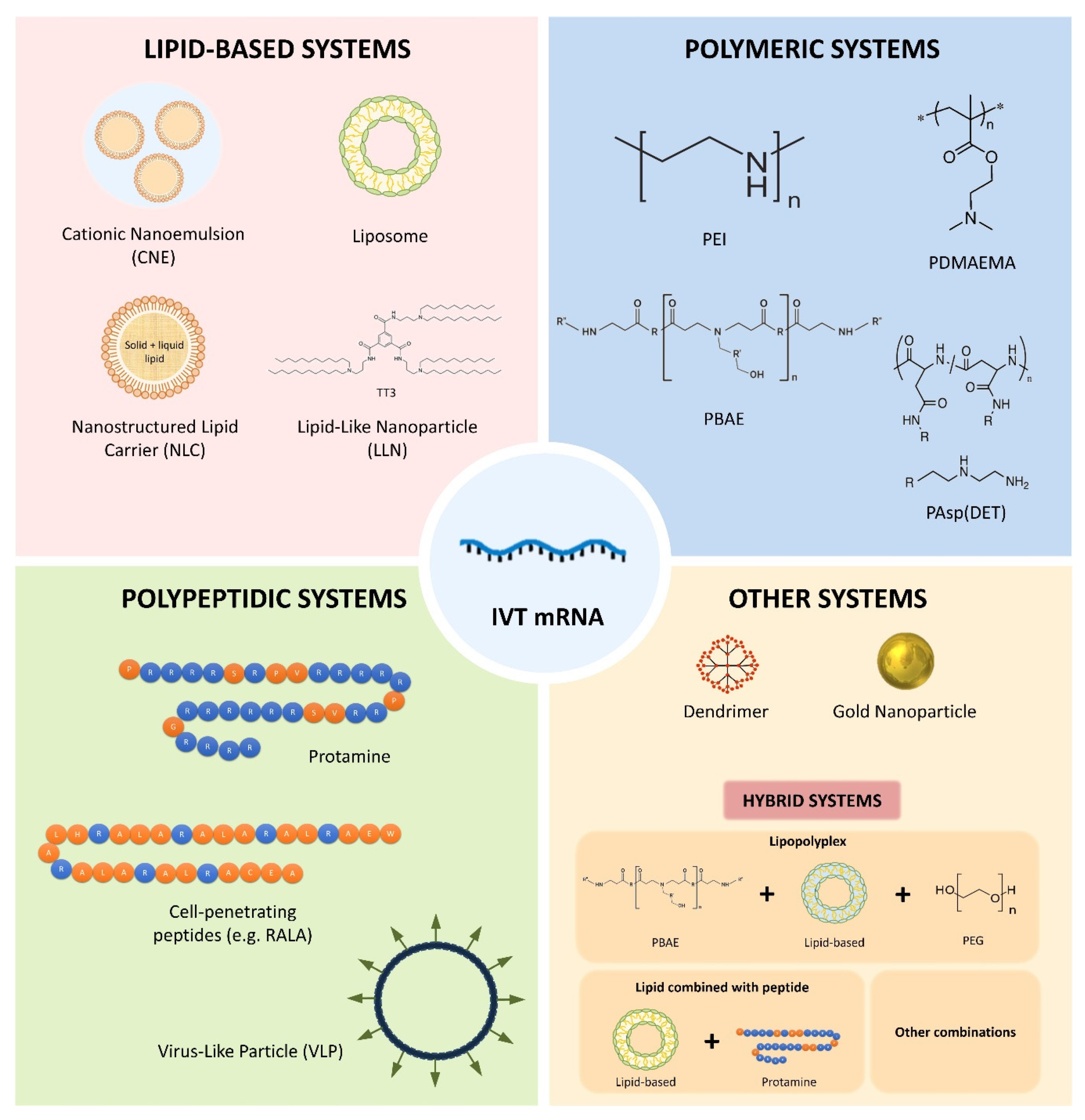
3. mRNA Nanomedicines
3.1. Lipid-Based Systems
3.2. Polymeric Systems
3.3. Polypeptidic Systems
3.4. Dendrimers
3.5. Gold Nanoparticles
3.6. Hybrid Systems
4. Therapeutic Applications of mRNA
4.1. Immunotherapy
4.1.1. mRNA Vaccines Against Infectious Diseases
4.1.2. Cancer Immunotherapy
4.2. Protein Replacement
4.3. Gene Editing
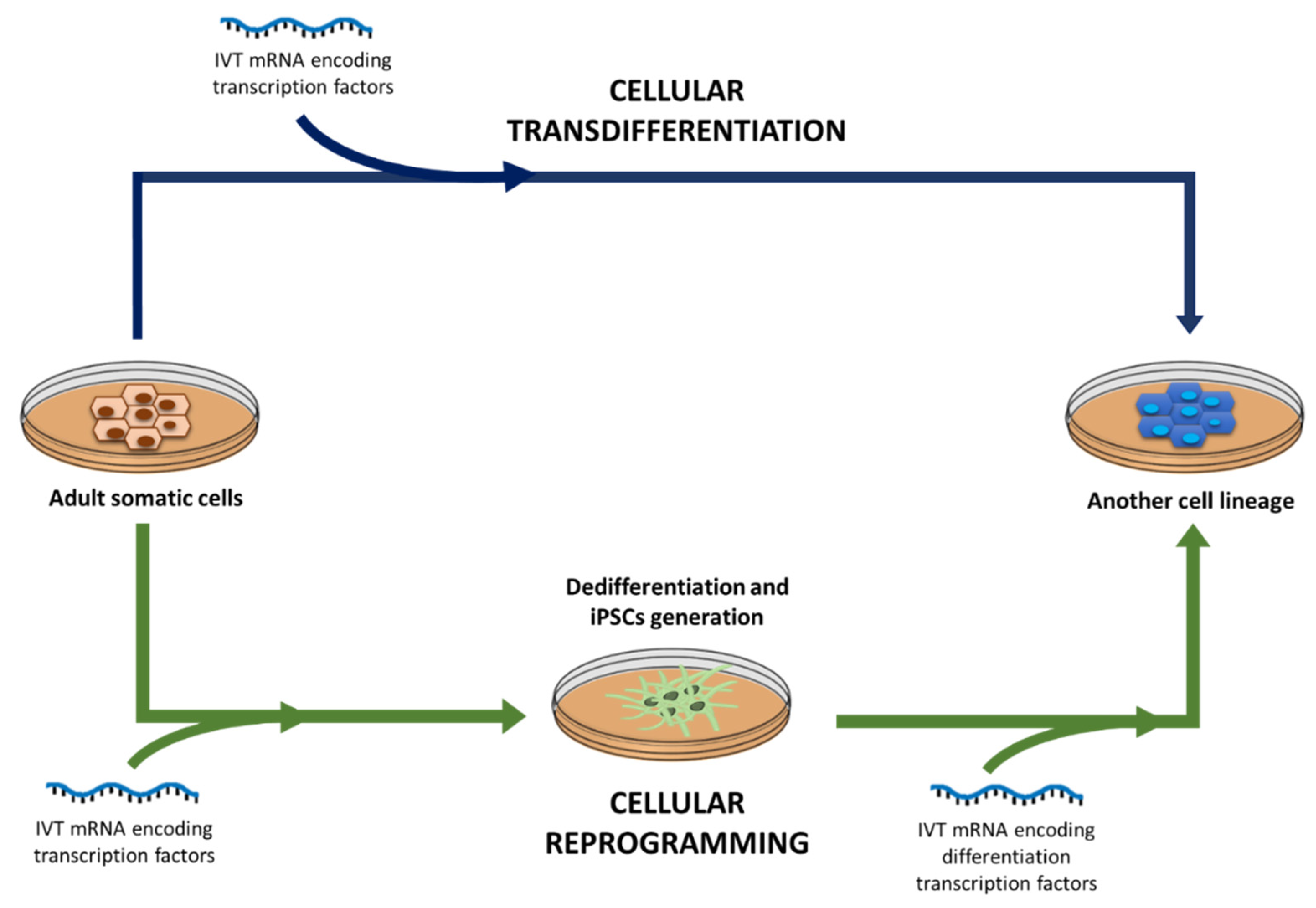
4.4. Regenerative Medicine and Cell Engineering
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- European Medicines Agency. Guideline on the quality, non-clinical and clinical aspects of gene therapy medicinal products. Eur. Med. Agency Guidel. 2015, 44, 1–41. [Google Scholar]
- del Pozo-Rodríguez, A.; Rodríguez-Gascón, A.; Rodríguez-Castejón, J.; Vicente-Pascual, M.; Gómez-Aguado, I.; Battaglia, L.S.; Solinís, M.A. Gene therapy. Adv. Biochem. Eng. Biotechnol. 2019, 171, 321–368. [Google Scholar]
- Thorne, B.; Takeya, R.; Vitelli, F.; Swanson, X. Gene Therapy. In New Bioprocessing Strategies: Development and Manufacturing of Recombinant Antibodies and Proteins. Advances in Biochemical Engineering/Biotechnology; Kiss, B., Gottschalk, U., Pohlscheidt, M., Eds.; Springer: Cham, Switzerland, 2017; Volume 165. [Google Scholar]
- Anguela, X.M.; High, K.A. Entering the Modern Era of Gene Therapy. Ann. Rev. Med. 2019, 70, 273–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajj, K.A.; Whitehead, K.A. Tools for translation: Non-viral materials for therapeutic mRNA delivery. Nat. Rev. Mater. 2017, 2, 17056. [Google Scholar] [CrossRef]
- Sahin, U.; Karikó, K.; Türeci, Ö. MRNA-based therapeutics-developing a new class of drugs. Nat. Rev. Drug Discov. 2014, 13, 759–780. [Google Scholar] [CrossRef]
- Meng, Z.; O’Keeffe-Ahern, J.; Lyu, J.; Pierucci, L.; Zhou, D.; Wang, W. A new developing class of gene delivery: Messenger RNA-based therapeutics. Biomater. Sci. 2017, 5, 2381–2392. [Google Scholar] [CrossRef]
- Zarghampoor, F.; Azarpira, N.; Khatami, S.R.; Behzad-Behbahani, A.; Foroughmand, A.M. Improved translation efficiency of therapeutic mRNA. Gene 2019, 707, 231–238. [Google Scholar] [CrossRef]
- Zhong, Z.; Mc Cafferty, S.; Combes, F.; Huysmans, H.; De Temmerman, J.; Gitsels, A.; Vanrompay, D.; Portela Catania, J.; Sanders, N.N. mRNA therapeutics deliver a hopeful message. Nano Today 2018, 23, 16–39. [Google Scholar] [CrossRef]
- Schlake, T.; Thess, A.; Thran, M.; Jordan, I. mRNA as novel technology for passive immunotherapy. Cell. Mol. Life Sci. 2019, 76, 301–328. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.; Athirasala, A.; Menezes, P.P.; Ashwanikumar, N.; Zou, T.; Sahay, G.; Bertassoni, L.E. Messenger RNA Delivery for Tissue Engineering and Regenerative Medicine Applications. Tissue Eng. Part A 2019, 25, 91–112. [Google Scholar] [CrossRef]
- Hecker, J.G. Non-Viral, Lipid-Mediated DNA and mRNA Gene Therapy of the Central Nervous System (CNS): Chemical-Based Transfection. In Gene Therapy for Neurological Disorders: Methods and Protocols; Manfredsson, F.P., Ed.; Springer: New York, NY, USA, 2016; pp. 307–324. [Google Scholar]
- Vallazza, B.; Petri, S.; Poleganov, M.A.; Eberle, F.; Kuhn, A.N.; Sahin, U. Recombinant messenger RNA technology and its application in cancer immunotherapy, transcript replacement therapies, pluripotent stem cell induction, and beyond. Wiley Interdiscip. Rev. RNA 2015, 6, 471–499. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, P.S.; Rudra, A.; Miao, L.; Anderson, D.G. Delivering the Messenger: Advances in Technologies for Therapeutic mRNA Delivery. Mol. Ther. 2019, 27, 710–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, Q.; Lee, G.Y.; Ding, J.; Li, W.; Shi, J. Biomedical applications of mRNA nanomedicine. Nano Res. 2018, 11, 5281–5309. [Google Scholar] [CrossRef] [PubMed]
- del Pozo-Rodríguez, A.; Delgado, D.; Solinís, M.A.; Gascón, A.R.; Pedraz, J.L. Solid lipid nanoparticles for retinal gene therapy: Transfection and intracellular trafficking in RPE cells. Int. J. Pharm. 2008, 360, 177–183. [Google Scholar] [CrossRef]
- Gan, L.; Wang, J.; Zhao, Y.; Chen, D.; Zhu, C.; Liu, J.; Gan, Y. Hyaluronan-modified core-shell liponanoparticles targeting CD44-positive retinal pigment epithelium cells via intravitreal injection. Biomaterials 2013, 34, 5978–5987. [Google Scholar] [CrossRef]
- Apaolaza, P.S.; del Pozo-Rodríguez, A.; Solinís, M.A.; Rodríguez, J.M.; Friedrich, U.; Torrecilla, J.; Weber, B.H.F.; Rodríguez-Gascón, A. Structural recovery of the retina in a retinoschisin-deficient mouse after gene replacement therapy by solid lipid nanoparticles. Biomaterials 2016, 90, 40–49. [Google Scholar] [CrossRef]
- Stewart, M.P.; Sharei, A.; Ding, X.; Sahay, G.; Langer, R.; Jensen, K.F. In vitro and ex vivo strategies for intracellular delivery. Nature 2016, 538, 183–192. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.; Ashwanikumar, N.; Robinson, E.; Duross, A.; Sun, C.; Murphy-Benenato, K.E.; Benenato, M.; Mihai, C.; Almarsson, O.; Sahay, G. Boosting Intracellular Delivery of Lipid Nanoparticle-Encapsulated mRNA. Nano Lett. 2017, 17, 5711–5718. [Google Scholar] [CrossRef]
- Steinle, H.; Behring, A.; Schlensak, C.; Wendel, H.P.; Avci-Adali, M. Concise Review: Application of In Vitro Transcribed Messenger RNA for Cellular Engineering and Reprogramming: Progress and Challenges. Stem Cells 2017, 35, 68–79. [Google Scholar] [CrossRef] [Green Version]
- Karikó, K.; Muramatsu, H.; Ludwig, J.; Weissman, D. Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic Acids Res. 2011, 39, e142. [Google Scholar] [CrossRef] [Green Version]
- Weissman, D. mRNA transcript therapy. Expert Rev. Vaccines 2014, 14, 265–281. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.A.; Reesor, E.K.G.; Xu, Y.; Zope, H.R.; Zetter, B.R.; Shi, J. Biomaterials for mRNA delivery. Biomater. Sci. 2015, 3, 1519–1533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramanathan, A.; Robb, G.B.; Chan, S.H. mRNA capping: Biological functions and applications. Nucleic Acids Res. 2016, 44, 7511–7526. [Google Scholar] [CrossRef] [PubMed]
- Muttach, F.; Muthmann, N.; Rentmeister, A. Synthetic mRNA capping. Beilstein J. Org. Chem. 2017, 13, 2819–2832. [Google Scholar] [CrossRef] [PubMed]
- Werner, M.; Purta, E.; Kaminska, K.H.; Cymerman, I.A.; Campbell, D.A.; Mittra, B.; Zamudio, J.R.; Sturm, N.R.; Jaworski, J.; Bujnicki, J.M. 2′-O-ribose methylation of cap2 in human: Function and evolution in a horizontally mobile family. Nucleic Acids Res. 2011, 39, 4756–4768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCracken, S.; Fong, N.; Rosonina, E.; Yankulov, K.; Brothers, G.; Siderovski, D.; Hessel, A.; Foster, S.; Shuman, S.; Bentley, D.L. 5′-Capping enzymes are targeted to pre-mRNA by binding to the phosphorylated carboxy-terminal domain of RNA polymerase II. Genes Dev. 1997, 11, 3306–3318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchs, A.-L.; Neu, A.; Sprangers, R. A general method for rapid and cost-efficient large-scale production of 5′ capped RNA. RNA 2016, 22, 1454–1466. [Google Scholar] [CrossRef] [Green Version]
- Trilink Biotechnologies. Available online: https://www.trilinkbiotech.com/cleancap (accessed on 30 January 2020).
- Warminski, M.; Kowalska, J.; Buck, J.; Zuberek, J.; Lukaszewicz, M.; Nicola, C.; Kuhn, A.N.; Sahin, U.; Darzynkiewicz, E.; Jemielity, J. The synthesis of isopropylidene mRNA cap analogs modified with phosphorothioate moiety and their evaluation as promoters of mRNA translation. Bioorg. Med. Chem. Lett. 2013, 23, 3753–3758. [Google Scholar] [CrossRef]
- Ziemniak, M.; Kowalska, J.; Lukaszewicz, M.; Zuberek, J.; Wnek, K.; Darzynkiewicz, E.; Jemielity, J. Phosphate-modified analogues of m7GTP and m7Gppppm7G - Synthesis and biochemical properties. Bioorg. Med. Chem. 2015, 23, 5369–5381. [Google Scholar] [CrossRef]
- Kuhn, A.N.; Diken, M.; Kreiter, S.; Selmi, A.; Kowalska, J.; Jemielity, J.; Darzynkiewicz, E.; Huber, C.; Türeci, Ö.; Sahin, U. Phosphorothioate cap analogs increase stability and translational efficiency of RNA vaccines in immature dendritic cells and induce superior immune responses in vivo. Gene Ther. 2010, 17, 961–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strenkowska, M.; Kowalska, J.; Lukaszewicz, M.; Zuberek, J.; Su, W.; Rhoads, R.E.; Darzynkiewicz, E.; Jemielity, J. Towards mRNA with superior translational activity: Synthesis and properties of ARCA tetraphosphates with single phosphorothioate modifications. New J. Chem. 2010, 34, 993–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zytek, M.; Kowalska, J.; Lukaszewicz, M.; Wojtczak, B.A.; Zuberek, J.; Ferenc-Mrozek, A.; Darzynkiewicz, E.; Niedzwiecka, A.; Jemielity, J. Towards novel efficient and stable nuclear import signals: Synthesis and properties of trimethylguanosine cap analogs modified within the 5′,5′-triphosphate bridge. Org. Biomol. Chem. 2014, 12, 9184–9199. [Google Scholar] [CrossRef] [PubMed]
- Rydzik, A.M.; Kulis, M.; Lukaszewicz, M.; Kowalska, J.; Zuberek, J.; Darzynkiewicz, Z.M.; Darzynkiewicz, E.; Jemielity, J. Synthesis and properties of mRNA cap analogs containing imidodiphosphate moiety—Fairly mimicking natural cap structure, yet resistant to enzymatic hydrolysis. Bioorg. Med. Chem. 2012, 20, 1699–1710. [Google Scholar] [CrossRef]
- Al-Saif, M.; Khabar, K.S.A. UU/UA dinucleotide frequency reduction in coding regions results in increased mRNA stability and protein expression. Mol. Ther. 2012, 20, 954–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauro, V.P.; Chappell, S.A. A critical analysis of codon optimization in human therapeutics. Trends Mol. Med. 2014, 20, 604–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexaki, A.; Hettiarachchi, G.K.; Athey, J.C.; Katneni, U.K.; Simhadri, V.; Hamasaki-Katagiri, N.; Nanavaty, P.; Lin, B.; Takeda, K.; Freedberg, D.; et al. Effects of codon optimization on coagulation factor IX translation and structure: Implications for protein and gene therapies. Sci. Rep. 2019, 9, 15449. [Google Scholar] [CrossRef] [Green Version]
- Presnyak, V.; Alhusaini, N.; Chen, Y.H.; Martin, S.; Morris, N.; Kline, N.; Olson, S.; Weinberg, D.; Baker, K.E.; Graveley, B.R.; et al. Codon optimality is a major determinant of mRNA stability. Cell 2015, 160, 1111–1124. [Google Scholar] [CrossRef] [Green Version]
- Hunt, R.C.; Simhadri, V.L.; Iandoli, M.; Sauna, Z.E.; Kimchi-Sarfaty, C. Exposing synonymous mutations. Trends Genet. 2014, 30, 308–321. [Google Scholar] [CrossRef]
- McCarthy, C.; Carrea, A.; Diambra, L. Bicodon bias can determine the role of synonymous SNPs in human diseases. BMC Genom. 2017, 18, 227. [Google Scholar] [CrossRef] [Green Version]
- Bali, V.; Bebok, Z. Decoding mechanisms by which silent codon changes influence protein biogenesis and function. Int. J. Biochem. Cell Biol. 2015, 64, 58–74. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Zhang, X.; Dong, Y. Nanoscale platforms for messenger RNA delivery. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2019, 11, e1530. [Google Scholar] [CrossRef] [PubMed]
- Jalkanen, A.L.; Coleman, S.J.; Wilusz, J. Determinants and implications of mRNA poly(A) tail size—Does this protein make my tail look big. In Seminars in Cell & Developmental Biology; Academic Press: Cambridge, MA, USA, 2014; Volume 34, pp. 24–32. [Google Scholar]
- Eckmann, C.R.; Rammelt, C.; Wahle, E. Control of poly(A) tail length. Wiley Interdiscip. Rev. RNA 2011, 2, 348–361. [Google Scholar] [CrossRef] [PubMed]
- Weill, L.; Belloc, E.; Bava, F.A.; Méndez, R. Translational control by changes in poly(A) tail length: Recycling mRNAs. Nat. Struct. Mol. Biol. 2012, 19, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Schoenberg, D.R. mRNA with a <20-nt poly(A) tail imparted by the poly(A)-limiting element is translated as efficiently in vivo as long poly(A) mRNA. RNA 2005, 11, 1131–1140. [Google Scholar] [PubMed] [Green Version]
- Holtkamp, S.; Kreiter, S.; Selmi, A.; Simon, P.; Koslowski, M.; Huber, C.; Türeci, O.; Sahin, U. Modification of antigen-encoding RNA increases stability, translational efficacy, and T-cell stimulatory capacity of dendritic cells. Blood 2006, 108, 4009–4017. [Google Scholar] [CrossRef] [PubMed]
- Mockey, M.; Gonçalves, C.; Dupuy, F.P.; Lemoine, F.M.; Pichon, C.; Midoux, P. mRNA transfection of dendritic cells: Synergistic effect of ARCA mRNA capping with Poly(A) chains in cis and in trans for a high protein expression level. Biochem. Biophys. Res. Commun. 2006, 340, 1062–1068. [Google Scholar] [CrossRef]
- Kwon, H.; Kim, M.; Seo, Y.; Moon, Y.S.; Lee, H.J.; Lee, K.; Lee, H. Emergence of synthetic mRNA: In vitro synthesis of mRNA and its applications in regenerative medicine. Biomaterials 2018, 156, 172–193. [Google Scholar] [CrossRef]
- Matoulkova, E.; Michalova, E.; Vojtesek, B.; Hrstka, R. The role of the 3′ untranslated region in post-transcriptional regulation of protein expression in mammalian cells. RNA Biol. 2012, 9, 563–576. [Google Scholar] [CrossRef] [Green Version]
- Hellen, C.U.T.; Sarnow, P. Internal ribosome entry sites in eukaryotic mRNA molecules. Genes Dev. 2001, 15, 1593–1612. [Google Scholar] [CrossRef] [Green Version]
- Johnson, A.G.; Grosely, R.; Petrov, A.N.; Puglisi, J.D.; Puglisi, J.D. Dynamics of IRES-mediated translation. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160177. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, Z. IRES-mediated cap-independent translation, a path leading to hidden proteome. J. Mol. Cell Biol. 2019, 11, 911–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozak, M. Point mutations close to the AUG initiator codon affect the efficiency of translation of rat preproinsulin in vivo. Nature 1984, 308, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Kozak, M. At least six nucleotides preceding the AUG initiator codon enhance translation in mammalian cells. J. Mol. Biol. 1987, 196, 947–950. [Google Scholar] [CrossRef]
- Jiang, Y.; Xu, X.-S.; Russell, J.E. A Nucleolin-Binding 3′ Untranslated Region Element Stabilizes β-Globin mRNA In Vivo. Mol. Cell. Biol. 2006, 26, 2419–2429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volloch, V.; Housman, D. Stability of globin mRNA in terminally differentiating murine erythroleukemia cells. Cell 1981, 23, 509–514. [Google Scholar] [CrossRef]
- Berkovits, B.D.; Mayr, C. Alternative 3′ UTRs act as scaffolds to regulate membrane protein localization. Nature 2015, 522, 363–367. [Google Scholar] [CrossRef] [Green Version]
- Karikó, K.; Muramatsu, H.; Welsh, F.A.; Ludwig, J.; Kato, H.; Akira, S.; Weissman, D. Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol. Ther. 2008, 16, 1833–1840. [Google Scholar] [CrossRef]
- Hornung, V.; Ellegast, J.; Kim, S.; Brzózka, K.; Jung, A.; Kato, H.; Poeck, H.; Akira, S.; Conzelmann, K.-K.; Schlee, M.; et al. 5′-Triphosphate RNA Is the Ligand for RIG-I. Science 2006, 314, 994–997. [Google Scholar] [CrossRef] [Green Version]
- Diebold, S.S.; Massacrier, C.; Akira, S.; Paturel, C.; Morel, Y.; Reis e Sousa, C. Nucleic acid agonists for Toll-like receptor 7 are defined by the presence of uridine ribonucleotides. Eur. J. Immunol. 2006, 36, 3256–3267. [Google Scholar] [CrossRef]
- Gorden, K.K.B.; Qiu, X.; Battiste, J.J.L.; Wightman, P.P.D.; Vasilakos, J.P.; Alkan, S.S. Oligodeoxynucleotides Differentially Modulate Activation of TLR7 and TLR8 by Imidazoquinolines. J. Immunol. 2006, 177, 8164–8170. [Google Scholar] [CrossRef] [Green Version]
- Karikó, K.; Buckstein, M.; Ni, H.; Weissman, D. Suppression of RNA recognition by Toll-like receptors: The impact of nucleoside modification and the evolutionary origin of RNA. Immunity 2005, 23, 165–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenz, C.; Fotin-Mleczek, M.; Roth, G.; Becker, C.; Dam, T.C.; Verdurmen, W.P.R.; Brock, R.; Probst, J.; Schlake, T. Protein expression from exogenous mRNA: Uptake by receptor-mediated endocytosis and trafficking via the lysosomal pathway. RNA Biol. 2011, 8, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Svitkin, Y.V.; Cheng, Y.M.; Chakraborty, T.; Presnyak, V.; John, M.; Sonenberg, N. N1-methyl-pseudouridine in mRNA enhances translation through eIF2α-dependent and independent mechanisms by increasing ribosome density. Nucleic Acids Res. 2017, 45, 6023–6036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andries, O.; Mc Cafferty, S.; De Smedt, S.C.; Weiss, R.; Sanders, N.N.; Kitada, T. N1-methylpseudouridine-incorporated mRNA outperforms pseudouridine-incorporated mRNA by providing enhanced protein expression and reduced immunogenicity in mammalian cell lines and mice. J. Control. Release 2015, 217, 337–344. [Google Scholar] [CrossRef]
- Martini, P.G.V.; Guey, L.T. A New Era for Rare Genetic Diseases: Messenger RNA Therapy. Hum. Gene Ther. 2019, 30, 1180–1189. [Google Scholar] [CrossRef]
- Meyer, K.D.; Patil, D.P.; Zhou, J.; Zinoviev, A.; Skabkin, M.A.; Elemento, O.; Pestova, T.V.; Qian, S.-B.; Jaffrey, S.R. 5′ UTR m6A Promotes Cap-Independent Translation. Cell 2015, 163, 999–1010. [Google Scholar] [CrossRef] [Green Version]
- Ulkoski, D.; Bak, A.; Wilson, J.T.; Krishnamurthy, V.R. Recent advances in polymeric materials for the delivery of RNA therapeutics. Expert Opin. Drug Deliv. 2019, 16, 1149–1167. [Google Scholar] [CrossRef]
- Rodríguez-Gascón, A.; del Pozo-Rodríguez, A.; Solinís, M.Á. Development of nucleic acid vaccines: Use of self-amplifying RNA in lipid nanoparticles. Int. J. Nanomed. 2014, 9, 1833–1843. [Google Scholar] [CrossRef] [Green Version]
- Gene Therapy Clinical Trials Worldwide, Provided by the Journal of Gene Medicine, Jon Wiley and Sons Ltd. 2019. Available online: http://www.abedia.com/wiley/vectors.php (accessed on 13 January 2020).
- Carvalho, M.; Sepodes, B.; Martins, A.P. Regulatory and Scientific Advancements in Gene Therapy: State-of-the-Art of Clinical Applications and of the Supporting European Regulatory Framework. Front. Med. 2017, 4, 182. [Google Scholar] [CrossRef] [Green Version]
- del Pozo-Rodríguez, A.; Solinís, M.Á.; Rodríguez-Gascón, A. Applications of lipid nanoparticles in gene therapy. Eur. J. Pharm. Biopharm. 2016, 109, 184–193. [Google Scholar] [CrossRef]
- Wang, Y.; Rajala, A.; Rajala, R. Lipid Nanoparticles for Ocular Gene Delivery. J. Funct. Biomater. 2015, 6, 379–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, H.; Kanasty, R.L.; Eltoukhy, A.A.; Vegas, A.J.; Dorkin, J.R.; Anderson, D.G. Non-viral vectors for gene-based therapy. Nat. Rev. Genet. 2014, 15, 541–555. [Google Scholar] [CrossRef] [PubMed]
- Guan, S.; Rosenecker, J. Nanotechnologies in delivery of mRNA therapeutics using nonviral vector-based delivery systems. Gene Ther. 2017, 24, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Gascón, A.; del Pozo-Rodríguez, A.; Isla, A.; Solinís, M.A. Vaginal gene therapy. Adv. Drug Deliv. Rev. 2015, 92, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Gascón, A.R.; del Pozo-Rodríguez, A.; Solinís, M.A. Non-Viral Delivery Systems in Gene Therapy. In Gene Therapy; Molina, F.M., Ed.; IntechOpen: Rijeka, Croatia, 2013. [Google Scholar]
- Van Tendeloo, V.F.I.; Ponsaerts, P.; Lardon, F.; Nijs, G.; Lenjou, M.; Van Broeckhoven, C.; Van Bockstaele, D.R.V.; Berneman, Z.N. Highly efficient gene delivery by mRNA electroporation in human hematopoietic cells: Superiority to lipofection and passive pulsing of mRNA and to electroporation of plasmid cDNA for tumor antigen loading of dendritic cells. Blood 2001, 98, 49–56. [Google Scholar] [CrossRef]
- Bugeon, S.; De Chevigny, A.; Boutin, C.; Coré, N.; Wild, S.; Bosio, A. Harold Cremer 3, Christophe Beclin Direct and efficient transfection of mouse neural stem cells and mature neurons by in vivo mRNA electroporation. Development 2017, 144, 3968–3977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golombek, S.; Pilz, M.; Steinle, H.; Kochba, E.; Levin, Y.; Lunter, D.; Schlensak, C.; Wendel, H.P.; Avci-Adali, M. Intradermal Delivery of Synthetic mRNA Using Hollow Microneedles for Efficient and Rapid Production of Exogenous Proteins in Skin. Mol. Ther Nucleic Acids 2018, 11, 382–392. [Google Scholar] [CrossRef] [Green Version]
- Moody, S.A. Microinjection of mRNAs and oligonucleotides. Cold Spring Harb. Protoc. 2018, 2018, 923–932. [Google Scholar] [CrossRef]
- Ainger, K.; Avossa, D.; Morgan, F.; Hill, S.J.; Barry, C.; Barbarese, E.; Carson, J.H. Transport and localization of exogenous myelin basic protein mRNA microinjected into oligodendrocytes. J. Cell Biol. 1993, 123, 431–441. [Google Scholar] [CrossRef]
- Belyantseva, I.A. Helios® Gene Gun-Mediated Transfection of the Inner Ear Sensory Epithelium: Recent Updates. In Auditory and Vestibular Research: Methods and Protocols; Sokolowski, B., Ed.; Springer: New York, NY, USA, 2016; pp. 3–26. [Google Scholar]
- Vassilev, V.B.; Gil, L.H.V.G.; Donis, R.O. Microparticle-mediated RNA immunization against bovine viral diarrhea virus. Vaccine 2001, 19, 2012–2019. [Google Scholar] [CrossRef]
- Ramezanpour, M.; Schmidt, M.L.; Bodnariuc, I.; Kulkarni, J.A.; Leung, S.S.W.; Cullis, P.R.; Thewalt, J.L.; Tieleman, D.P. Ionizable amino lipid interactions with POPC: Implications for lipid nanoparticle function. Nanoscale 2019, 11, 14141–14146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Islam, M.A.; Xu, Y.; Tao, W.; Ubellacker, J.M.; Lim, M.; Aum, D.; Lee, G.Y.; Zhou, K.; Zope, H.; Yu, M.; et al. Restoration of tumour-growth suppression in vivo via systemic nanoparticle-mediated delivery of PTEN mRNA. Nat. Biomed. Eng. 2018, 2, 850–864. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, K.J.; Webber, M.J.; Anderson, D.G. Materials for non-viral intracellular delivery of messenger RNA therapeutics. J. Control. Release 2016, 240, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Phua, K.K.L.; Leong, K.W.; Nair, S.K. Transfection Efficiency and Transgene Expression Kinetics of mRNA Delivered in Naked and Nanoparticle Format. J. Control. Release 2013, 166, 227–233. [Google Scholar] [CrossRef] [Green Version]
- Slivac, I.; Guay, D.; Mangion, M.; Champeil, J.; Gaillet, B. Non-viral nucleic acid delivery methods. Expert Opin. Biol. Ther. 2017, 17, 105–118. [Google Scholar] [CrossRef]
- Aderibigbe, B.A.; Ray, S.S. Preparation, characterization and in vitro release kinetics of polyaspartamide-based conjugates containing antimalarial and anticancer agents for combination therapy. J. Drug Deliv. Sci. Technol. 2016, 36, 34–45. [Google Scholar] [CrossRef]
- Malone, R.W.; Felgner, P.L.; Verma, I.M. Cationic liposome-mediated RNA transfection. Proc. Natl. Acad. Sci. USA 1989, 86, 6077–6081. [Google Scholar] [CrossRef] [Green Version]
- Koltover, I.; Salditt, T.; Rädler, J.O.; Safinya, C.R. An inverted hexagonal phase of cationic liposome-DNA complexes related to DNA release and delivery. Science 1998, 281, 78–81. [Google Scholar] [CrossRef] [Green Version]
- Sayour, E.J.; De Leon, G.; Pham, C.; Grippin, A.; Kemeny, H.; Chua, J.; Huang, J.; Sampson, J.H.; Sanchez-Perez, L.; Flores, C.; et al. Systemic activation of antigen-presenting cells via RNA-Loaded nanoparticles. Oncoimmunology 2017, 6, e1256527. [Google Scholar] [CrossRef] [Green Version]
- Kranz, L.M.; Diken, M.; Haas, H.; Kreiter, S.; Loquai, C.; Reuter, K.C.; Meng, M.; Fritz, D.; Fulvia, V.; Hefesha, H.; et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 2016, 534, 396–401. [Google Scholar] [CrossRef]
- Kulkarni, J.A.; Cullis, P.R.; Van Der Meel, R. Lipid Nanoparticles Enabling Gene Therapies: From Concepts to Clinical Utility. Nucleic Acid Ther. 2018, 28, 146–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bogers, W.M.; Oostermeijer, H.; Mooij, P.; Koopman, G.; Verschoor, E.J.; Davis, D.; Ulmer, J.B.; Brito, L.A.; Cu, Y.; Banerjee, K.; et al. Potent immune responses in rhesus macaques induced by nonviral delivery of a self-amplifying RNA vaccine expressing HIV type 1 envelope with a cationic nanoemulsion. J. Infect. Dis. 2015, 211, 947–955. [Google Scholar] [CrossRef] [PubMed]
- Brito, L.A.; Chan, M.; Shaw, C.A.; Hekele, A.; Carsillo, T.; Schaefer, M.; Archer, J.; Seubert, A.; Otten, G.R.; Beard, C.W.; et al. A cationic nanoemulsion for the delivery of next-generation RNA vaccines. Mol. Ther. 2014, 22, 2118–2129. [Google Scholar] [CrossRef] [Green Version]
- Kauffman, K.J.; Dorkin, J.R.; Yang, J.H.; Heartlein, M.W.; Derosa, F.; Mir, F.F.; Fenton, O.S.; Anderson, D.G. Optimization of Lipid Nanoparticle Formulations for mRNA Delivery in Vivo with Fractional Factorial and Definitive Screening Designs. Nano Lett. 2015, 15, 7300–7306. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Goel, V.; Robbie, G.J. Pharmacokinetics of Patisiran, the First Approved RNA Interference Therapy in Patients with Hereditary Transthyretin-Mediated Amyloidosis. J. Clin. Pharmacol. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sedic, M.; Senn, J.J.; Lynn, A.; Laska, M.; Smith, M.; Platz, S.J.; Bolen, J.; Hoge, S.; Bulychev, A.; Jacquinet, E.; et al. Safety Evaluation of Lipid Nanoparticle–Formulated Modified mRNA in the Sprague-Dawley Rat and Cynomolgus Monkey. Vet. Pathol. 2018, 55, 341–354. [Google Scholar] [CrossRef]
- Lu, D.; Benjamin, R.; Kim, M.; Conry, R.M.; Curiel, D.T. Optimization of methods to achieve mRNA-mediated transfection of tumor cells in vitro and in vivo employing cationic liposome vectors. Cancer Gene Ther. 1994, 1, 245–252. [Google Scholar]
- Pardi, N.; Hogan, M.J.; Pelc, R.S.; Muramatsu, H.; Andersen, H.; DeMaso, C.R.; Dowd, K.A.; Sutherland, L.L.; Scearce, R.M.; Parks, R.; et al. Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature 2017, 543, 248–251. [Google Scholar] [CrossRef]
- Hekele, A.; Bertholet, S.; Archer, J.; Gibson, D.G.; Palladino, G.; Brito, L.A.; Hekele, A.; Bertholet, S.; Archer, J.; Gibson, D.G.; et al. Rapidly produced SAM vaccine against H7N9 influenza is immunogenic in mice. Emerg. Microbes Infect. 2013, 2, e52. [Google Scholar] [CrossRef]
- Zhou, W.Z.; Hoon, D.S.B.; Huang, S.K.S.; Fujii, S.; Hashimoto, K.; Morishita, R.; Kaneda, Y. RNA melanoma vaccine: Induction of antitumor immunity by human glycoprotein 100 mRNA immunization. Hum. Gene. Ther. 1999, 10, 2719–2724. [Google Scholar] [CrossRef]
- Finn, J.D.; Smith, A.R.; Patel, M.C.; Shaw, L.; Youniss, M.R.; van Heteren, J.; Dirstine, T.; Ciullo, C.; Lescarbeau, R.; Seitzer, J.; et al. A Single Administration of CRISPR/Cas9 Lipid Nanoparticles Achieves Robust and Persistent In Vivo Genome Editing. Cell Rep. 2018, 22, 2227–2235. [Google Scholar] [CrossRef] [Green Version]
- Kormann, M.S.D.; Hasenpusch, G.; Aneja, M.K.; Nica, G.; Flemmer, A.W.; Herber-Jonat, S.; Huppmann, M.; Mays, L.E.; Illenyi, M.; Schams, A.; et al. Expression of therapeutic proteins after delivery of chemically modified mRNA in mice. Nat. Biotechnol. 2011, 29, 154–159. [Google Scholar] [CrossRef]
- Akinc, A.; Zumbuehl, A.; Goldberg, M.; Leshchiner, E.S.; Busini, V.; Hossain, N.; Bacallado, S.A.; Nguyen, D.N.; Fuller, J.; Alvarez, R.; et al. A combinatorial library of lipid-like materials for delivery of RNAi therapeutics. Nat. Biotechnol. 2008, 26, 561–569. [Google Scholar] [CrossRef]
- Miller, J.B.; Zhang, S.; Kos, P.; Xiong, H.; Zhou, K.; Perelman, S.S.; Zhu, H.; Siegwart, D.J. Non-Viral CRISPR/Cas Gene Editing In Vitro and In Vivo Enabled by Synthetic Nanoparticle Co-Delivery of Cas9 mRNA and sgRNA. Angew. Chem. Int. Ed. 2017, 56, 1059–1063. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Luo, X.; Deng, B.; Wang, J.; McComb, D.W.; Shi, Y.; Gaensler, K.M.L.; Tan, X.; Dunn, A.L.; Kerlin, B.A.; et al. An Orthogonal Array Optimization of Lipid-like Nanoparticles for mRNA Delivery in Vivo. Nano Lett. 2015, 15, 8099–8107. [Google Scholar] [CrossRef] [Green Version]
- Nagpal, G.; Chaudhary, K.; Dhanda, S.K.; Pal, G.; Raghava, S. Computational Prediction of the Immunomodulatory Potential of RNA Sequences. Methods Mol. Biol. 2017, 1632, 75–90. [Google Scholar]
- Beloqui, A.; Solinís, M.Á.; Rodríguez-Gascón, A.; Almeida, A.J.; Préat, V. Nanostructured lipid carriers: Promising drug delivery systems for future clinics. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 143–161. [Google Scholar] [CrossRef]
- Beloqui, A.; del Pozo-Rodríguez, A.; Isla, A.; Rodríguez-Gascón, A.; Solinís, M.Á. Nanostructured lipid carriers as oral delivery systems for poorly soluble drugs. J. Drug Deliv. Sci. Technol. 2017, 42, 144–154. [Google Scholar] [CrossRef]
- Beloqui, A.; Solinís, M.Á.; des Rieux, A.; Préat, V.; Rodríguez-Gascón, A. Dextran–protamine coated nanostructured lipid carriers as mucus-penetrating nanoparticles for lipophilic drugs. Int. J. Pharm. 2014, 468, 105–111. [Google Scholar] [CrossRef]
- Erasmus, J.H.; Khandhar, A.P.; Guderian, J.; Granger, B.; Archer, J.; Archer, M.; Gage, E.; Fuerte-Stone, J.; Larson, E.; Lin, S.; et al. A Nanostructured Lipid Carrier for Delivery of a Replicating Viral RNA Provides Single, Low-Dose Protection against Zika. Mol. Ther. 2018, 26, 2507–2522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godbey, W.T.; Wu, K.K.; Mikos, A.G. Size matters: Molecular weight affects the efficiency of poly(ethylenimine) as a gene delivery vehicle. J. Biomed. Mater. Res. 1999, 45, 268–275. [Google Scholar] [CrossRef]
- Lv, H.; Zhang, S.; Wang, B.; Cui, S.; Yan, J. Toxicity of cationic lipids and cationic polymers in gene delivery. J. Control. Release 2006, 114, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Rejman, J.; Tavernier, G.; Bavarsad, N.; Demeester, J.; De Smedt, S.C. MRNA transfection of cervical carcinoma and mesenchymal stem cells mediated by cationic carriers. J. Control. Release 2010, 147, 385–391. [Google Scholar] [CrossRef]
- Sultana, N.; Magadum, A.; Hadas, Y.; Kondrat, J.; Singh, N.; Youssef, E.; Calderon, D.; Chepurko, E.; Dubois, N.; Hajjar, R.J.; et al. Optimizing Cardiac Delivery of Modified mRNA. Mol. Ther. 2017, 25, 1306–1315. [Google Scholar] [CrossRef] [Green Version]
- Üzgün, S.; Nica, G.; Pfeifer, C.; Bosinco, M.; Michaelis, K.; Lutz, J.F.; Schneider, M.; Rosenecker, J.; Rudolph, C. PEGylation improves nanoparticle formation and transfection efficiency of messenger RNA. Pharm. Res. 2011, 28, 2223–2232. [Google Scholar]
- Cheng, C.; Convertine, A.J.; Stayton, P.S.; Bryers, J.D. Multifunctional triblock copolymers for intracellular messenger RNA delivery. Biomaterials 2012, 33, 6868–6876. [Google Scholar] [CrossRef] [Green Version]
- Jarzębińska, A.; Pasewald, T.; Lambrecht, J.; Mykhaylyk, O.; Kümmerling, L.; Beck, P.; Hasenpusch, G.; Rudolph, C.; Plank, C.; Dohmen, C. A Single Methylene Group in Oligoalkylamine-Based Cationic Polymers and Lipids Promotes Enhanced mRNA Delivery. Angew. Chem. Int. Ed. 2016, 55, 9591–9595. [Google Scholar]
- Liu, Y.; Li, Y.; Keskin, D.; Shi, L. Poly (β-Amino Esters): Synthesis, Formulations, and Their Biomedical Applications. Adv. Healthc. Mater. 2019, 8, 1801359. [Google Scholar] [CrossRef]
- Kaczmarek, J.C.; Patel, A.K.; Kauffman, K.J.; Fenton, O.S.; Webber, M.J.; Heartlein, M.W.; DeRosa, F. Daniel G Anderson. Polymer–Lipid Nanoparticles for Systemic Delivery of mRNA to the Lungs. Angew. Chem. Int. Ed. 2016, 55, 13808–13812. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.K.; Kaczmarek, J.C.; Bose, S.; Kauffman, K.J.; Mir, F.; Heartlein, M.W.; DeRosa, F.; Langer, R.; Anderson, D.G. Inhaled Nanoformulated mRNA Polyplexes for Protein Production in Lung Epithelium. Adv. Mater. 2019, 31, 1805116. [Google Scholar] [CrossRef] [Green Version]
- Uchida, S.; Kataoka, K. Design concepts of polyplex micelles for in vivo therapeutic delivery of plasmid DNA and messenger RNA. J. Biomed. Mater. Res. Part A 2019, 107, 978–990. [Google Scholar] [CrossRef]
- Uchida, H.; Itaka, K.; Nomoto, T.; Ishii, T.; Suma, T.; Ikegami, M.; Miyata, K.; Oba, M.; Nishiyama, N.; Kataoka, K. Modulated protonation of side chain aminoethylene repeats in N-substituted polyaspartamides promotes mRNA transfection. J. Am. Chem. Soc. 2014, 136, 12396–12405. [Google Scholar] [CrossRef] [Green Version]
- Baba, M.; Itaka, K.; Kondo, K.; Yamasoba, T.; Kataoka, K. Treatment of neurological disorders by introducing mRNA in vivo using polyplex nanomicelles. J. Control. Release 2015, 201, 41–48. [Google Scholar] [CrossRef] [Green Version]
- Matsui, A.; Uchida, S.; Ishii, T.; Itaka, K.; Kataoka, K. Messenger RNA-based therapeutics for the treatment of apoptosis-associated diseases. Sci. Rep. 2015, 5, 15810. [Google Scholar] [CrossRef]
- Palamà, I.E.; Cortese, B.; D’Amone, S.; Gigli, G. MRNA delivery using non-viral PCL nanoparticles. Biomater. Sci. 2015, 3, 144–151. [Google Scholar] [CrossRef]
- Lallana, E.; Rios de la Rosa, J.M.; Tirella, A.; Pelliccia, M.; Gennari, A.; Stratford, I.J.; Puri, S.; Ashford, M.; Tirelli, N. Chitosan/Hyaluronic Acid Nanoparticles: Rational Design Revisited for RNA Delivery. Mol. Pharm. 2017, 14, 2422–2436. [Google Scholar] [CrossRef]
- McKinlay, C.J.; Vargas, J.R.; Blake, T.R.; Hardy, J.W.; Kanada, M.; Contag, C.H.; Wender, P.A.; Waymouth, R.M. Charge-altering releasable transporters (CARTs) for the delivery and release of mRNA in living animals. Proc. Natl. Acad. Sci. USA 2017, 114, E448–E456. [Google Scholar] [CrossRef] [Green Version]
- McKinlay, C.J.; Benner, N.L.; Haabeth, O.A.; Waymouth, R.M.; Wender, P.A. Enhanced mRNA delivery into lymphocytes enabled by lipid-varied libraries of charge-altering releasable transporters. Proc. Natl. Acad. Sci. USA 2018, 115, E5859–E5866. [Google Scholar] [CrossRef] [Green Version]
- Haabeth, O.A.W.; Blake, T.R.; McKinlay, C.J.; Waymouth, R.M.; Wender, P.A.; Levy, R. mRNA vaccination with charge-altering releasable transporters elicits human T cell responses and cures established tumors in mice. Proc. Natl. Acad. Sci. USA 2018, 115, E9153–E9161. [Google Scholar] [CrossRef] [Green Version]
- Armbruster, N.; Jasny, E.; Petsch, B. Advances in RNA Vaccines for Preventive Indications: A Case Study of A Vaccine Against Rabies. Vaccines 2019, 7, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amos, H. Protamine enhancement of RNA uptake by cultured chick cells. Biochem. Biophys. Res. Commun. 1961, 5, 1–4. [Google Scholar] [CrossRef]
- Scheel, B.; Teufel, R.; Probst, J.; Carralot, J.P.; Geginat, J.; Radsak, M.; Jarrossay, D.; Wagner, H.; Jung, G.; Rammensee, H.-G.; et al. Toll-like receptor-dependent activation of several human blood cell types by protamine-condensed mRNA. Eur. J. Immunol. 2005, 35, 1557–1566. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, M.; Schröder, A.; Scheel, B.; Hong, H.S.; Muth, A.; von Boehmer, L.; Zippelius, A.; Mayer, F.; Reck, M.; Atanackovic, D.; et al. A phase I/IIa study of the mRNA-based cancer immunotherapy CV9201 in patients with stage IIIB/IV non-small cell lung cancer. Cancer Immunol. Immunother. 2019, 68, 799–812. [Google Scholar] [CrossRef]
- Kübler, H.; Scheel, B.; Gnad-Vogt, U.; Miller, K.; Schultze-Seemann, W.; Dorp, F.; Parmiani, G.; Hampel, C.; Wedel, S.; Trojan, L.; et al. Self-adjuvanted mRNA vaccination in advanced prostate cancer patients: A first-in-man phase I/IIa study. J. Immunother. Cancer 2015, 3, 26. [Google Scholar] [CrossRef] [Green Version]
- Petsch, B.; Schnee, M.; Vogel, A.B.; Lange, E.; Hoffmann, B.; Voss, D.; Schlake, T.; Thess, A.; Kallen, K.J.; Stitz, L.; et al. Protective efficacy of in vitro synthesized, specific mRNA vaccines against influenza A virus infection. Nat. Biotechnol. 2012, 30, 1210–1216. [Google Scholar] [CrossRef]
- Udhayakumar, V.K.; De Beuckelaer, A.; McCaffrey, J.; McCrudden, C.M.; Kirschman, J.L.; Vanover, D.; Hoecke, L.V.; Roose, K.; Deswarte, K.; De Geest, B.G.; et al. Arginine-Rich Peptide-Based mRNA Nanocomplexes Efficiently Instigate Cytotoxic T Cell Immunity Dependent on the Amphipathic Organization of the Peptide. Adv. Healthc. Mater. 2017, 6, 1601412. [Google Scholar] [CrossRef]
- Li, J.; Sun, Y.; Jia, T.; Zhang, R.; Zhang, K.; Wang, L. Messenger RNA vaccine based on recombinant MS2 virus-like particles against prostate cancer. Int. J. Cancer 2014, 134, 1683–1694. [Google Scholar] [CrossRef]
- Jekhmane, S.; de Haas, R.; da Silva Filho, O.; van Asbeck, A.H.; Favretto, M.E.; Hernandez Garcia, A.; Brock, R. Renko de Vries Virus-Like Particles of mRNA with Artificial Minimal Coat Proteins: Particle Formation, Stability, and Transfection Efficiency. Nucleic Acid Ther. 2017, 27, 159–167. [Google Scholar] [CrossRef]
- Zhitnyuk, Y.; Gee, P.; Lung, M.S.Y.; Sasakawa, N.; Xu, H.; Saito, H.; Hotta, A. Efficient mRNA delivery system utilizing chimeric VSVG-L7Ae virus-like particles. Biochem. Biophys. Res. Commun. 2018, 505, 1097–1102. [Google Scholar] [CrossRef]
- Palmerston Mendes, L.; Pan, J.; Torchilin, V.P. Dendrimers as Nanocarriers for Nucleic Acid and Drug Delivery in Cancer Therapy. Molecules 2017, 22, 1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chahal, J.S.; Khan, O.F.; Cooper, C.L.; McPartlan, J.S.; Tsosie, J.K.; Tilley, L.D.; Sidik, S.M.; Lourido, S.; Langer, R.; Bavari, S.; et al. Dendrimer-RNA nanoparticles generate protective immunity against lethal Ebola, H1N1 influenza, and Toxoplasma gondii challenges with a single dose. Proc. Natl. Acad. Sci. USA 2016, 113, E4133–E4142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chahal, J.S.; Fang, T.; Woodham, A.W.; Khan, O.F.; Ling, J.; Anderson, D.G.; Ploegh, H.L. An RNA nanoparticle vaccine against Zika virus elicits antibody and CD8+ T cell responses in a mouse model. Sci. Rep. 2017, 7, 252. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Jiang, Z.; Saha, K.; Kim, C.S.; Kim, S.T.; Landis, R.F.; Rotello, V.M. Gold Nanoparticles for Nucleic Acid Delivery. Mol. Ther. 2014, 22, 1075–1083. [Google Scholar] [CrossRef] [Green Version]
- Yeom, J.H.; Ryou, S.M.; Won, M.; Park, M.; Bae, J.; Lee, K. Inhibition of Xenograft Tumor Growth by Gold Nanoparticle-DNA Oligonucleotide Conjugates-Assisted Delivery of BAX mRNA. PLoS ONE 2013, 8, e75369. [Google Scholar] [CrossRef] [Green Version]
- Hoerr, I.; Obst, R.; Rammensee, H.G.; Jung, G. In vivo application of RNA leads to induction of specific cytotoxic T lymphocytes and antibodies. Eur. J. Immunol. 2000, 30, 1–7. [Google Scholar] [CrossRef]
- Wang, Y.; Su, H.H.; Yang, Y.; Hu, Y.; Zhang, L.; Blancafort, P.; Huang, L. Systemic delivery of modified mRNA encoding herpes simplex virus 1 thymidine kinase for targeted cancer gene therapy. Mol. Ther. 2013, 21, 358–367. [Google Scholar] [CrossRef]
- Lee, K.; Yu, P.; Lingampalli, N.; Kim, H.J.; Tang, R.; Murthy, N. Peptide-enhanced mRNA transfection in cultured mouse cardiac fibroblasts and direct reprogramming towards cardiomyocyte-like cells. Int. J. Nanomed. 2015, 10, 1841–1854. [Google Scholar]
- Zohra, F.T.; Chowdhury, E.H.; Akaike, T. High performance mRNA transfection through carbonate apatite-cationic liposome conjugates. Biomaterials 2009, 30, 4006–4013. [Google Scholar] [CrossRef]
- Mockey, M.; Bourseau, E.; Chandrashekhar, V.; Chaudhuri, A.; Lafosse, S.; Le Cam, E.; Quesniaux, V.F.; Ryffel, B.; Pichon, C.; Midoux, P. mRNA-based cancer vaccine: Prevention of B16 melanoma progression and metastasis by systemic injection of MART1 mRNA histidylated lipopolyplexes. Cancer Gene Ther. 2007, 14, 802–814. [Google Scholar] [CrossRef] [Green Version]
- Pichon, C.; Midoux, P. Mannosylated and Histidylated LPR Technology for Vaccination with Tumor Antigen mRNA. In Synthetic Messenger RNA and Cell Metabolism Modulation: Methods and Protocols; Rabinovich, P.M., Ed.; Humana Press: Totowa, NJ, USA, 2013; pp. 247–274. [Google Scholar]
- Uchida, S.; Kinoh, H.; Ishii, T.; Matsui, A.; Tockary, T.A.; Takeda, K.M.; Uchida, H.; Osada, K.; Itaka, K.; Kataoka, K. Systemic delivery of messenger RNA for the treatment of pancreatic cancer using polyplex nanomicelles with a cholesterol moiety. Biomaterials 2016, 82, 221–228. [Google Scholar] [CrossRef]
- Dong, Y.; Dorkin, J.R.; Wang, W.; Chang, P.H.; Webber, M.J.; Tang, B.C.; Yang, J.; Abutbul-lonita, I.; Danino, D.; DeRosa, F.; et al. Poly (glycoamidoamine) Brushes Formulated Nanomaterials for Systemic siRNA and mRNA Delivery in Vivo. Nano Lett. 2016, 16, 842–848. [Google Scholar] [CrossRef] [Green Version]
- Lacroix, C.; Humanes, A.; Coiffier, C.; Gigmes, D.; Verrier, B.; Trimaille, T. Polylactide-Based Reactive Micelles as a Robust Platform for mRNA Delivery. Pharm. Res. 2020, 37, 30. [Google Scholar] [CrossRef]
- Fishman, S.; Lewis, M.D.; Siew, L.K.; Leenheer EDe Kakabadse, D.; Davies, J.; Ziv, D.; Margalit, A.; Karin, N.; Gross, G. Adoptive Transfer of mRNA-Transfected T Cells Redirected against Diabetogenic CD8 T Cells Can Prevent Diabetes. Mol. Ther. 2017, 25, 456–464. [Google Scholar] [CrossRef]
- Van Hoecke, L.; Roose, K. How mRNA therapeutics are entering the monoclonal antibody field. J. Transl. Med. 2019, 17, 54. [Google Scholar] [CrossRef] [Green Version]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines-a new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [Green Version]
- Liu, A.M. A Comparison of Plasmid DNA and mRNA as Vaccine Technologies. Vaccines 2019, 7, 37. [Google Scholar] [CrossRef] [Green Version]
- Maruggi, G.; Zhang, C.; Li, J.; Ulmer, J.B.; Yu, D. mRNA as a Transformative Technology for Vaccine Development to Control Infectious Diseases. Mol. Ther. 2019, 27, 757–772. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Berglund, P.; Rhodes, G.; Parker, S.E.; Jondal, M.; Liljeström, P. Self-replicating Semliki Forest virus RNA as recombinant vaccine. Vaccine 1994, 12, 1510–1514. [Google Scholar] [CrossRef]
- Fleeton, M.N.; Chen, M.; Berglund, P.; Rhodes, G.; Parker, S.E.; Murphy, M.; Atkins, G.J.; Liljeström, P. Self-Replicative RNA Vaccines Elicit Protection against Influenza A Virus, Respiratory Syncytial Virus, and a Tickborne Encephalitis Virus. J. Infect. Dis. 2001, 183, 1395–1398. [Google Scholar] [CrossRef] [Green Version]
- Geall, A.J.; Verma, A.; Otten, G.R.; Shaw, C.A.; Hekele, A.; Banerjee, K.; Cu, Y.; Beard, C.W.; Brito, L.A.; Krucker, T.; et al. Nonviral delivery of self-amplifying RNA vaccines. Proc. Natl. Acad. Sci. USA 2012, 109, 14604–14609. [Google Scholar] [CrossRef] [Green Version]
- Bernstein, D.I.; Reap, E.A.; Katen, K.; Watson, A.; Smith, K.; Norberg, P.; Olmsted, R.A.; Hoeper, A.; Morris, J.; Negri, S.; et al. Randomized, double-blind, Phase 1 trial of an alphavirus replicon vaccine for cytomegalovirus in CMV seronegative adult volunteers. Vaccine 2009, 28, 484–493. [Google Scholar] [CrossRef]
- AlphaVax. Available online: https://www.alphavax.com/clinical-experience.html (accessed on 30 January 2020).
- Wecker, M.; Gilbert, P.; Russell, N.; Hural, J.; Allen, M.; Pensiero, M.; Chulay, J.; Chiu, Y.L.; Abdool Karim, S.S.; Burke, D.S.; et al. Phase I Safety and Immunogenicity Evaluations of an Alphavirus Replicon HIV-1 Subtype C gag vaccine in healthy HIV-1-uninfected adults. Clin. Vaccine Immunol. 2012, 19, 1651–1660. [Google Scholar] [CrossRef] [Green Version]
- Schnee, M.; Vogel, A.B.; Voss, D.; Petsch, B.; Baumhof, P.; Kramps, T.; Stitz, L. An mRNA Vaccine Encoding Rabies Virus Glycoprotein Induces Protection against Lethal Infection in Mice and Correlates of Protection in Adult and Newborn Pigs. PLoS Negl. Trop. Dis. 2016, 10, e0004746. [Google Scholar] [CrossRef] [PubMed]
- Alberer, M.; Gnad-Vogt, U.; Hong, H.S.; Mehr, K.T.; Backert, L.; Finak, G.; Gottardo, R.; Bica, M.A.; Garofano, A.; Koch, S.D.; et al. Safety and immunogenicity of a mRNA rabies vaccine in healthy adults: An open-label, non-randomised, prospective, first-in-human phase 1 clinical trial. Lancet 2017, 390, 1511–1520. [Google Scholar] [CrossRef]
- Richner, J.M.; Himansu, S.; Dowd, K.A.; Butler, S.L.; Salazar, V.; Fox, J.M.; Julander, J.G.; Tang, W.W.; Shresta, S.; Pierson, T.C.; et al. Modified mRNA Vaccines Protect against Zika Virus Infection. Cell 2017, 168, 1114–1125.e10. [Google Scholar] [CrossRef]
- Bahl, K.; Senn, J.J.; Yuzhakov, O.; Bulychev, A.; Brito, L.A.; Hassett, K.J.; Laska, M.E.; Smith, M.; Almarsson, Ö.; Thompson, J.; et al. Preclinical and Clinical Demonstration of Immunogenicity by mRNA Vaccines against H10N8 and H7N9 Influenza Viruses. Mol. Ther. 2017, 25, 1316–1327. [Google Scholar] [CrossRef] [Green Version]
- Feldman, R.A.; Fuhr, R.; Smolenov, I.; (Mick) Ribeiro, A.; Panther, L.; Watson, M.; Senn, J.J.; Smith, M.; Almarsson, ö.; Pujar, H.S.; et al. mRNA vaccines against H10N8 and H7N9 influenza viruses of pandemic potential are immunogenic and well tolerated in healthy adults in phase 1 randomized clinical trials. Vaccine 2019, 37, 3326–3334. [Google Scholar] [CrossRef]
- Leal, L.; Guardo, A.C.; Morón-López, S.; Salgado, M.; Mothe, B.; Heirman, C.; Pannus, P.; Vanham, G.; van den Ham, H.J.; Gruters, R.; et al. Phase I clinical trial of an intranodally administered mRNA-based therapeutic vaccine against HIV-1 infection. AIDS 2018, 32, 2533. [Google Scholar] [CrossRef] [Green Version]
- Van Lint, S.; Renmans, D.; Broos, K.; Goethals, L.; Maenhout, S.; Benteyn, D.; Goyvaerts, C.; Du Four, S.; Van der Jeught, K.; Bialkowski, L.; et al. Intratumoral Delivery of TriMix mRNA Results in T-cell Activation by Cross-Presenting Dendritic Cells. Cancer Immunol. Res. 2016, 4, 146–156. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, R.T.; Kwon, D.S.; Macklin, E.A.; Shopis, J.R.; McLean, A.P.; McBrine, N.; Flynn, T.; Peter, L.; Sbrolla, A.; Kaufmann, D.E.; et al. Immunization of HIV-1-Infected Persons with Autologous Dendritic Cells Transfected With mRNA Encoding HIV-1 Gag and Nef: Results of a Randomized, Placebo-Controlled Clinical Trial. J. Acquir. Immune Defic. Syndr. 2016, 71, 246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNamara, M.A.; Nair, S.K.; Holl, E.K. RNA-Based Vaccines in Cancer Immunotherapy. J. Immunol. Res. 2015, 2015, 794528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kavanagh, D.G.; Kaufmann, D.E.; Sunderji, S.; Frahm, N.; Le Gall, S.; Boczkowski, D.; Rosenberg, E.S.; Stone, D.R.; Johnston, M.N.; Wagner, B.S.; et al. Expansion of HIV-specific CD4+ and CD8+ T cells by dendritic cells transfected with mRNA encoding cytoplasm- or lysosome-targeted Nef. Blood 2006, 107, 1963–1969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melhem, N.M.; Liu, X.D.; Boczkowski, D.; Gilboa, E.; Barratt-Boyes, S.M. Robust CD4+ and CD8+ T cell responses to SIV using mRNA-transfected DC expressing autologous viral Ag. Eur. J. Immunol. 2007, 37, 2164–2173. [Google Scholar] [CrossRef]
- Van Nuffel, A.M.T.; Benteyn, D.; Wilgenhof, S.; Pierret, L.; Corthals, J.; Heirman, C.; van der Bruggen, P.; Coulie, P.G.; Neyns, B.; Thielemans, K.; et al. Dendritic Cells Loaded with mRNA Encoding Full-length Tumor Antigens Prime CD4+ and CD8+ T Cells in Melanoma Patients. Mol. Ther. 2012, 20, 1063–1074. [Google Scholar] [CrossRef] [Green Version]
- Eisenächer, K.; Steinberg, C.; Reindl, W.; Krug, A. The role of viral nucleic acid recognition in dendritic cells for innate and adaptive antiviral immunity. Immunobiology 2007, 212, 701–714. [Google Scholar] [CrossRef]
- Wirth, T.C.; Kühnel, F. Neoantigen Targeting—Dawn of a New Era in Cancer Immunotherapy? Front. Immunol. 2017, 8, 1848. [Google Scholar] [CrossRef] [Green Version]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.P.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef]
- Conry, R.M.; LoBuglio, A.F.; Wright, M.; Sumerel, L.; Pike, M.J.; Johanning, F.; Benjamis, R.; Lu, D.; Curiel, D.T. Characterization of a Messenger RNA Polynucleotide Vaccine Vector. Cancer Res. 1995, 55, 1397–1400. [Google Scholar]
- Papachristofilou, A.; Hipp, M.M.; Klinkhardt, U.; Früh, M.; Sebastian, M.; Weiss, C.; Pless, M.; Cathomas, R.; Hilbe, W.; Pall, G.; et al. Phase Ib evaluation of a self-adjuvanted protamine formulated mRNA-based active cancer immunotherapy, BI1361849 (CV9202), combined with local radiation treatment in patients with stage IV non-small cell lung cancer. J. Immunother. Cancer 2019, 7, 38. [Google Scholar] [CrossRef]
- Boczkowski, B.D.; Nair, S.K.; Snyder, D.; Gilboa, E. Dendritic Cells Pulsed with RNA are Potent Antigen-presenting Cells In Vitro and In Vivo. J. Exp. Med. 1996, 184, 465–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyte, J.A.; Mu, L.; Aamdal, S.; Kvalheim, G.; Dueland, S.; Hauser, M.; Gullestad, H.P.; Ryder, T.; Lislerud, K.; Hammerstad, H.; et al. Phase I/II trial of melanoma therapy with dendritic cells transfected with autologous tumor-mRNA. Cancer Gene Ther. 2006, 13, 905–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilgenhof, S.; Van Nuffel, A.M.T.; Benteyn, D.; Corthals, J.; Aerts, C.; Heirman, C.; Van Riet, I.; Bonehill, A.; Thielemans, K.; Neyns, B. A phase IB study on intravenous synthetic mRNA electroporated dendritic cell immunotherapy in pretreated advanced melanoma patients. Ann. Oncol. 2013, 24, 2686–2693. [Google Scholar] [CrossRef] [PubMed]
- Wilgenhof, S.; Corthals, J.; Heirman, C.; Van Baren, N.; Lucas, S.; Kvistborg, P.; Thielemans, K.; Neyns, B. Phase II study of autologous monocyte-derived mRNA electroporated dendritic cells (TriMixDC-MEL) plus ipilimumab in patientswith pretreated advanced melanoma. J. Clin. Oncol. 2016, 34, 1330–1338. [Google Scholar] [CrossRef] [PubMed]
- Nogrady, B. Gene therapy delivers hope. Nature 2018, 563, S42–S43. [Google Scholar] [CrossRef] [PubMed]
- Rabinovich, P.M.; Komarovskaya, M.E.; Ye, Z.J.; Imai, C.; Campana, D.; Bahceci, E.; Weissman, S.M. Synthetic messenger RNA as a tool for gene therapy. Hum. Gene Ther. 2006, 17, 1027–1035. [Google Scholar] [CrossRef]
- Foster, J.B.; Barrett, D.M.; Karikó, K. The Emerging Role of In Vitro-Transcribed mRNA in Adoptive T Cell Immunotherapy. Mol. Ther. 2019, 27, 747–756. [Google Scholar] [CrossRef] [Green Version]
- Maus, M.V.; Haas, A.R.; Beatty, G.L.; Albelda, S.M.; Levine, B.L.; Liu, X.; Zhao, Y.; Kalos, M.; June, C.H. T Cells Expressing Chimeric Antigen Receptors Can Cause Anaphylaxis in Humans. Cancer Immunol. Res. 2013, 1, 26–31. [Google Scholar] [CrossRef] [Green Version]
- Svoboda, J.; Rheingold, S.R.; Gill, S.I.; Grupp, S.A.; Lacey, S.F.; Kulikovskaya, I.; Suhoski, M.M.; Melenhorst, J.J.; Loudon, B.; Mato, A.R.; et al. Nonviral RNA chimeric antigen receptor–modified T cells in patients with Hodgkin lymphoma. Blood 2018, 132, 1022–1026. [Google Scholar] [CrossRef]
- Tchou, J.; Zhao, Y.; Levine, B.L.; Zhang, P.J.; Davis, M.M.; Melenhorst, J.J.; Kulikovskaya, I.; Brennan, A.L.; Liu, X.; Lacey, S.F.; et al. Safety and Efficacy of Intratumoral Injections of Chimeric Antigen Receptor (CAR) T Cells in Metastatic Breast Cancer. Cancer Immunol. Res. 2017, 5, 1152–1161. [Google Scholar] [CrossRef] [Green Version]
- Jirikowski, G.F.; Sanna, P.P.; Maciejewski-Lenoir, D.; Bloom, F.E. Reversal of diabetes insipidus in Brattleboro tats: Intrahypothalamic injection of vasopressin mRNA. Science 1992, 255, 996–998. [Google Scholar] [CrossRef]
- Trepotec, Z.; Lichtenegger, E.; Plank, C.; Aneja, M.K.; Rudolph, C. Delivery of mRNA Therapeutics for the Treatment of Hepatic Diseases. Mol. Ther. 2019, 27, 794–802. [Google Scholar] [CrossRef] [Green Version]
- Sahu, I.; Haque, A.K.M.A.; Weidensee, B.; Weinmann, P.; Kormann, M.S.D. Recent Developments in mRNA-Based Protein Supplementation Therapy to Target Lung Diseases. Mol. Ther. 2019, 27, 803–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magadum, A.; Kaur, K.; Zangi, L. mRNA-Based Protein Replacement Therapy for the Heart. Mol. Ther. 2019, 27, 785–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lescan, M.; Perl, R.M.; Golombek, S.; Pilz, M.; Hann, L.; Yasmin, M.; Behring, A.; Keller, T.; Nolte, A.; Gruhn, F.; et al. De Novo Synthesis of Elastin by Exogenous Delivery of Synthetic Modified mRNA into Skin and Elastin-Deficient Cells. Mol. Ther. Nucleic Acids 2018, 11, 475–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, S.; Ryals, R.C.; Weller, K.K.; Pennesi, M.E.; Sahay, G. Lipid nanoparticles for delivery of messenger RNA to the back of the eye. J. Control. Release 2019, 303, 91–100. [Google Scholar] [CrossRef] [PubMed]
- DeRosa, F.; Guild, B.; Karve, S.; Smith, L.; Love, K.; Dorkin, J.R.; Kauffman, K.J.; Zhang, J.; Yahalom, B.; Anderson, D.G.; et al. Therapeutic efficacy in a hemophilia B model using a biosynthetic mRNA liver depot system. Gene Ther. 2016, 23, 699–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.; Yin, L.; Theisen, M.; Zhuo, J.; Siddiqui, S.; Levy, B.; Presnyak, V.; Frassetto, A.; Milton, J.; Salerno, T.; et al. Systemic mRNA Therapy for the Treatment of Fabry Disease: Preclinical Studies in Wild-Type Mice, Fabry Mouse Model, and Wild-Type Non-human Primates. Am. J. Hum. Genet. 2019, 104, 625–637. [Google Scholar] [CrossRef] [Green Version]
- An, D.; Schneller, J.L.; Frassetto, A.; Liang, S.; Zhu, X.; Park, J.S.; Theisen, M.; Hong, S.J.; Zhou, J.; Rajendran, R.; et al. Systemic Messenger RNA Therapy as a Treatment for Methylmalonic Acidemia. Cell Rep. 2017, 21, 3548–3558. [Google Scholar] [CrossRef] [Green Version]
- Moderna. Cambridge: Moderna, Inc. Available online: https://investors.modernatx.com/news-releases/news-release-details/moderna-announces-open-ind-propionic-acidemia-program-mrna-3927/ (accessed on 30 January 2020).
- Jiang, L.; Berraondo, P.; Jericó, D.; Guey, L.T.; Sampedro, A.; Frassetto, A.; Benenato, K.E.; Burke, K.; Santamaría, E.; Alegre, M.; et al. Systemic messenger RNA as an etiological treatment for acute intermittent porphyria. Nat. Med. 2018, 24, 1899–1909. [Google Scholar] [CrossRef]
- Berraondo, P.; Martini, P.G.V.; Avila, M.A.; Fontanellas, A. Messenger RNA therapy for rare genetic metabolic diseases. Gut 2019, 68, 1323–1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, E.; MacDonald, K.D.; Slaughter, K.; McKinney, M.; Patel, S.; Sun, C.; Sahay, G. Lipid Nanoparticle-Delivered Chemically Modified mRNA Restores Chloride Secretion in Cystic Fibrosis. Mol. Ther. 2018, 26, 2034–2046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlsson, L.; Clarke, J.C.; Yen, C.; Gregoire, F.; Albery, T.; Billger, M.; Egnell, A.C.; Gan, L.M.; Jennbacken, K.; Johansson, E.; et al. Biocompatible, Purified VEGF-A mRNA Improves Cardiac Function after Intracardiac Injection 1 Week Post-myocardial Infarction in Swine. Mol. Ther. Methods Clin. Dev. 2018, 9, 330–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gan, L.-M.; Lagerström-Fermér, M.; Carlsson, L.G.; Arfvidsson, C.; Egnell, A.-C.; Rudvik, A.; Kjaer, M.; Collén, A.; Thompson, J.D.; Joyal, J.; et al. Intradermal delivery of modified mRNA encoding VEGF-A in patients with type 2 diabetes. Nat. Commun. 2019, 10, 871. [Google Scholar] [CrossRef] [PubMed]
- Genetics Home reference. U.S. National Library of Medicine. Bethesda: USA.gov. Available online: https://ghr.nlm.nih.gov/condition/propionic-acidemia (accessed on 30 January 2020).
- Fraser, J.L.; Venditti, C.P. Methylmalonic and propionic acidemias: Clinical management update. Curr. Opin. Pediatr. 2016, 28, 682–693. [Google Scholar] [CrossRef]
- Critelli, K.; McKiernan, P.; Vockley, J.; Mazariegos, G.; Squires, R.H.; Soltys, K.; Squires, J.E. Liver Transplantation for Propionic Acidemia and Methylmalonic Acidemia: Perioperative Management and Clinical Outcomes. Liver Transpl. 2018, 24, 1260–1270. [Google Scholar] [CrossRef] [Green Version]
- Human Genomics in Global Health. Available online: https://www.who.int/genomics/public/geneticdiseases/en/index2.html#CF (accessed on 30 January 2020).
- Zuckerman, J.B.; McCoy, K.; Schechter, M.S.; Dorgan, D.; Jain, M.; MacDonald, K.D.; Callison, C.; Walker, S.; Bodie, S.; Barbier, A.; et al. Safety and Tolerability of a Single Dose of MRT5005, a Inhaled CFTR mRNA Therapeutic, in Adult CF Patients (Poster 515). Pediatric Pulmonol. 2019, 54, S350. [Google Scholar]
- Waisbren, S.E.; Gropman, A.L.; Batshaw, M.L.; Members of the Urea Cycle Disorders Consortium (UCDC). Improving long term outcomes in urea cycle disorders-report from the Urea Cycle Disorders Consortium. J. Inherit. Metab. Dis. 2016, 39, 573–584. [Google Scholar] [CrossRef] [Green Version]
- Brassier, A.; Gobin, S.; Arnoux, J.B.; Valayannopoulos, V.; Habarou, F.; Kossorotoff, M.; Servais, A.; Barbier, V.; Dubois, S.; Touati, G.; et al. Long-term outcomes in Ornithine Transcarbamylase deficiency: A series of 90 patients. Orphanet J. Rare Dis. 2015, 10, 58. [Google Scholar] [CrossRef] [Green Version]
- Intrado. Available online: https://www.globenewswire.com/news-release/2019/09/09/1913059/0/en/Translate-Bio-Announces-Pipeline-Program-Update.html (accessed on 30 January 2020).
- Shim, G.; Kim, D.; Park, G.T.; Jin, H.; Suh, S.K.; Oh, Y.K. Therapeutic gene editing: Delivery and regulatory perspectives. Acta Pharmacol. Sin. 2017, 38, 738–753. [Google Scholar] [CrossRef] [Green Version]
- Conway, A.; Mendel, M.; Kim, K.; McGovern, K.; Boyko, A.; Zhang, L.; Miller, J.C.; DeKelver, R.C.; Paschon, D.E.; Mui, B.L.; et al. Non-viral Delivery of Zinc Finger Nuclease mRNA Enables Highly Efficient In Vivo Genome Editing of Multiple Therapeutic Gene Targets. Mol. Ther. 2019, 27, 866–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gersbach, C.A.; Gaj, T.; Barbas, C.F. Synthetic zinc finger proteins: The advent of targeted gene regulation and genome modification technologies. Acc. Chem. Res. 2014, 47, 2309–2318. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Treen, N. TALEN-Based Knockout System. Adv. Exp. Med. Biol. 2018, 1029, 131–139. [Google Scholar] [PubMed]
- Wang, H.; Yang, H.; Shivalila, C.S.; Dawlaty, M.M.; Cheng, A.W.; Zhang, F.; Jaenisch, R. One-step generation of mice carrying mutations in multiple genes by CRISPR/cas-mediated genome engineering. Cell 2013, 153, 910–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.-X.; Zhang, Y.; Yin, H. Genome Editing with mRNA Encoding ZFN, TALEN, and Cas9. Mol. Ther. 2019, 27, 735–746. [Google Scholar] [CrossRef] [Green Version]
- Crudele, J.M.; Chamberlain, J.S. Cas9 immunity creates challenges for CRISPR gene editing therapies. Nat. Commun. 2018, 9, 3497. [Google Scholar] [CrossRef]
- Wang, J.; Exline, C.M.; DeClercq, J.J.; Llewellyn, G.N.; Hayward, S.B.; Li, P.W.-L.; Shivak, D.A.; Surosky, R.T.; Gregory, P.D.; Holmes, M.C.; et al. Homology-driven genome editing in hematopoietic stem and progenitor cells using ZFN mRNA and AAV6 donors. Nat. Biotechnol. 2015, 33, 1256–1263. [Google Scholar] [CrossRef] [Green Version]
- De Ravin, S.S.; Reik, A.; Liu, P.-Q.; Li, L.; Wu, X.; Su, L.; Raley, C.; Theobald, N.; Choi, U.; Song, A.H.; et al. Targeted gene addition in human CD34+ hematopoietic cells for correction of X-linked chronic granulomatous disease. Nat. Biotechnol. 2016, 34, 424–429. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; DeClercq, J.J.; Hayward, S.B.; Li, P.W.-L.; Shivak, D.A.; Gregory, P.D.; Lee, G.; Holmes, M.C. Highly efficient homology-driven genome editing in human T cells by combining zinc-finger nuclease mRNA and AAV6 donor delivery. Nucleic Acids Res. 2015, 44, e30. [Google Scholar] [CrossRef] [Green Version]
- Wefers, B.; Panda, S.K.; Ortiz, O.; Brandl, C.; Hensler, S.; Hansen, J.; Wurst, W.; Kühn, R. Generation of targeted mouse mutants by embryo microinjection of TALEN mRNA. Nat. Protoc. 2013, 8, 2355–2379. [Google Scholar] [CrossRef]
- Hwang, W.Y.; Fu, Y.; Reyon, D.; Maeder, M.L.; Tsai, S.Q.; Sander, J.D.; Peterson, R.T.; Yeh, J.-R.J.; Joung, J.K. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol. 2013, 31, 227–229. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Nakano, K.; Matsunari, H.; Matsuda, T.; Maehara, M.; Kanai, T.; Kobayashi, M.; Matsumura, Y.; Sakai, R.; Kuramoto, M.; et al. Generation of Interleukin-2 Receptor Gamma Gene Knockout Pigs from Somatic Cells Genetically Modified by Zinc Finger Nuclease-Encoding mRNA. PLoS ONE 2013, 8, e76478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Chang, J.; Jiang, Y.; Meng, X.; Sun, T.; Mao, L.; Xu, Q.; Wang, M. Fast and Efficient CRISPR/Cas9 Genome Editing In Vivo Enabled by Bioreducible Lipid and Messenger RNA Nanoparticles. Adv. Mater. 2019, 31, 1902575. [Google Scholar] [CrossRef]
- Samson, M.; Libert, F.; Doranz, B.J.; Rucker, J.; Liesnard, C.; Farber, C.-M.; Saragosti, S.; Lapoumeroulie, C.; Cognaux, J.; Forceille, C.; et al. Resistance to HIV-1 infection in Caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature 1996, 382, 722–725. [Google Scholar] [CrossRef] [PubMed]
- Ware, R.E.; de Montalembert, M.; Tshilolo, L.; Abboud, M.R. Sickle cell disease. Lancet 2017, 390, 311–323. [Google Scholar] [CrossRef]
- Smith, E.C.; Luc, S.; Croney, D.M.; Woodworth, M.B.; Greig, L.C.; Fujiwara, Y.; Nguyen, M.; Sher, F.; Macklis, J.D.; Bauer, D.E.; et al. Strict in vivo specificity of the Bcl11a erythroid enhancer. Blood 2016, 128, 2338–2342. [Google Scholar] [CrossRef] [PubMed]
- Qasim, W.; Zhan, H.; Samarasinghe, S.; Adams, S.; Amrolia, P.; Stafford, S.; Butler, K.; Rivat, C.; Wright, G.; Somana, K.; et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci. Transl. Med. 2017, 9, eaaj2013. [Google Scholar] [CrossRef]
- Jo, J.-I.; Gao, J.-Q.; Tabata, Y. Biomaterial-based delivery systems of nucleic acid for regenerative research and regenerative therapy. Regen. Ther. 2019, 11, 123–130. [Google Scholar] [CrossRef]
- Jopling, C.; Boue, S.; Belmonte, J.C.I. Dedifferentiation, transdifferentiation and reprogramming: Three routes to regeneration. Nat. Rev. Mol. Cell Biol. 2011, 12, 79–89. [Google Scholar] [CrossRef]
- Eguizabal, C.; Carlos, J.; Belmonte, I. Reprogramming: Future Directions in Regenerative Medicine. Semin. Reprod. Med. 2013, 31, 82–94. [Google Scholar] [CrossRef] [Green Version]
- Yakubov, E.; Rechavi, G.; Rozenblatt, S.; Givol, D. Reprogramming of human fibroblasts to pluripotent stem cells using mRNA of four transcription factors. Biochem. Biophys. Res. Commun. 2010, 394, 189–193. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warren, L.; Manos, P.D.; Ahfeldt, T.; Loh, Y.H.; Li, H.; Lau, F.; Ebina, W.; Mandal, P.K.; Smith, Z.D.; Meissner, A.; et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 2010, 7, 618–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corritore, E.; Lee, Y.-S.; Pasquale, V.; Liberati, D.; Hsu, M.-J.; Lombard, C.A.; Van Der Smissen, P.; Vetere, A.; Bonner-Weir, S.; Piemonti, L.; et al. V-Maf Musculoaponeurotic Fibrosarcoma Oncogene Homolog A Synthetic Modified mRNA Drives Reprogramming of Human Pancreatic Duct-Derived Cells into Insulin-Secreting Cells. Stem Cells Transl. Med. 2016, 5, 1525–1537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orlando, G.; Gianello, P.; Salvatori, M.; Stratta, R.J.; Soker, S.; Ricordi, C.; Domínguez-Bendala, J. Cell Replacement Strategies Aimed at Reconstitution of the β-Cell Compartment in Type 1 Diabetes. Diabetes 2014, 63, 1433–1444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koblas, T.; Leontovyc, I.; Loukotova, S.; Kosinova, L.; Saudek, F. Reprogramming of Pancreatic Exocrine Cells AR42J Into Insulin-producing Cells Using mRNAs for Pdx1, Ngn3, and MafA Transcription Factors. Mol. Ther. Nucleic Acids 2016, 5, e320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elangovan, S.; Khorsand, B.; Do, A.V.; Hong, L.; Dewerth, A.; Komann, M.; Ross, R.D.; Sumner, D.R.; Allamargot, C.; Salem, A.K. Chemically modified RNA activated matrices enhance bone regeneration. J. Control. Release 2015, 218, 22–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Badieyan, Z.S.; Berezhanskyy, T.; Utzinger, M.; Aneja, M.K.; Emrich, D.; Erben, R.; Schüler, C.; Altpeter, P.; Ferizi, M.; Hasenpusch, G.; et al. Transcript-activated collagen matrix as sustained mRNA delivery system for bone regeneration. J. Control. Release 2016, 239, 137–148. [Google Scholar] [CrossRef]
- Park, J.S.; Suryaprakash, S.; Lao, Y.H.; Leong, K.W. Engineering mesenchymal stem cells for regenerative medicine and drug delivery. Methods 2015, 84, 3–16. [Google Scholar] [CrossRef] [Green Version]
- Ryser, M.F.; Ugarte, F.; Thieme, S.; Bornhäuser, M.; Roesen-Wolff, A.; Brenner, S. mRNA Transfection of CXCR4-GFP Fusion—Simply Generated by PCR—Results in Efficient Migration of Primary Human Mesenchymal Stem Cells. Tissue Eng. Part C Methods 2008, 14, 179–184. [Google Scholar] [CrossRef]
- Nowakowski, A.; Andrzejewska, A.; Boltze, J.; Nitzsche, F.; Cui, L.L.; Jolkkonen, J.; Walczak, P.; Lukomska, B.; Janowski, M. Translation, but not transfection limits clinically relevant, exogenous mRNA based induction of alpha-4 integrin expression on human mesenchymal stem cells. Sci. Rep. 2017, 7, 1103. [Google Scholar] [CrossRef] [PubMed]
- Levy, O.; Zhao, W.; Mortensen, L.J.; Leblanc, S.; Tsang, K.; Fu, M. mRNA-engineered mesenchymal stem cells for targeted delivery of interleukin-10 to sites of inflammation. Blood 2013, 122, e23–e32. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Infectious Disease | Biological Active/Encoding Sequence | Strategy/Delivery System | Administration Route | NCT Number/Phase |
|---|---|---|---|---|
| Rabies | CV7201 mRNA/Rabies virus glycoprotein (RABV-G) | In vivo/Polypeptide system | i.d. or i.m. | NCT02241135/Phase I |
| CV7202 mRNA/Rabies virus glycoprotein (RABV-G) | In vivo/Lipid nanosystem | i.m. | NCT03713086/Phase I | |
| Zika Virus | mRNA-1893/Structural proteins of Zika virus | In vivo/Lipid nanosystem | NCT04064905/Phase I | |
| mRNA-1325/Zika virus antigen | In vivo/Lipid nanosystem | i.d. | NCT03014089/Phase I | |
| Cytomegalovirus (CMV) | mRNA-1647 and mRNA-1443/Pentamer complex and full-length membrane-bound glycoprotein B (gB) and pp65 T cell antigen of CMV | In vivo/Lipid nanosystem | i.d. | NCT03382405/Phase I |
| AVX601/Alphavirus replicon vaccine expressing gB, pp65 and IE1 proteins of CMV | In vivo/Viral vector | i.m. or s.c. | NCT00439803/Phase I | |
| hMPV and PIV3 | mRNA-1653: Fusion proteins of hMPV and PIV3 | In vivo/Lipid nanosystems | i.d. | NCT03392389/Phase I |
| Tuberculosis | GSK 692,342/Immunogenic fusion protein (M72) derived from Mycobacterium tuberculosis | In vivo/Lipid nanosytems | i.m. | NCT01669096/Phase II |
| HIV-1 | HIV-1 Gag and Nef | Ex vivo/mRNA transfected autologous DCs | i.d. | NCT00833781/Phase I |
| iHIVARNA: TriMix and HTI/APC activation molecules (CD40L+CD70+caTLR4) and HIV immunogen sequences (Gag, Pol, Vif and Nef) | In vivo/Naked mRNA | Inguinal intranodal | NCT02413645 and NCT02888756/Phase I and Phase II | |
| AVX101/Alphavirus replicon vaccine expressing HIV Gag antigen | In vivo/Viral vector | s.c. | NCT00097838 and NCT00063778/Phase I | |
| Influenza | VAL-506440/H10N8 antigen | In vivo/Lipid nanosystems | i.d. or i.m. | NCT03076385/Phase I |
| VAL-339851/H7N9 antigen | In vivo/Lipid nanosystems | i.d. or i.m. | NCT03345043/Phase I | |
| AVX502/Alphavirus replicon vaccine expressing Influenza A/Wyoming/03/2003 Hemagglutinin | In vivo/Viral vector | i.m. or s.c. | NCT00440362 and NCT00706732/Phase I/II |
| Type of Cancer | Biological Active/Encoding Sequence | Strategy/Delivery System | Administration Route | NCT Number/Phase |
|---|---|---|---|---|
| Non-small-cell lung carcinoma (NSCLC) | CV9201/five mRNAs encoding antigens which are overexpressed or exclusively expressed in NSCLC cells | In vivo/Polypeptide system | i.d. | NCT00923312/Phase I/II |
| CV9202/six mRNAs encoding antigens which are overexpressed in NSCLC compared to healthy tissue | In vivo/Polypeptide system | i.d. | NCT01915524/Phase I | |
| Metastatic NSCLC | BI 1,361,849/NSCLC-associated antigens (NY-ESO-1, MAGE-C1, MAGE-C2, 5T4, and MUC-1) | In vivo/Polypeptide system | i.d. | NCT03164772/Phase I and II |
| Esophageal Cancer and NSCLC | Personalized mRNA Tumor Vaccine/Neoantigen (tumor associated specific antigens) | In vivo/- | s.c. | NCT03908671/NotA |
| Malignant Melanoma | mRNA coding for melanoma associated antigens | In vivo/Naked mRNA | s.c. | NCT00204516/Phase I/II |
| mRNA coding melanoma associated antigens (Melan-A, Mage-A1, Mage-A3, Survivin, GP100 and Tyrosinase) | In vivo/Polypeptide system | i.d. | NCT00204607/Phase I/II | |
| mRNA coding the unique spectrum of tumor antigens in each patient | Ex vivo/mRNA transfected DCs | i.d. or intranodal | NCT01278940/Phase I/II | |
| Malignant Melanoma III and IV | TriMix-DC encoding melamona tumor-associated antigens (MAGE-A3, MAGE-C2, tyrosinase and gp100) | Ex vivo/autologous TriMix-DC | i.v. | NCT01302496/Phase II |
| Melanoma | mRNA-4157/personalized cancer vaccine targeting twenty tumor-associated antigens | In vivo/Lipid nanosystems | i.d. | NCT03897881/Phase II |
| (RBL001; RBL002)/malignant melanoma associated antigens | In vivo/Naked mRNA | intranodal | NCT01684241/Phase I | |
| IVAC MUTANOME/poly-neo-epitopic personalized cancer vaccine targeting tumor-associated antigens (with or without initial treatment with RBL001/RBL002) | In vivo/Naked mRNA | intranodal | NCT02035956/Phase I | |
| RBL001.1; RBL002.2; RBL003.1; RBL004.1/malignant melanoma-associated antigens | In vivo/Lipid nanosystems | i.v. | NCT02410733/Phase I | |
| mRNA encoding TriMix | Ex vivo/mRNA-transfected autologous DCs | i.d. and i.v. | NCT01066390/Phase I | |
| mRNA encoding melanoma-associated tumor antigens (gp100 and tyrosinase) and TriMix | Ex vivo/mRNA-transfected autologous DCs | intranodal | NCT01530698/Phase I/II | |
| Melanoma Stage III or IV | mRNA encoding melanoma associated antigens (gp100 and tyrosinase) | Ex vivo/mRNA-transfected DCs | i.v., i.d., intranodal | NCT00243529/Phase I/II |
| Metastatic Malignant Melanoma | hTERT-, Survivin- and tumor cell derived mRNA + ex vivo T cell expansion | Ex vivo/mRNA-transfected DCs | i.d. and i.v. | NCT00961844/Phase I/II |
| Uveal Melanoma | mRNA coding tumor associated antigens | Ex vivo/mRNA-transfected DCs | i.d./i.v. | NCT00929019/Phase I/II |
| Acute Myeloid Leukemia (AML) | mRNA coding for the Wilms’ tumor protein (WT1) | Ex vivo/mRNA transfected autologous DCs | i.d. | NCT00834002/Phase I |
| AML-specific mRNA | Ex vivo/mRNA transfected autologous DCs | i.d. | NCT00514189/Phase I | |
| mRNA encoding WT1, PRAME, and CMVpp65 | Ex vivo/mRNA transfected autologous DCs | i.d. | NCT01734304/Phase I/II | |
| Relapsed or Refractory AML | Autologous Anti-CD 123 CAR TCR/4-1BB-expressing T-lymphocytes/anti-CD123 chimeric antigen receptors expressing tandem TCR and 4-1BB (TCR/4-1BB) costimulatory domains | Ex vivo/mRNA transfected autologous CAR T cells | iv | NCT02623582/Early Phase I |
| Multiple Myeloma | mRNA encoding CT7, MAGE-A3, and WT1 | Ex vivo/mRNA-transfected autologous Langerhans-type DCs | i.d. | NCT01995708/Phase I |
| Prostate Cancer | CV9104/mRNAs encoding PSA, PSCA, PSMA, STEAP1, PAP and Mucin 1 antigens | In vivo/Polypeptide system | i.d. | NCT01817738 and NCT02140138/Phase I/II and Phase II |
| mRNA coding tumor associated antigens | Ex vivo/mRNA-transfected DCs | i.d. | NCT01278914/Phase I/II | |
| mRNA extracted from Primary Prostate Cancer Tissue, combined with mRNA encoding hTERT and Survivin | Ex vivo/mRNA-transfected DCs | i.d. | NCT01197625/Phase I/II | |
| Metastatic Prostate Cancer | mRNA derived from the patient’s own tumor | Ex vivo/mRNA-transfected autologous DCs | i.d. | NCT01153113/Phase I/II (withdrawn) |
| Hormonal Refractory Prostate Cancer | CV9103/mRNAs encoding PSA, PSCA, PSMA and STEAP1 antigens | In vivo/Polypeptide system | i.d. | NCT00831467 (eudract 2008-003967-37) and NCT00906243/Phase I/II |
| Glioblastoma | mRNA encoding Survivin, hTERT or autologous tumor stem cells derived from tumorspheres | Ex vivo/mRNA-transfected autologous DCs | id | NCT03548571/Phase II/III |
| Ovarian Cancer | W_ova1 vaccine: Three ovarian cancer tumor associated antigens mRNAs | In vivo/Lipid nanosystems | i.v. | NCT04163094/Phase I |
| Recurrent Epithelial Ovarian Cancer | mRNA encoding hTERT and Survivin in addition to amplified cancer stem cell mRNA | Ex vivo/mRNA-transfected DCs | i.d. | NCT01334047/Phase I/II |
| Breast Cancer | cMet RNA CAR T cells | Ex vivo/mRNA transfected autologous CAR T cells | intratumoral | NCT01837602/Phase I |
| Early Breast Cancer | mRNA encoding TriMix | In vivo/naked mRNA | intratumoral | NCT03788083/Phase I |
| Triple-negative breast cancer | IVAC_WAREHOUSE_bre1_uID; IVAC MUTANOME _uID/personalized cancer vaccine targeting tumor-associated antigens | In vivo/Lipid nanosystems | i.v. | NCT02316457/Phase I |
| Solid tumors | mRNA-4157/personalized cancer vaccine targeting twenty tumor-associated antigens | In vivo/Lipid nanosystem | i.m. | NCT03313778/Phase I |
| Hodgkin Lymphoma | RNA anti-CD19 CAR T cells/CD19 chimeric antigen receptors expressing tandem TCR/4-1BB costimulatory domains | Ex vivo/mRNA transfected autologous CAR T cells | i.v. | NCT02277522 and NCT02624258/Early Phase I |
| Metastatic Pancreatic Ductal Adenocarcinoma | RNA mesothelin re-directed autologous T cell/chimeric anti-mesothelin immunoreceptor SS1 | Ex vivo/mRNA transfected autologous CAR T cells | i.v. | NCT01897415/Phase I |
| Malignant Pleural Mesothelioma | Autologous anti-mesothelin CAR T cells/chimeric anti-mesothelin immunoreceptor | Ex vivo/mRNA transfected autologous CAR T cells | i.v. | NCT01355965/Phase I |
| Malignant Melanoma, Breast Cancer | RNA CART-cMET/MET chimeric antigen receptors with tandem TCRζ and 4-1BB (TCRζ/4-1BB) co-stimulatory domains | Ex vivo/mRNA transfected autologous CAR T cells | i.v. | NCT03060356/Early Phase I |
| Brain Cancer, Neoplasm Metastases | Personalized cellular vaccine/tumor associated antigen mRNA | Ex vivo/mRNA transfected autologous DCs | NA | NCT02808416/Phase I |
| Advanced Esophageal Squamous Carcinoma, Gastric Adenocarcinoma, Pancreatic Adenocarcinoma, Colorectal Adenocarcinoma | Personalized mRNA Tumor Vaccine/Neoantigen (tumor associated specific antigens) | In vivo/- | s.c. | NCT03468244/NotA |
| Melanoma, Colon cancer, Gastrointestinal cancer, Genitourinary cancer, hepatocellular cancer | NCI-4650/mRNA-based, Personalized Cancer Vaccine | In vivo/Lipid nanosystems | i.m. | NCT03480152/Phase I/II |
| Melanoma, NSCLC, Bladder Cancer, Colorectal Cancer, Triple Negative Breast Cancer, Renal Cancer, Head and Neck Cancer, Other Solid Cancers | RO7198457/personalized cancer vaccine targeting tumor-associated antigens | In vivo/Lipid nanosystem | i.v. | NCT03289962/Phase I |
| Relapsed/Refractory Solid Tumor Malignancies or Lymphoma, Ovarian Cancer | mRNA-2416/OX40L | In vivo/Lipid nanosystems | Intratumoral | NCT03323398/Phase I and II |
| Squamous Cell Carcinoma, Head and Neck Neoplasm, Cervical Neoplasm, Penile Neoplasms Malignant | human papillomavirus (HPV16) mRNA vaccine/HPV16-derived E6, E7 tumor antigens | In vivo/Naked mRNA | i.d. | NCT03418480/Phase I and II |
| Advanced or Metastatic Malignancies Expressing CEA (Colorectal Cancer, Breast Cancer, Lung Cancer, Pancreatic Cancer) or Stage III Colon Cancer | AVX701/Alphaviral replicon particle vaccine expressing Carcinoembryonic Antigen Gene (CEA(6D)). | In vivo/Viral vector | i.m. | NCT00529984, NCT01890213/Phase I and II, Phase I |
| Glioblastoma, Renal Cell Carcinoma, Sarcomas, Breast Cancers, Malignant Mesothelioma, Colorectal Tumor | mRNA encoding WT1 | Ex vivo/mRNA-transfected autologous DCs | i.d. | NCT01291420/Phase I/I, |
| Disease | Biological Active/Encoding Sequence | Strategy/Delivery System | Administration Route | NCT Number/Phase |
|---|---|---|---|---|
| Heart Failure | AZD8601/Vascular endothelial growth factor-A (VEGF-A) | Naked mRNA | Epicardial injection | NCT03370887/Phase II |
| Ulcers associated with type II diabetes | AZD8601/Vascular endothelial growth factor-A (VEGF-A) | Naked mRNA | Intradermal | NCT02935712/Phase I |
| Propionic Acidemia | mRNA-3927/alpha and beta subunits of the mitochondrial enzyme propionyl-CoA carboxylase | In vivo/Lipid nanosytems | Intravenous | NCT04159103/Phase I and II |
| Isolated Methylmalonic Acidemia | mRNA-3704/methylmalonyl-coenzyme A mutase (MUT) | In vivo/Lipid nanosytems | Intravenous | NCT03810690/Phase I and II |
| Ornithine Transcarbamylase Deficiency | MRT5201/Ornithine transcarbamylase | In vivo/Lipid nanosystems | Intravenous | NCT03767270/Phase I and II |
| Cystic Fibrosis | MRT5005/Human Cystic Fibrosis Transmembrane Regulator protein (CFTR) | In vivo/Lipid nanosystems | Nebulization | NCT03375047/Phase I and II |
| Disease | Biological Active | Therapeutic mRNA | Target Protein | Strategy/Delivery System | Administration Route | NCT Number/Phase |
|---|---|---|---|---|---|---|
| HIV | SB-728mR | ZFN mRNA | CCR5 | Ex vivo/Autologous CD4+ T Cells | Intravenous | NCT02388594/Phase I |
| SB-728mR | ZFN mRNA | CCR5 | Ex vivo/Autologous CD4 CAR+ T cells | Intravenous | NCT03617198/Phase I | |
| SB-728mR-T | ZFN mRNA | CCR5 | Ex vivo/Autologous T cells | Intravenous | NCT02225665, NCT04201782,/Phase I, Phase I/II | |
| SB-728mR-HSPC | ZFN mRNA | CCR5 | Ex vivo/Autologous CD34+ hHSPCs | Intravenous | NCT02500849/Phase I | |
| Sickle Cell Disease | BIVV003 | ZFN mRNA | B-cell lymphoma/leukemia 11A (BCL11A) | Ex vivo/Autologous CD34 + hematopoietic stem cells (HSPC) | Intravenous | NCT03653247/Phase I/II |
| B acute lymphoblastic leukemia | UCART19 | TALEN mRNA | TCR and CD52 | Ex vivo/Allogenic T cells | Intravenous | NCT02808442, NCT02746952, NCT02735083/Phase I |
| B cell leukemia and B cell lymphoma | UCART019 | CRISPR/Cas9 mRNA | TCR, B2M | Ex vivo/Allogenic T cells | Intravenous | NCT03166878/Phase I/II |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gómez-Aguado, I.; Rodríguez-Castejón, J.; Vicente-Pascual, M.; Rodríguez-Gascón, A.; Solinís, M.Á.; del Pozo-Rodríguez, A. Nanomedicines to Deliver mRNA: State of the Art and Future Perspectives. Nanomaterials 2020, 10, 364. https://0-doi-org.brum.beds.ac.uk/10.3390/nano10020364
Gómez-Aguado I, Rodríguez-Castejón J, Vicente-Pascual M, Rodríguez-Gascón A, Solinís MÁ, del Pozo-Rodríguez A. Nanomedicines to Deliver mRNA: State of the Art and Future Perspectives. Nanomaterials. 2020; 10(2):364. https://0-doi-org.brum.beds.ac.uk/10.3390/nano10020364
Chicago/Turabian StyleGómez-Aguado, Itziar, Julen Rodríguez-Castejón, Mónica Vicente-Pascual, Alicia Rodríguez-Gascón, María Ángeles Solinís, and Ana del Pozo-Rodríguez. 2020. "Nanomedicines to Deliver mRNA: State of the Art and Future Perspectives" Nanomaterials 10, no. 2: 364. https://0-doi-org.brum.beds.ac.uk/10.3390/nano10020364