Reduction of the Diazo Functionality of α-Diazocarbonyl Compounds into a Methylene Group by NH3BH3 or NaBH4 Catalyzed by Au Nanoparticles



Abstract
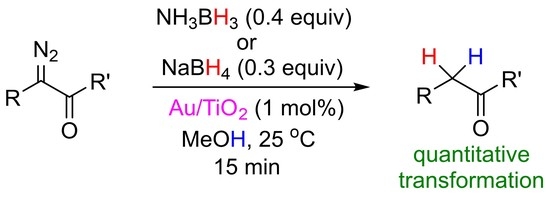
:
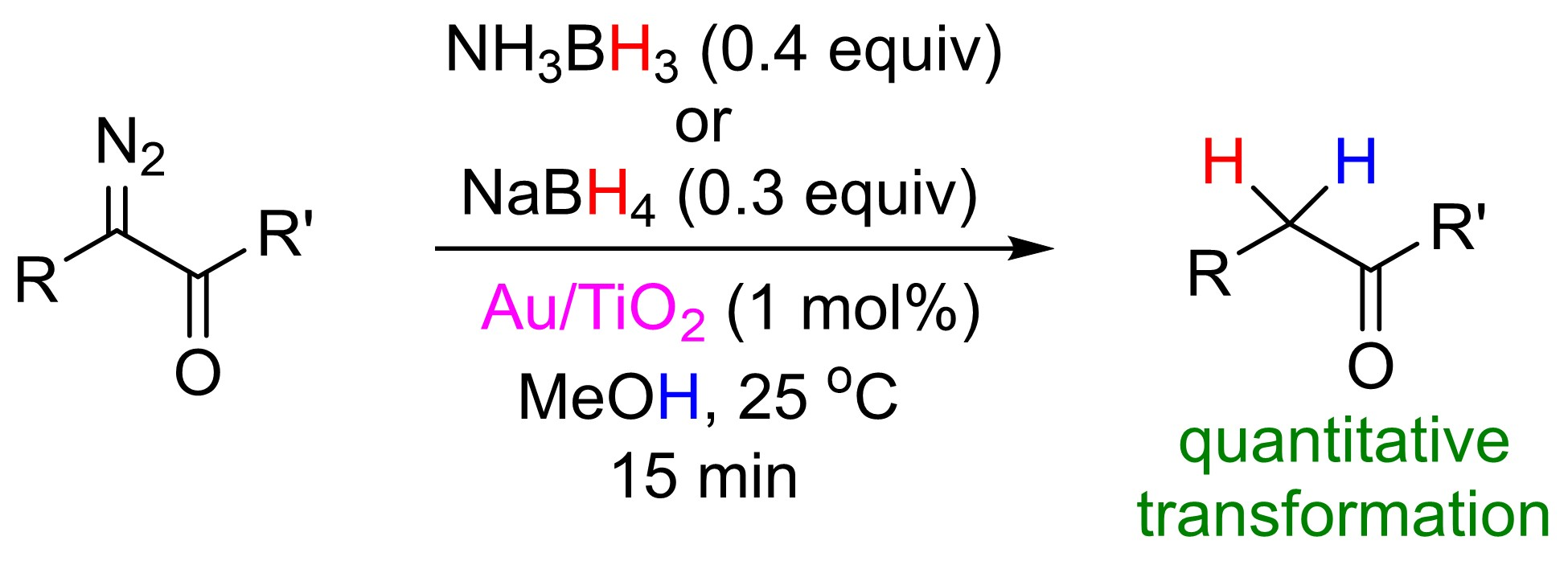{kind=link}
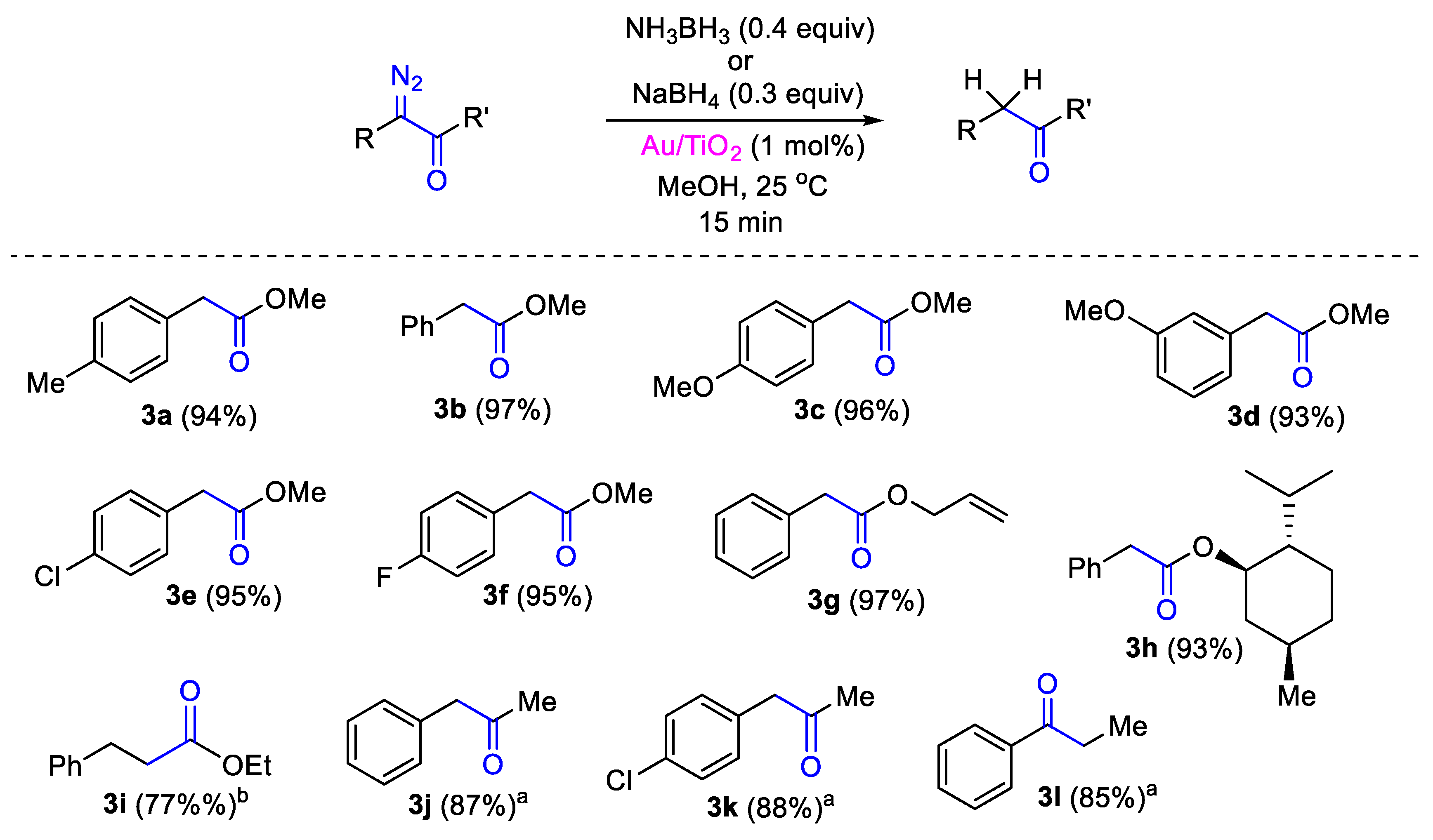{kind=link}
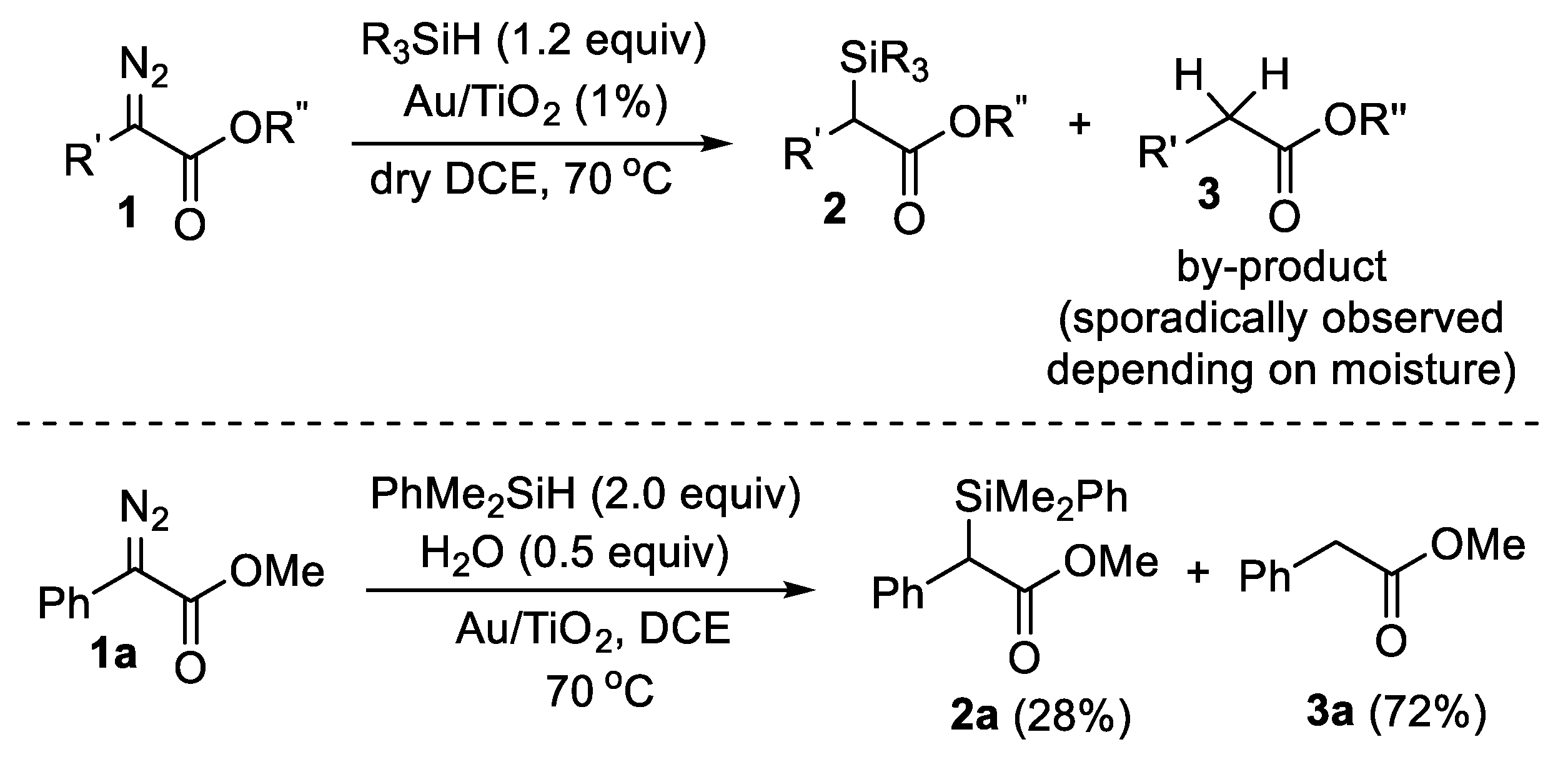{kind=link}
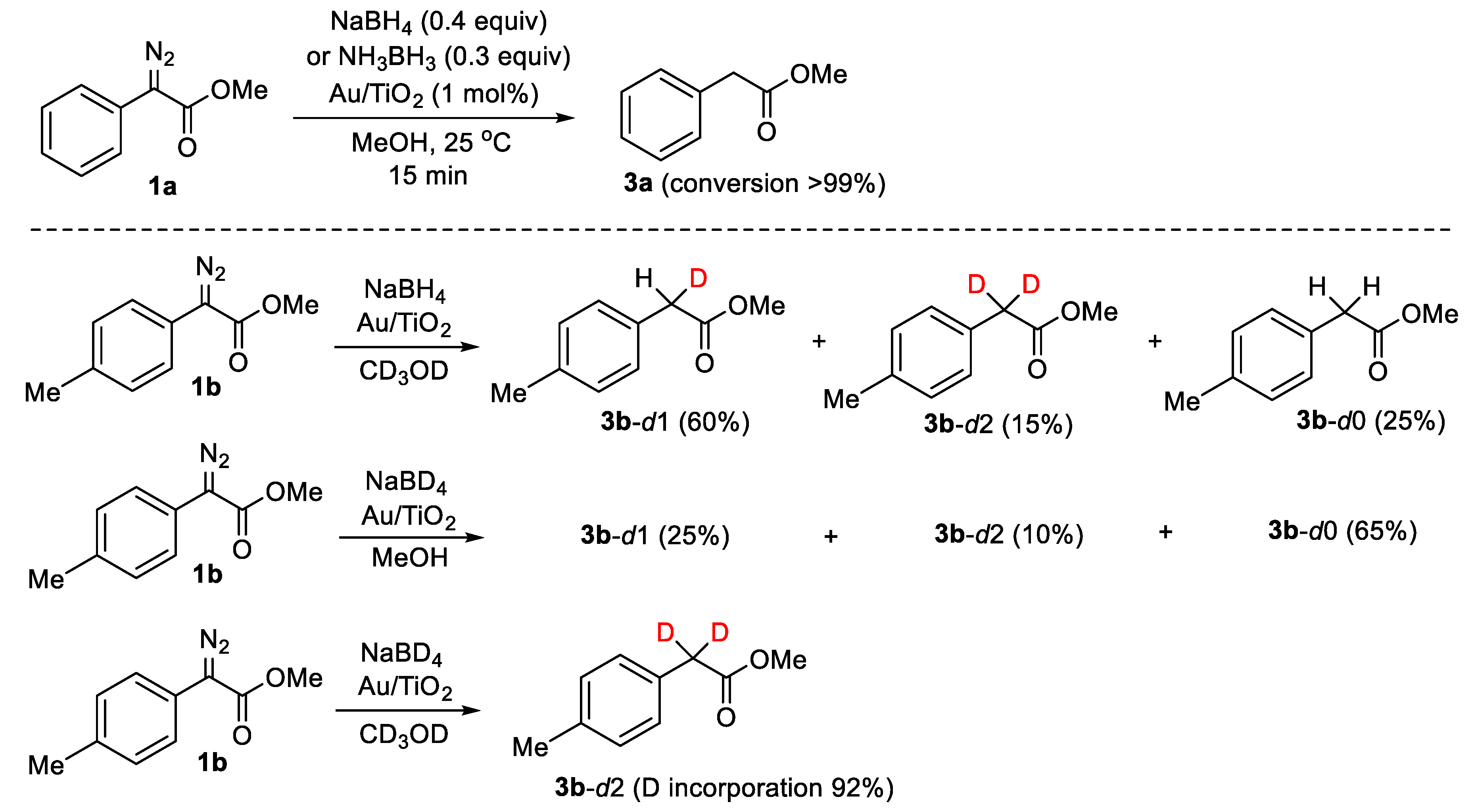{kind=link}
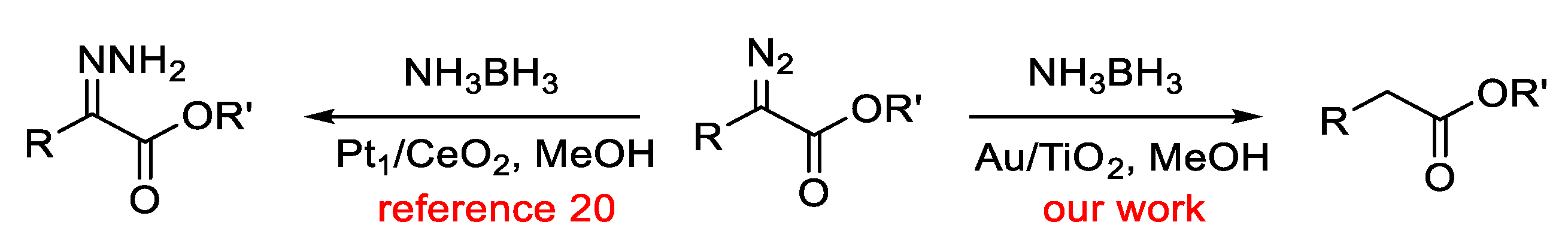{kind=link}
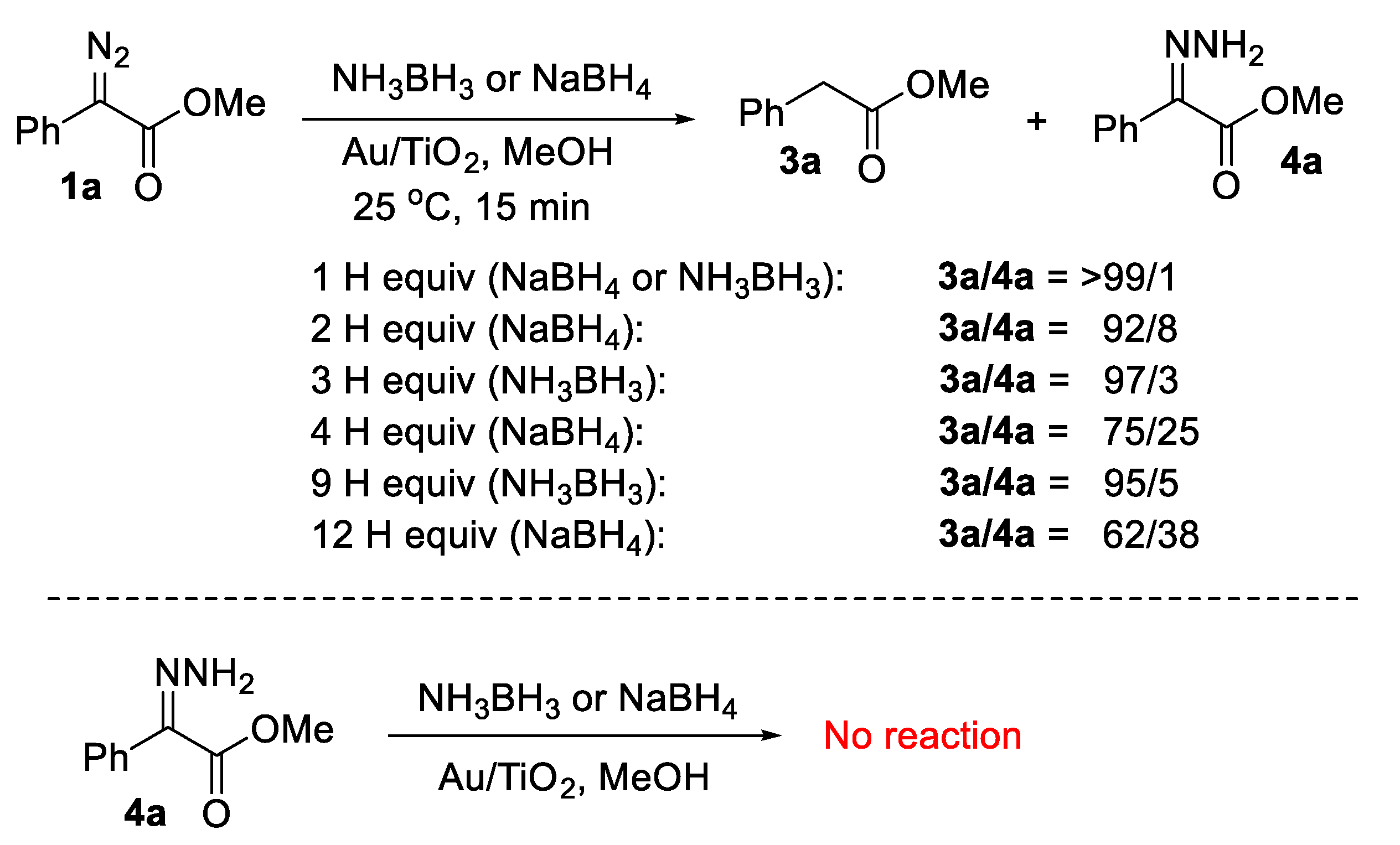{kind=link}
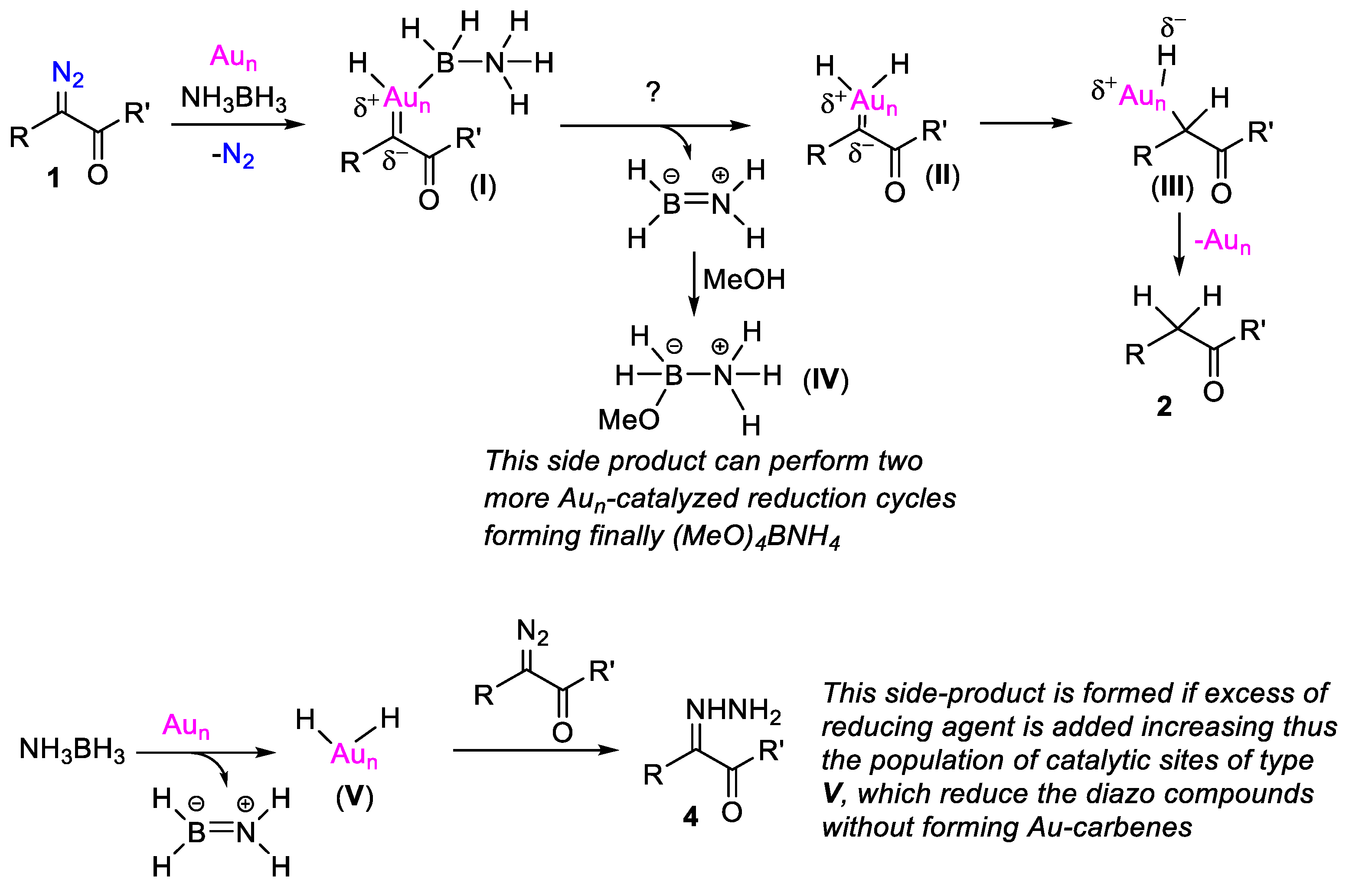{kind=link}
1. Introduction
2. Results
3. Experimental Section
3.1. Catalyst
3.2. Reactants
3.3. Catalytic Reactions
3.4. Characterization of Products
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References and Note
- Wolfrom, M.L.; Brown, R.L. The action of diazomethane upon acyclic sugar derivatives. V. Halogen derivatives. J. Am. Chem. Soc. 1943, 65, 1516–1521. [Google Scholar] [CrossRef]
- Horner, L.; Schwarz, H. Die durch radicale ausgelgste “reductive eliminierung von diazostickstoff” aus diazocarbonyl und diazosulfonylvebbindungen. Tetrahedron Lett. 1966, 30, 3579–3583. [Google Scholar] [CrossRef]
- Pellicciari, R.; Fringuelli, R.; Ceccherelli, P.; Sisani, E. β-Keto esters from the rhodium(II) acetate catalysed conversion of α-diazo-β-hydroxy esters. J. Chem. Soc. Chem. Commun. 1979, 959–960. [Google Scholar] [CrossRef]
- Ye, T.; McKewey, M.A. Synthesis of chiral N-protected α-amino-β-diketones from α-diazoketones derived from natural amino acids. Tetrahedron 1992, 37, 8007–8022. [Google Scholar] [CrossRef]
- Tan, Z.; Qu, Z.; Chen, B.; Wang, J. Diazo decomposition in the presence of tributyltin hydride. Reduction of α-diazo carbonyl compounds. Tetrahedron 2000, 56, 7457–7461. [Google Scholar] [CrossRef]
- Schobert, R.; Hohlein, U. Reduction and isomerization of oxiranes and α-diazoketones by various early transition metallocenes. Synlett 1990, 8, 465–466. [Google Scholar] [CrossRef]
- Pellicciari, R.; Natalini, B.; Cecchetti, S.; Fringuelli, R. Reduction of α-diazo-β-hydroxy esters to β-hydroxy esters: Application in one of two convergent syntheses of a (22S)-22-hydroxy bile acid from fish bile and its (22R)-epimer. J. Chem. Soc. Perkin Trans. I 1985, 493–497. [Google Scholar] [CrossRef]
- Kidonakis, M.; Stratakis, M. Au nanoparticle-catalyzed insertion of carbenes from α-diazocarbonyl compounds into hydrosilanes. Org. Lett. 2018, 20, 4086–4089. [Google Scholar] [CrossRef]
- Oliver-Meseguer, J.; Boronat, M.; Vidal-Moya, A.; Concepcion, P.; Rivero-Crespo, M.A.; Leyva-Perez, A.; Corma, A. Generation and reactivity of electron-rich carbenes on the surface of catalytic gold nanoparticles. J. Am. Chem. Soc. 2018, 140, 3215–3218. [Google Scholar] [CrossRef] [Green Version]
- Stratakis, M.; Garcia, H. Catalysis by supported gold nanoparticles: Beyond aerobic oxidative processes. Chem. Rev. 2012, 112, 4469–4506. [Google Scholar] [CrossRef]
- Stratakis, M.; Lykakis, I.N. Nanogold(0)-catalyzed addition of heteroelement σ linkages to functional groups. Synthesis 2019, 51, 2435–2454. [Google Scholar] [CrossRef]
- Takale, B.S.; Bao, M.; Yamamoto, Y. Gold nanoparticle (AuNPs) and gold nanopore (AuNPore) catalysts in organic synthesis. Org. Biomol. Chem. 2014, 12, 2005–2027. [Google Scholar] [CrossRef] [PubMed]
- Jin, T.; Terada, M.; Bao, M.; Yamamoto, Y. Catalytic performance of nanoporous metal skeleton catalysts for molecular transformations. ChemSusChem 2019, 12, 2936–2954. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Ge, L.; Yuan, H.; Liu, Y.; Gui, Y.; Zhang, B.; Zhou, L.; Fang, S. Heterogeneous gold catalysts for selective hydrogenation: From nanoparticles to atomically precise nanoclusters. Nanoscale 2019, 11, 11429–11436. [Google Scholar] [CrossRef]
- Mitsudome, T.; Kaneda, K. Gold nanoparticle catalysts for selective hydrogenations. Green Chem. 2013, 15, 2636–2654. [Google Scholar] [CrossRef]
- Vasilikogiannaki, E.; Titilas, I.; Vassilikogiannakis, G.; Stratakis, M. cis-Semihydrogenation of alkynes with amine borane complexes catalyzed by gold nanoparticles under mild conditions. Chem. Commun. 2015, 51, 2384–2387. [Google Scholar] [CrossRef]
- Vasilikogiannaki, E.; Gryparis, C.; Kotzabasaki, V.; Lykakis, I.N.; Stratakis, M. Activation of ammonia-borane complex by gold nanoparticles: Facile reduction of nitroarenes into anilines and nitroalkanes into hydroxylamines. Adv. Synth. Catal. 2013, 355, 907–911. [Google Scholar] [CrossRef]
- Fountoulaki, S.; Daikopoulou, V.; Gkizis, P.L.; Tamiolakis, I.; Armatas, G.S.; Lykakis, I.N. Mechanistic studies of the reduction of nitroarenes by NaBH4 or hydrosilanes catalyzed by supported gold nanoparticles. ACS Catal. 2014, 4, 3504–3511. [Google Scholar] [CrossRef]
- Zhou, L.; Liu, Z.; Liu, Y.; Zhang, Y.; Wang, J. N-Tosylhydrazine-mediated deoxygenative hydrogenation of aldehydes and ketones catalyzed by Pd/C. Tetrahedron 2013, 69, 6083–6087. [Google Scholar] [CrossRef]
- Liu, C.; Chen, Z.; Yan, H.; Xi, S.; Yam, K.M.; Gao, J.; Du, Y.; Li, J.; Zhao, X.; Xie, K.; et al. Expedient synthesis of E-hydrazone esters and 1H-indazole scaffolds through heterogeneous single-atom platinum catalysis. Sci. Adv. 2019, 5, eaay1537. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Granda, S.; Costin, T.A.; Sa, M.M.; Gotor-Fernandez, V. Stereoselective bioreduction of α-diazo-β-keto esters. Molecules 2020, 25, 931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittmann, E.; Hu, Y.; Peschke, T.; Rabe, K.S.; Niemeyer, C.M.; Brase, S. Chemoenzymatic synthesis of O-containing heterocycles from α-diazo esters. ChemCatChem 2019, 11, 5519–5523. [Google Scholar] [CrossRef] [Green Version]
- Yasui, E.; Wada, M.; Nagumo, S.; Takamura, N. A novel method for the synthesis of 3,4-disubstituted pyrrole-2,5-dicarboxylates from hydrazones derived from α-diazo esters. Tetrahedron 2013, 69, 4325–4330. [Google Scholar] [CrossRef]
- In contrast to the Fischer metal-carbenes where the carbon atom that is bonded to the metal is electrophilic, in Schrock-type carbenes the carbon atom is nucleophilic. Chemisorbed α-diazocarbonyl compounds on Au nanoparticles are Schrock-type carbenes (see references 8 and 9).
- Ramachandran, P.V.; Gagare, P.D. Preparation of ammonia borane in high yield and purity, methanolysis, and regeneration. Inorg. Chem. 2007, 46, 7810–7817. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Shi, X.; Qiao, X.; Wu, Z.; Bai, G. Ligand-free nickel-catalyzed semihydrogenation of alkynes with sodium borohydride: A highly efficient and selective process for cis-alkenes under ambient conditions. Chem. Commun. 2017, 53, 5372–5375. [Google Scholar] [CrossRef] [PubMed]
- Louka, A.; Gryparis, C.; Stratakis, M. Reduction of quinolines to 1,2,3,4-tetrahydroquinolines with hydrosilane/ethanol catalyzed by TiO2-supported gold nanoparticles under solvent free conditions. Arkivoc 2015, 2015, 38–51. [Google Scholar] [CrossRef] [Green Version]
- Yan, M.; Jin, T.; Ishikawa, Y.; Minato, T.; Fujita, T.; Chen, L.-Y.; Bao, M.; Asao, N.; Chen, M.-W.; Yamamoto, Y. Nanoporous gold catalyst for highly selective semihydrogenation of alkynes: Remarkable effect of amine additives. J. Am. Chem. Soc. 2012, 134, 17536–17542. [Google Scholar] [CrossRef]
- Keipour, H.; Jalba, A.; Delage-Laurin, L.; Ollevier, T. Copper-catalyzed carbenoid insertion reactions of α-diazoesters and α-diazoketones into Si−H and S−H Bonds. J. Org. Chem. 2017, 82, 3000–3010. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kidonakis, M.; Stratakis, M. Reduction of the Diazo Functionality of α-Diazocarbonyl Compounds into a Methylene Group by NH3BH3 or NaBH4 Catalyzed by Au Nanoparticles. Nanomaterials 2021, 11, 248. https://0-doi-org.brum.beds.ac.uk/10.3390/nano11010248
Kidonakis M, Stratakis M. Reduction of the Diazo Functionality of α-Diazocarbonyl Compounds into a Methylene Group by NH3BH3 or NaBH4 Catalyzed by Au Nanoparticles. Nanomaterials. 2021; 11(1):248. https://0-doi-org.brum.beds.ac.uk/10.3390/nano11010248
Chicago/Turabian StyleKidonakis, Marios, and Manolis Stratakis. 2021. "Reduction of the Diazo Functionality of α-Diazocarbonyl Compounds into a Methylene Group by NH3BH3 or NaBH4 Catalyzed by Au Nanoparticles" Nanomaterials 11, no. 1: 248. https://0-doi-org.brum.beds.ac.uk/10.3390/nano11010248