
Bioactivity of PEGylated Graphene Oxide Nanoparticles Combined with Near-Infrared Laser Irradiation Studied in Colorectal Carcinoma Cells

 , , and
, , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of Poly(Ethylene Glycol)-Modified Graphene Oxide (GO–PEG) and Physicochemical Characterization of NPs
2.2. Cell Cultures, Media and Treatment Protocols
2.3. Near-Infrared Irradiation
2.4. Cell Proliferation Assays (WST-1)
2.5. Fluorescence-Activated Cell Sorting (FACS) of Cells
2.5.1. Cell Cycle Analyses after Staining with Propidium Iodide (PI)
2.5.2. Mitochondrial Activity Analyses after Staining with Rhodamine 123 (Rh123)
2.6. Genotoxicity Analysis by Single Cell Gel Electrophoresis (SCGE)
2.7. Fluorescent Microscopy Analysis of Mitochondria after Staining with Rh123
2.8. Gene Expression Analysis by RT-qPCR
2.9. Statistical Analysis
3. Results and Discussion
3.1. PEGylated Graphene Oxide Nanoparticles with Near-Infrared Laser Irradiation Proved Non-Toxic for Colorectal Carcinoma Cells
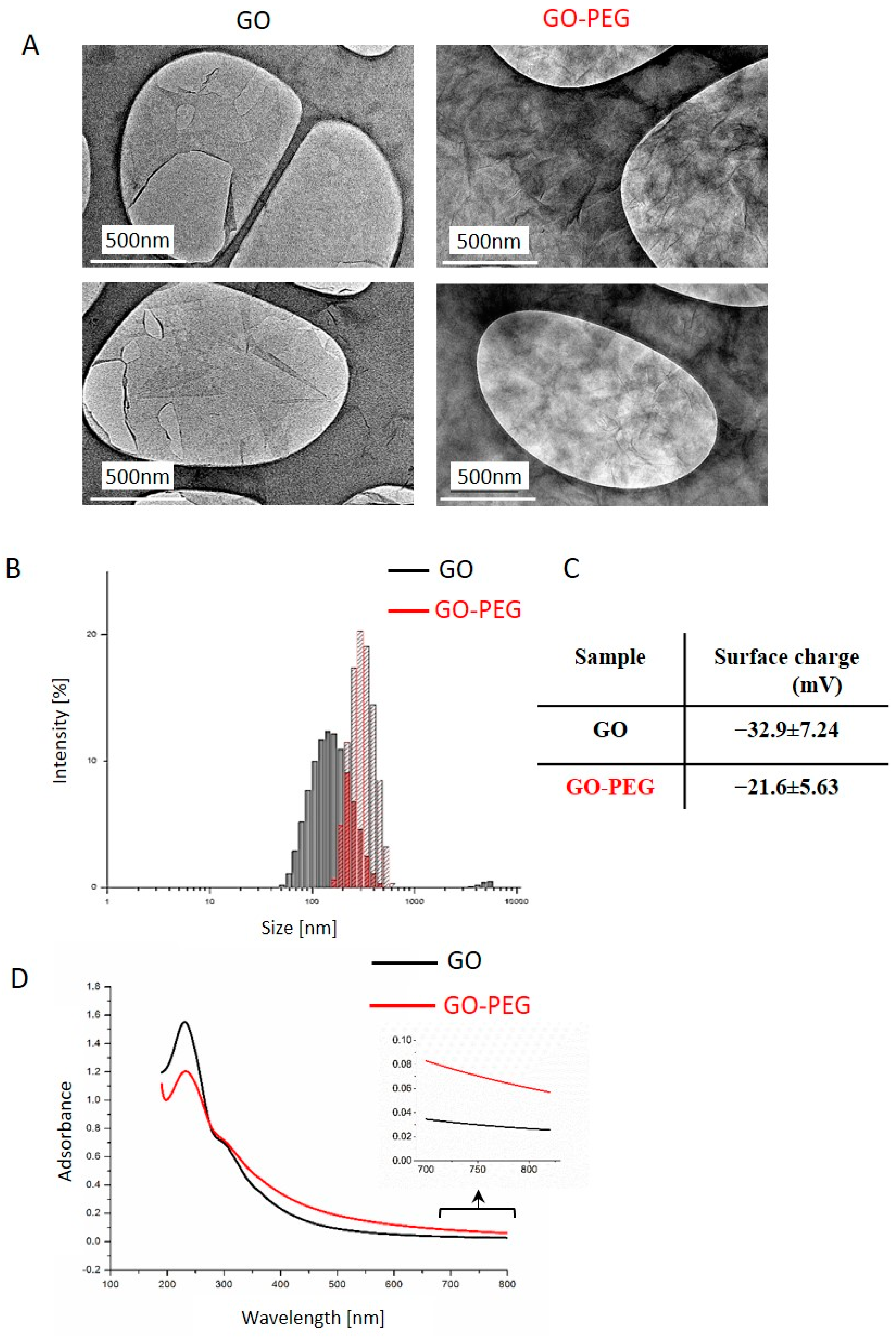
3.1.1. Physicochemical and Biophysical Characteristics of GO and GO–PEG NPs
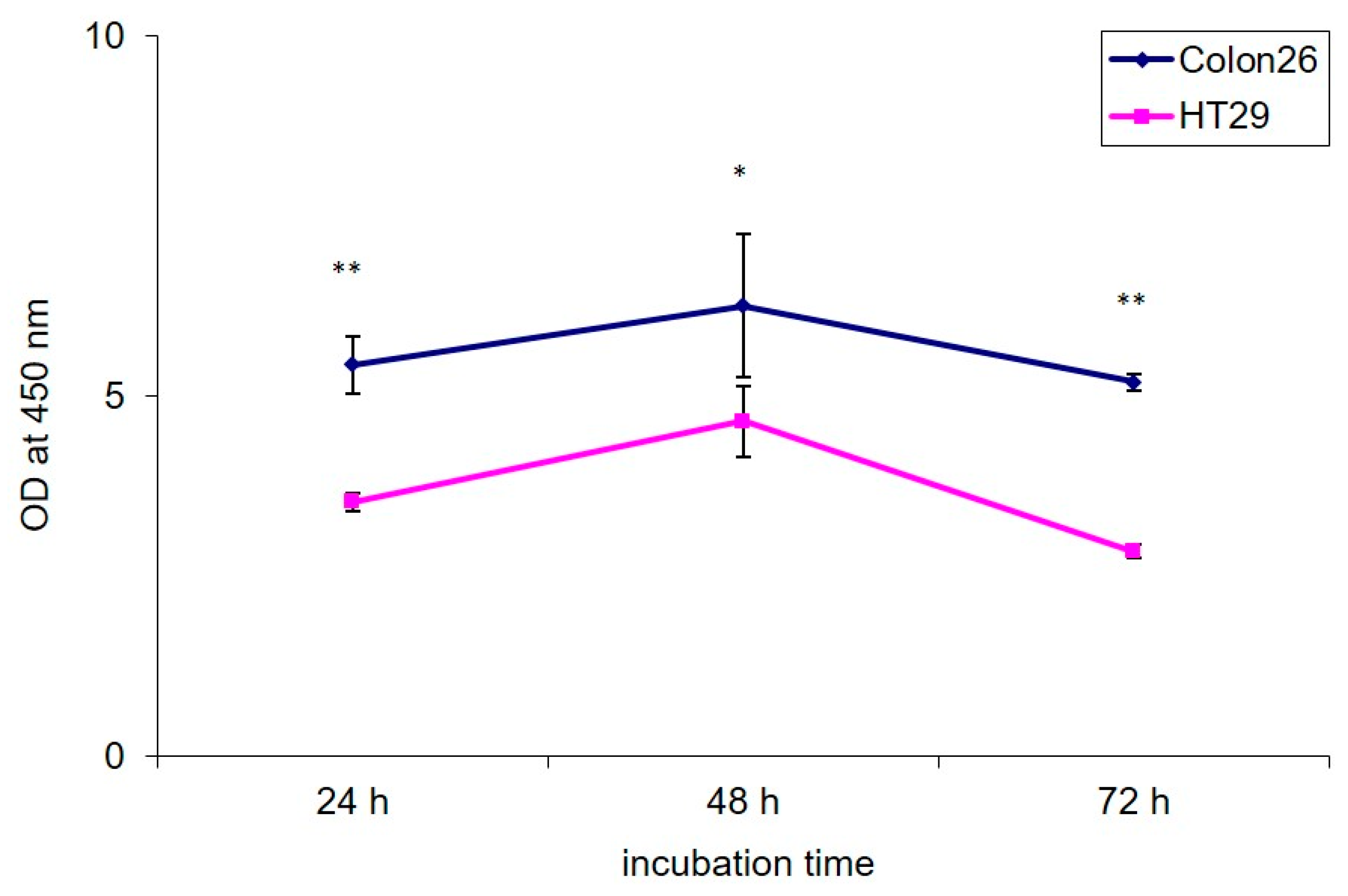
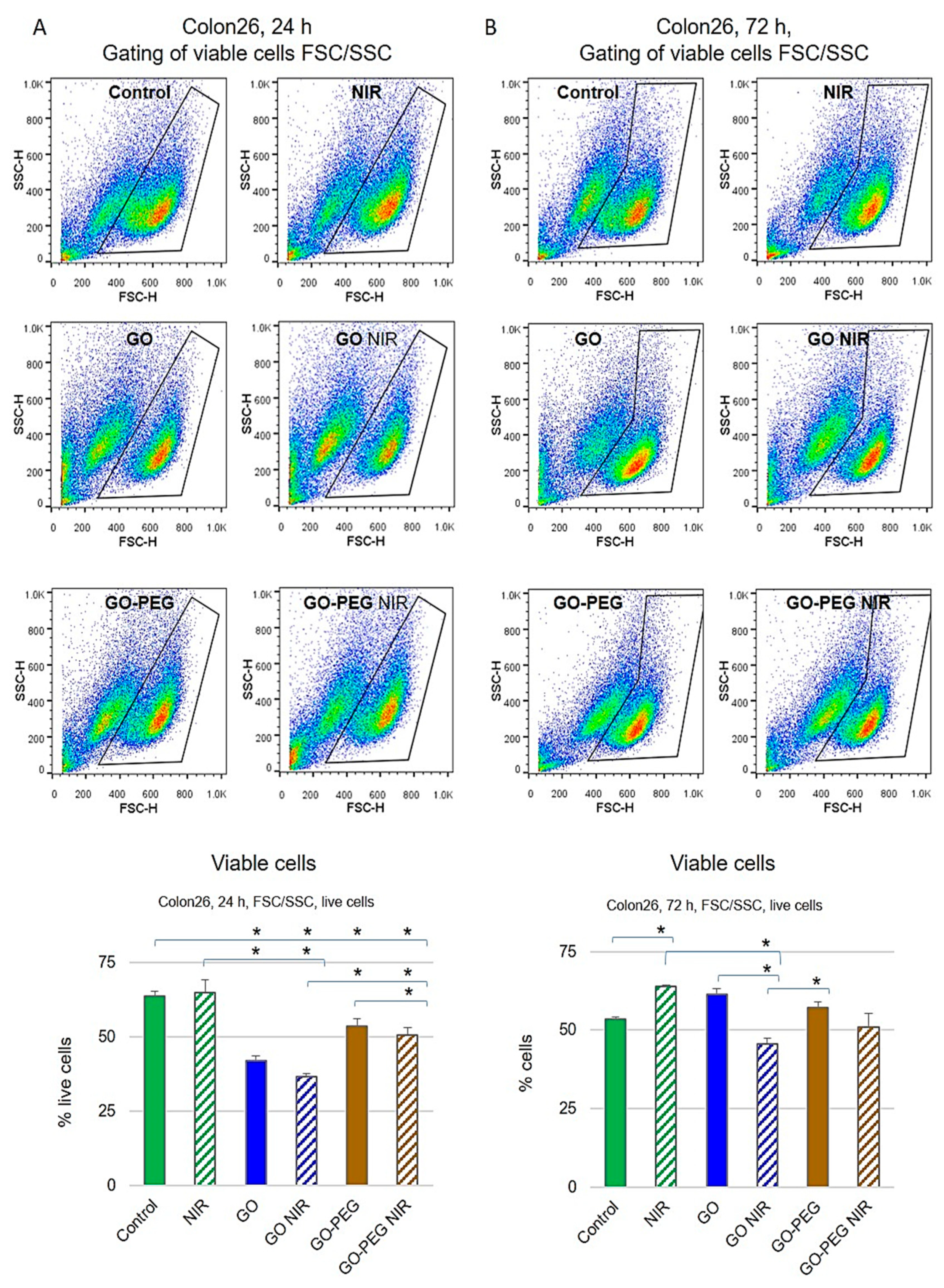
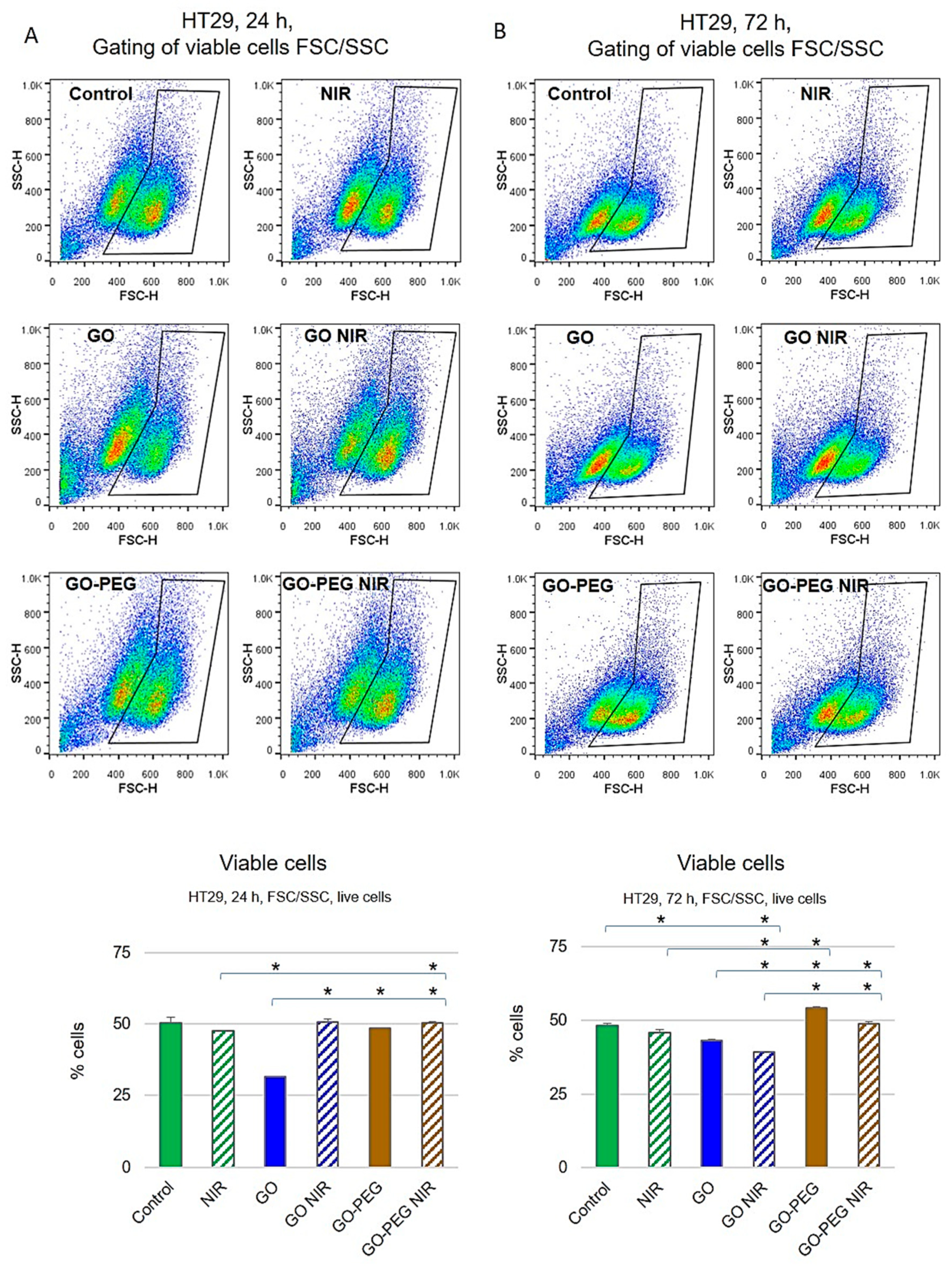
3.1.2. PEGylated GO, Combined with NIR Irradiation, Are Non-Toxic for Colorectal Carcinoma Cells Regardless of the Cultivation Time
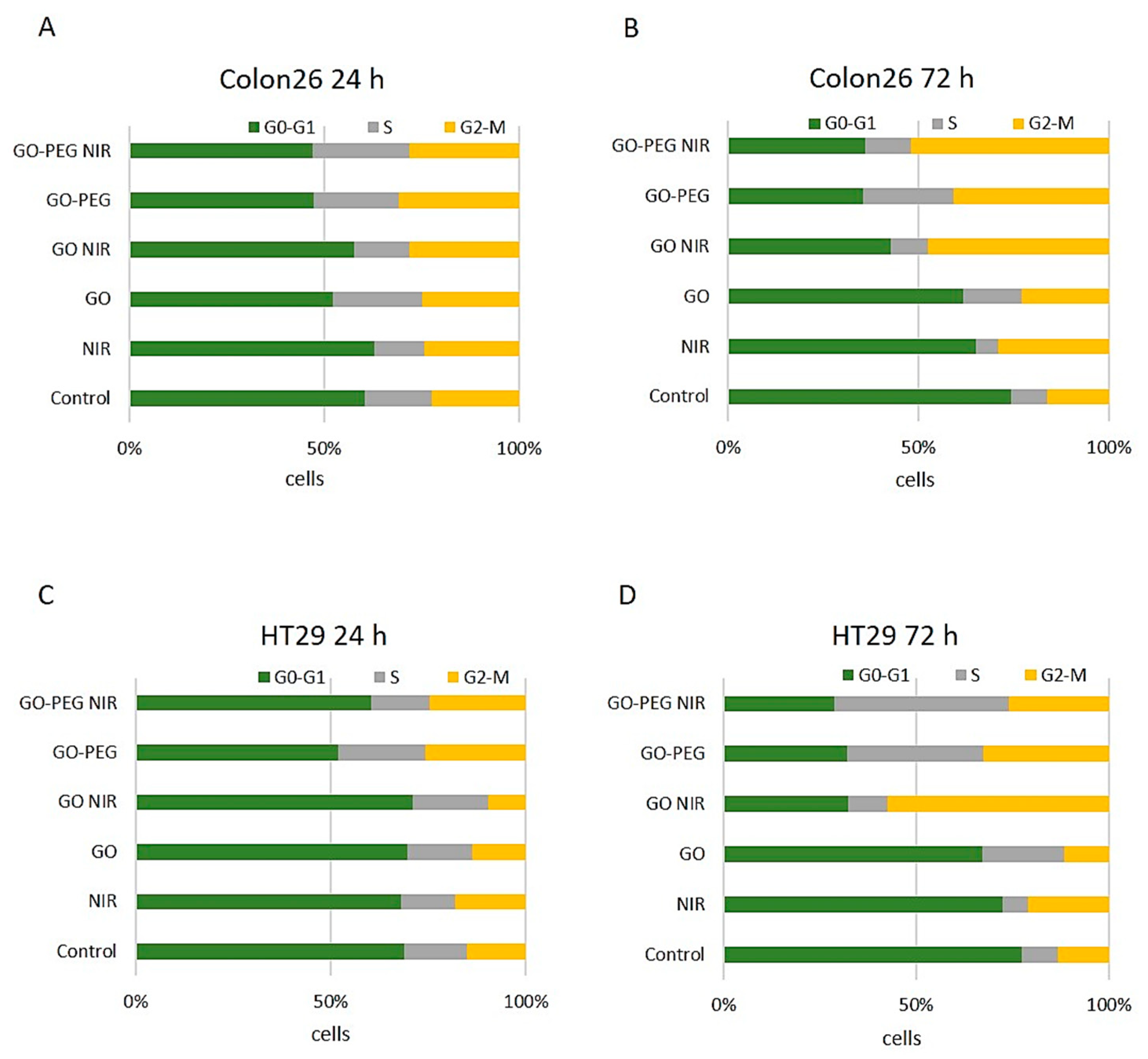
3.1.3. Treatment with GO–PEG with and without NIR Slightly Impaired the Cell Cycle of Colorectal Cell Carcinoma Cells at 24 h of Incubation
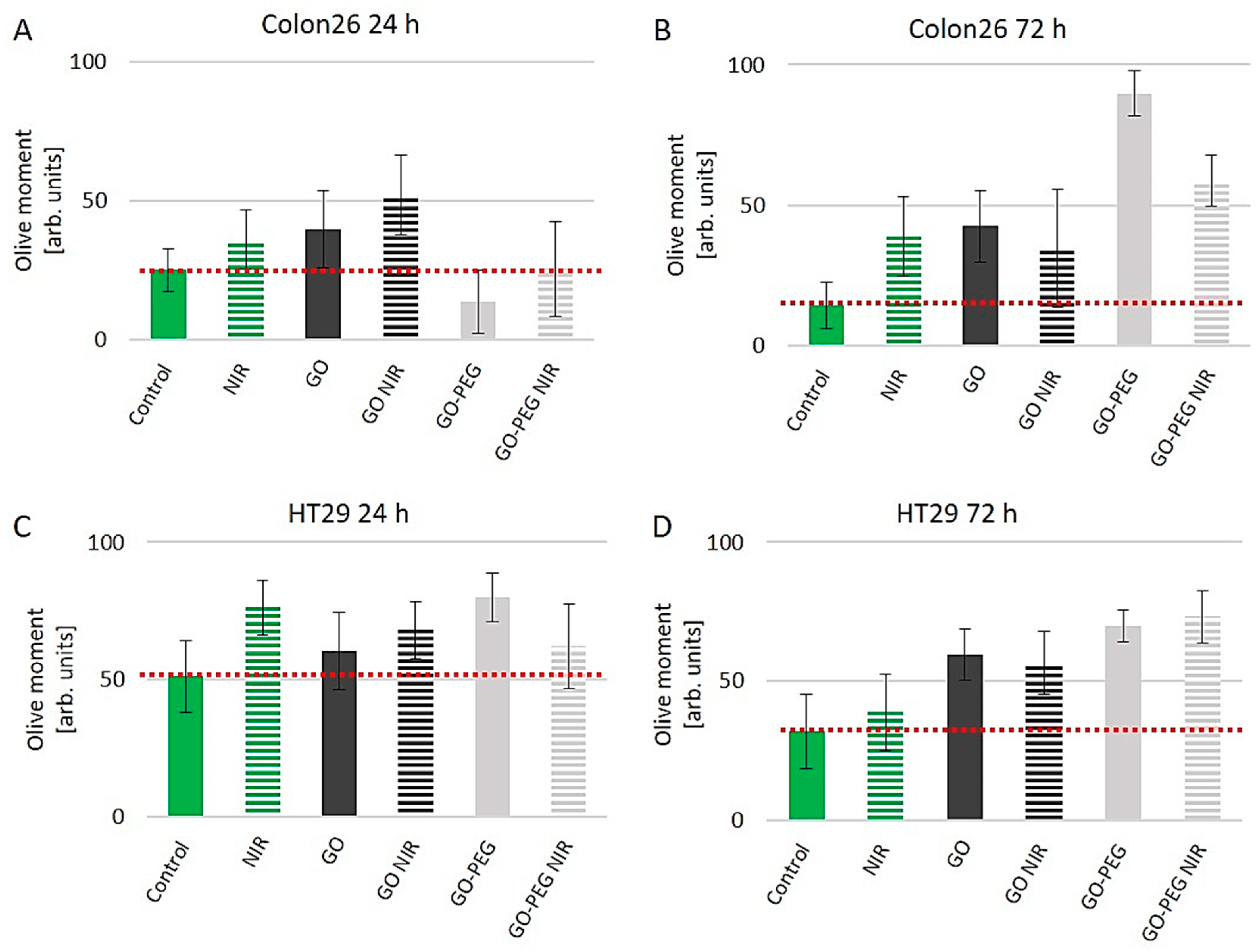
3.2. Insignificant Genotoxicity of GO–PEG NPs in Combination with NIR for Colon26 and HT29 Cells after 24 h of Cultivation
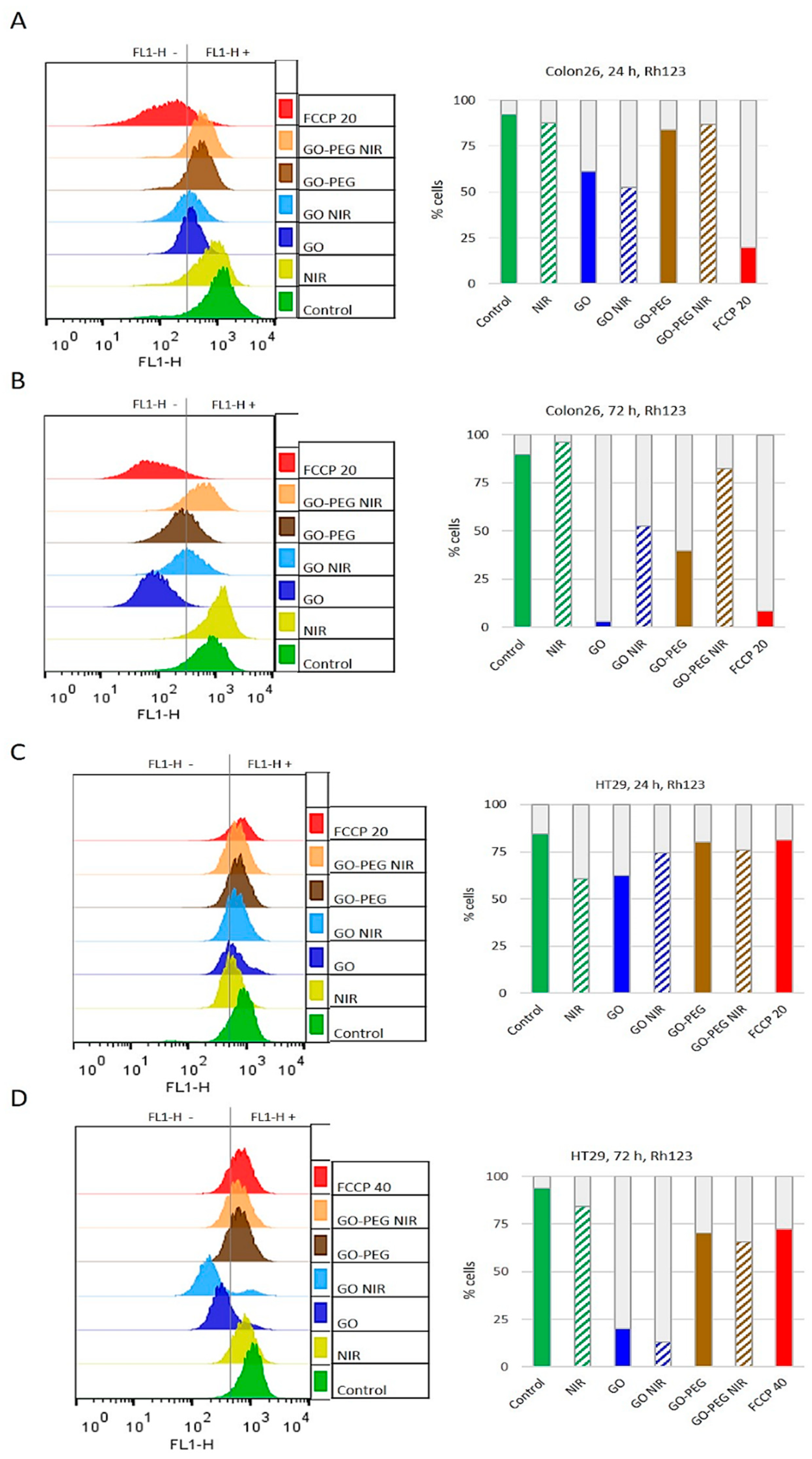
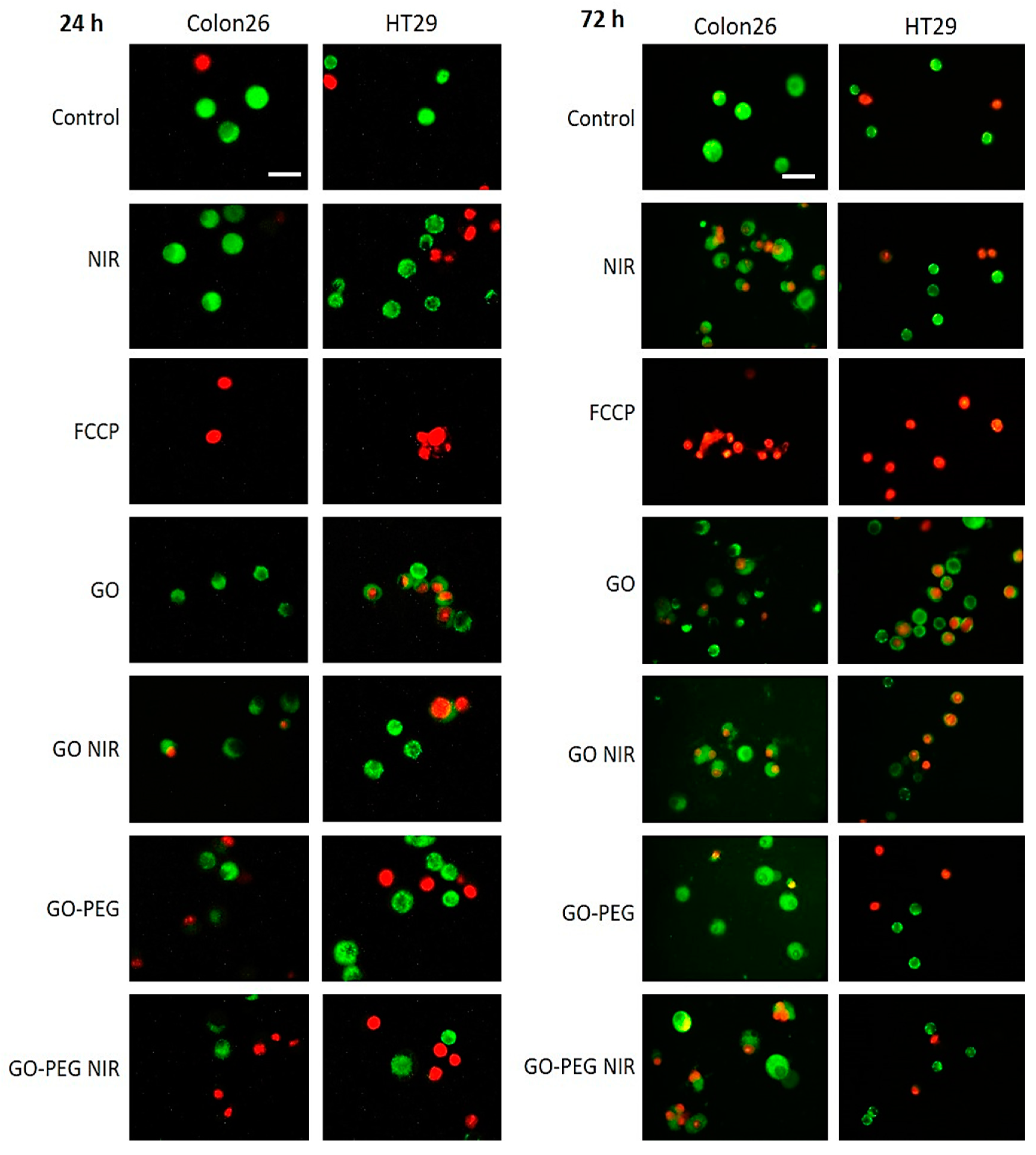
3.3. PEGylated Graphene Oxide Nanoparticles Combined with Near-Infrared Laser Irradiation Has Little Mitotoxicity in Colorectal Carcinoma Cells
| For Colon26 at 24 h: | GO NIR > GO > GO–PEG > GO–PEG NIR > NIR |
| For Colon26 at 72 h: | GO > GO–PEG > GO NIR > GO–PEG NIR > NIR |
| For HT29 at 24 h: | NIR > GO > GO NIR > GO–PEG NIR > GO–PEG |
| For HT29 at 72 h: | GO NIR > GO > GO–PEG NIR > GO–PEG > NIR |
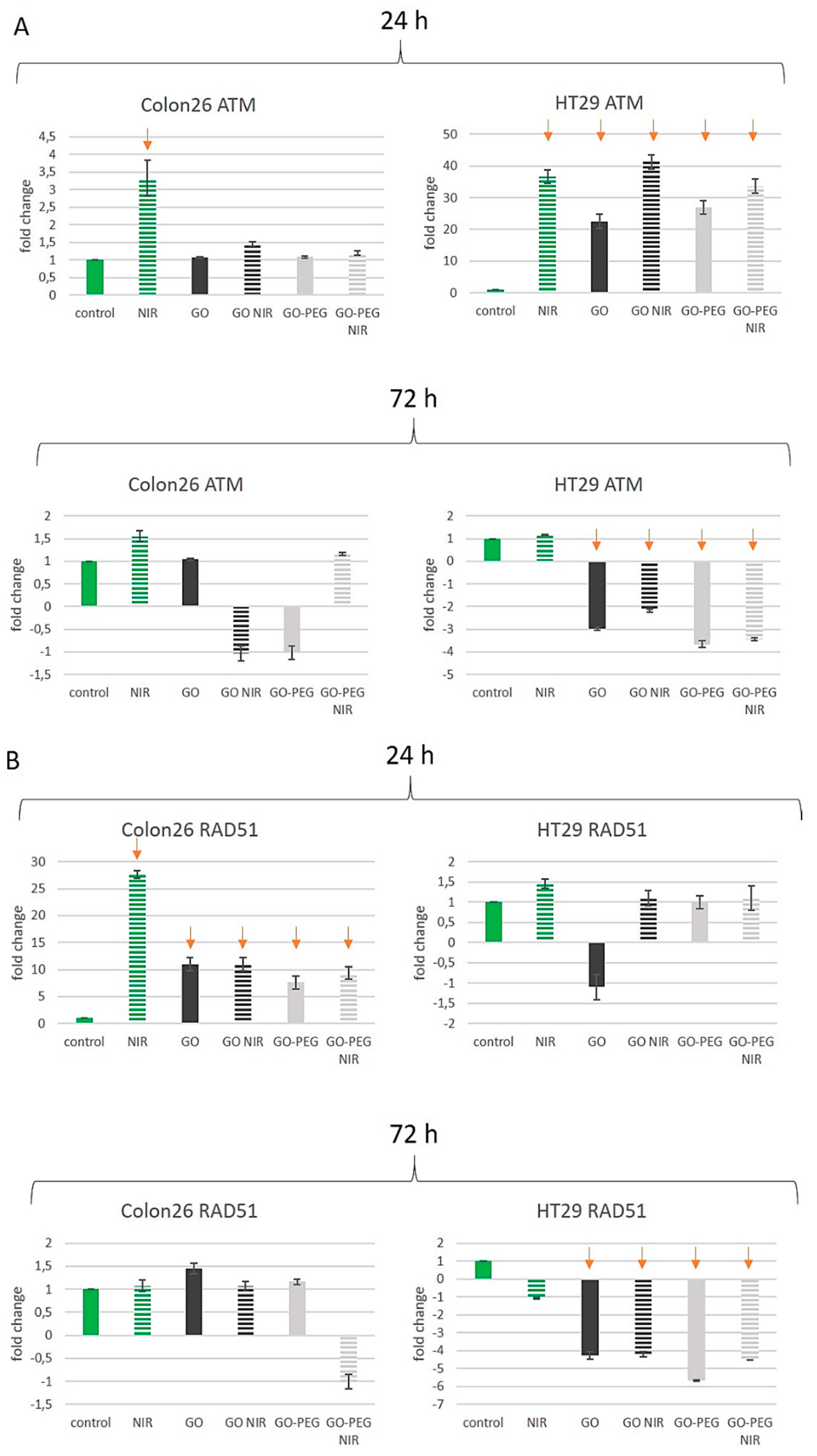
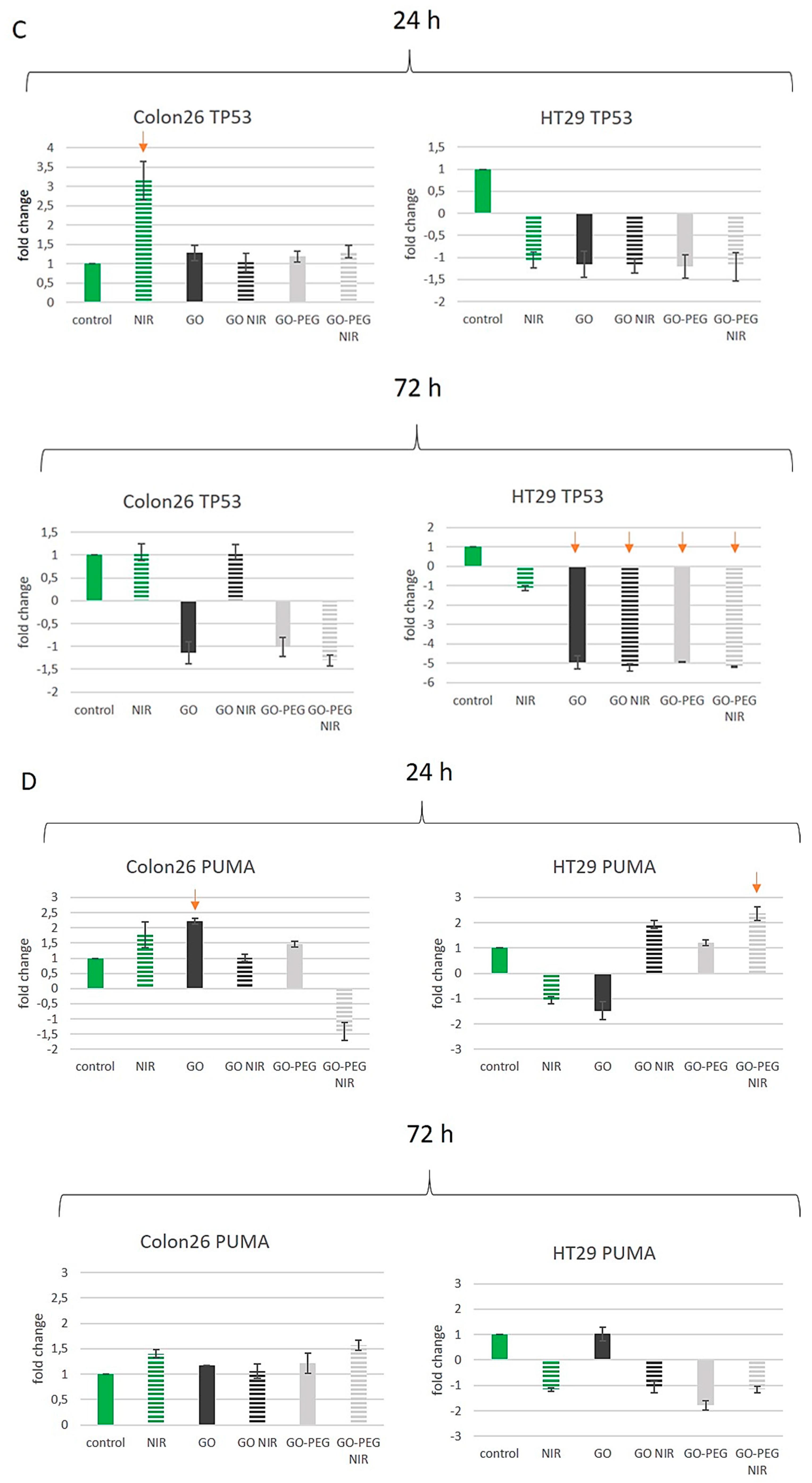
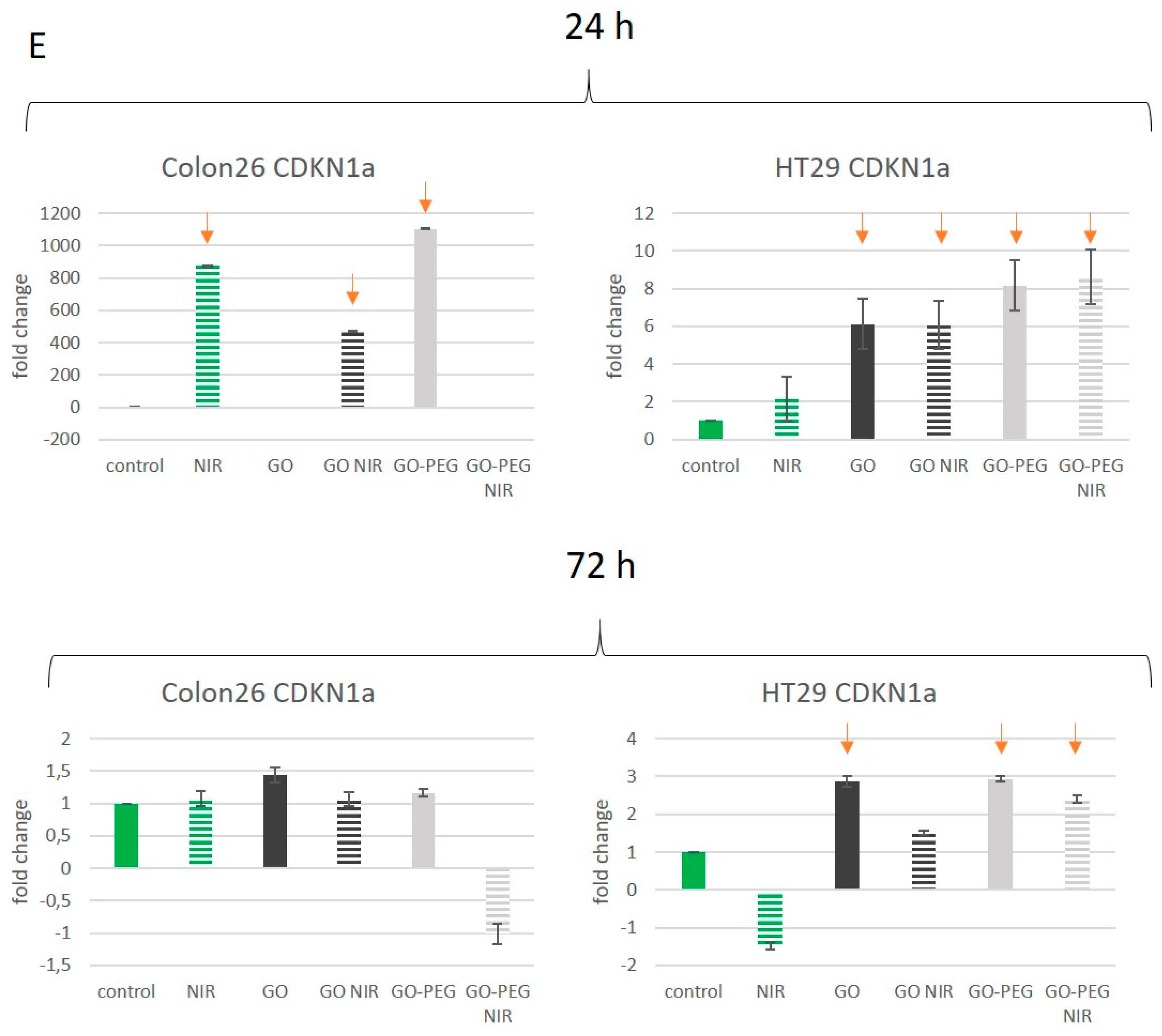
3.4. PEGylated Graphene Oxide Nanoparticles with Near-Infrared Laser Irradiation Modulate the Activity of Stress-Responsive Genes in Colorectal Carcinoma Cells
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Wathoni, N.; Nguyen, A.N.; Rusdin, A.; Umar, A.K.; Mohammed, A.F.A.; Motoyama, K.; Joni, I.M.; Muchtaridi, M. Enteric-Coated Strategies in Colorectal Cancer Nanoparticle Drug Delivery System. Drug Des. Dev. Ther. 2020, 14, 4387–4405. [Google Scholar] [CrossRef] [PubMed]
- Han, H.S.; Choi, K.Y. Advances in Nanomaterial-Mediated Photothermal Cancer Therapies: Toward Clinical Applications. Biomedicines 2021, 9, 305. [Google Scholar] [CrossRef] [PubMed]
- Trakarnsanga, A.; Ithimakin, S.; Weiser, M.R. Treatment of locally advanced rectal cancer: Controversies and questions. World J. Gastroenterol. 2012, 18, 5521–5532. [Google Scholar] [CrossRef]
- Liang, X.J.; Chen, C.; Zhao, Y.; Wang, P.C. Circumventing tumor resistance to chemotherapy by nanotechnology. Methods Mol. Biol. 2010, 596, 467–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [Green Version]
- Hu, Q.; Sun, W.; Wang, C.; Gu, Z. Recent advances of cocktail chemotherapy by combination drug delivery systems. Adv. Drug Deliv. Rev. 2016, 98, 19–34. [Google Scholar] [CrossRef] [Green Version]
- Zou, L.; Wang, H.; He, B.; Zeng, L.; Tan, T.; Cao, H.; He, X.; Zhang, Z.; Guo, S.; Li, Y. Current Approaches of Photothermal Therapy in Treating Cancer Metastasis with Nanotherapeutics. Theranostics 2016, 6, 762–772. [Google Scholar] [CrossRef]
- Doughty, A.C.V.; Hoover, A.R.; Layton, E.; Murray, C.K.; Howard, E.W.; Chen, W.R. Nanomaterial Applications in Photothermal Therapy for Cancer. Materials 2019, 12, 779. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Chen, Y.; Yang, Y.; Yu, Y.; Zhang, Y.; Zhu, D.; Yu, X.; Ouyang, X.; Xie, Z.; Zhao, Y.; et al. Recent Advances in Nanomaterials-Based Chemo-Photothermal Combination Therapy for Improving Cancer Treatment. Front. Bioeng. Biotechnol. 2019, 7, 293. [Google Scholar] [CrossRef] [PubMed]
- Hauck, T.S.; Jennings, T.L.; Yatsenko, T.; Kumaradas, J.C.; Chan, W.C.W. Enhancing the Toxicity of Cancer Chemotherapeutics with Gold Nanorod Hyperthermia. Adv. Mater. 2008, 20, 3832–3838. [Google Scholar] [CrossRef]
- Park, H.; Yang, J.; Lee, J.; Haam, S.; Choi, I.H.; Yoo, K.H. Multifunctional nanoparticles for combined doxorubicin and photothermal treatments. ACS Nano 2009, 3, 2919–2926. [Google Scholar] [CrossRef] [PubMed]
- Weissleder, R. A clearer vision for in vivo imaging. Nat. Biotechnol. 2001, 19, 316–317. [Google Scholar] [CrossRef]
- Weissleder, R.; Ntziachristos, V. Shedding light onto live molecular targets. Nat. Med. 2003, 9, 123–128. [Google Scholar] [CrossRef]
- Zou, J.; Li, L.; Yang, Z.; Chen, X. Phototherapy meets immunotherapy: A win–win strategy to fight against cancer. Nanophotonics 2021, 10, 3229–3245. [Google Scholar] [CrossRef]
- Wei, Y.; Zhou, F.; Zhang, D.; Chen, Q.; Xing, D. A graphene oxide based smart drug delivery system for tumor mitochondria-targeting photodynamic therapy. Nanoscale 2016, 8, 3530–3538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, P.; Sun, L.; Wang, F.; Liu, X.; Yan, Z.; Wang, M.; Zhang, L.; Tian, Z.; Liu, Z.; You, J. Highly fluorescent N-doped carbon dots with two-photon emission for ultrasensitive detection of tumor marker and visual monitor anticancer drug loading and delivery. Chem. Eng. J. 2019, 356, 994–1002. [Google Scholar] [CrossRef]
- Zeng, W.N.; Yu, Q.P.; Wang, D.; Liu, J.L.; Yang, Q.J.; Zhou, Z.K.; Zeng, Y.P. Mitochondria-targeting graphene oxide nanocomposites for fluorescence imaging-guided synergistic phototherapy of drug-resistant osteosarcoma. J. Nanobiotechnol. 2021, 19, 79. [Google Scholar] [CrossRef]
- Chen, S.; Lei, Q.; Qiu, W.-X.; Liu, L.-H.; Zheng, D.-W.; Fan, J.-X.; Rong, L.; Sun, Y.-X.; Zhang, X.-Z. Mitochondria-targeting “Nanoheater” for enhanced photothermal/chemo-therapy. Biomaterials 2017, 117, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Jeena, M.T.; Kim, S.; Jin, S.; Ryu, J.H. Recent Progress in Mitochondria-Targeted Drug and Drug-Free Agents for Cancer Therapy. Cancers 2019, 12, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Zhang, Z.; Xing, D. Cell death via mitochondrial apoptotic pathway due to activation of Bax by lysosomal photodamage. Free Radic. Biol. Med. 2011, 51, 53–68. [Google Scholar] [CrossRef] [PubMed]
- Teiten, M.H.; Bezdetnaya, L.; Morliere, P.; Santus, R.; Guillemin, F. Endoplasmic reticulum and Golgi apparatus are the preferential sites of Foscan® localisation in cultured tumour cells. Br. J. Cancer 2003, 88, 146–152. [Google Scholar] [CrossRef] [Green Version]
- Zaharie-Butucel, D.; Potara, M.; Suarasan, S.; Licarete, E.; Astilean, S. Efficient combined near-infrared-triggered therapy: Phototherapy over chemotherapy in chitosan-reduced graphene oxide-IR820 dye-doxorubicin nanoplatforms. J. Colloid Interface Sci. 2019, 552, 218–229. [Google Scholar] [CrossRef]
- Konan, Y.N.; Gurny, R.; Allemann, E. State of the art in the delivery of photosensitizers for photodynamic therapy. J. Photochem. Photobiol. B Biol. 2002, 66, 89–106. [Google Scholar] [CrossRef]
- Rong, P.; Yang, K.; Srivastan, A.; Kiesewetter, D.O.; Yue, X.; Wang, F.; Nie, L.; Bhirde, A.; Wang, Z.; Liu, Z.; et al. Photosensitizer loaded nano-graphene for multimodality imaging guided tumor photodynamic therapy. Theranostics 2014, 4, 229–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, B.; Wang, C.; Zhang, S.; Feng, L.; Liu, Z. Photothermally enhanced photodynamic therapy delivered by nano-graphene oxide. ACS Nano 2011, 5, 7000–7009. [Google Scholar] [CrossRef] [PubMed]
- Dimiev, A.; Kosynkin, D.V.; Alemany, L.B.; Chaguine, P.; Tour, J.M. Pristine Graphite Oxide. J. Am. Chem. Soc. 2012, 134, 2815–2822. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Liu, Z.; Welsher, K.; Robinson, J.T.; Goodwin, A.; Zaric, S.; Dai, H. Nano-Graphene Oxide for Cellular Imaging and Drug Delivery. Nano Res. 2008, 1, 203–212. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Robinson, J.T.; Sun, X.; Dai, H. PEGylated nanographene oxide for delivery of water-insoluble cancer drugs. J. Am. Chem. Soc. 2008, 130, 10876–10877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Zhang, X.; Liu, Z.; Ma, Y.; Huang, Y.; Chen, Y. High-Efficiency Loading and Controlled Release of Doxorubicin Hydrochloride on Graphene Oxide. J. Phys. Chem. C 2008, 112, 17554–17558. [Google Scholar] [CrossRef]
- Loh, K.P.; Bao, Q.; Eda, G.; Chhowalla, M. Graphene oxide as a chemically tunable platform for optical applications. Nat. Chem. 2010, 2, 1015–1024. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.; Yang, X.; Shi, X.; Tan, X.; Peng, R.; Wang, J.; Liu, Z. Polyethylene glycol and polyethylenimine dual-functionalized nano-graphene oxide for photothermally enhanced gene delivery. Small 2013, 9, 1989–1997. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Guo, Z.; Huang, D.; Liu, Z.; Guo, X.; Zhong, H. Synergistic effect of chemo-photothermal therapy using PEGylated graphene oxide. Biomaterials 2011, 32, 8555–8561. [Google Scholar] [CrossRef]
- Yang, K.; Wan, J.; Zhang, S.; Zhang, Y.; Lee, S.T.; Liu, Z. In vivo pharmacokinetics, long-term biodistribution, and toxicology of PEGylated graphene in mice. ACS Nano 2011, 5, 516–522. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Chen, H.; Sutrisno, L.; Kawazoe, N.; Chen, G. Nanomaterials and their composite scaffolds for photothermal therapy and tissue engineering applications. Sci. Technol Adv. Mater. 2021, 22, 404–428. [Google Scholar] [CrossRef] [PubMed]
- Georgieva, M.; Gospodinova, Z.; Keremidarska-Markova, M.; Kamenska, T.; Gencheva, G.; Krasteva, N. PEGylated Nanographene Oxide in Combination with Near-Infrared Laser Irradiation as a Smart Nanocarrier in Colon Cancer Targeted Therapy. Pharmaceutics 2021, 13, 424. [Google Scholar] [CrossRef]
- Kamenska, T.; Abrashev, M.; Georgieva, M.; Krasteva, N. Impact of Polyethylene Glycol Functionalization of Graphene Oxide on Anticoagulation and Haemolytic Properties of Human Blood. Materials 2021, 14, 4853. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Liu, H.; Zhao, C.; Qin, G.; Xi, G.; Li, T.; Wang, X.; Chen, T. One-step reduction and PEGylation of graphene oxide for photothermally controlled drug delivery. Biomaterials 2014, 35, 4986–4995. [Google Scholar] [CrossRef]
- Gospodinova, Z.; Kamenska, T.; Gencheva, G.; Georgieva, M.; Krasteva, N. PEGylation of graphene oxide nanosheets modulate cancer cell motility and proliferative ability. J. Phys. Conf. Ser. 2021, 1762, 012001. [Google Scholar] [CrossRef]
- Krasteva, N.; Keremidarska-Markova, M.; Hristova-Panusheva, K.; Andreeva, T.; Speranza, G.; Wang, D.; Draganova-Filipova, M.; Miloshev, G.; Georgieva, M. Aminated Graphene Oxide as a Potential New Therapy for Colorectal Cancer. Oxidative Med. Cell. Longev. 2019, 2019, 3738980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferlini, C.; Biselli, R.; Nisini, R.; Fattorossi, A. Rhodamine 123: A useful probe for monitoring T cell activation. Cytometry 1995, 21, 284–293. [Google Scholar] [CrossRef]
- Johnson, L.V.; Walsh, M.L.; Chen, L.B. Localization of mitochondria in living cells with rhodamine 123. Proc. Natl. Acad. Sci. USA 1980, 77, 990–994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, L.V.; Walsh, M.L.; Bockus, B.J.; Chen, L.B. Monitoring of relative mitochondrial membrane potential in living cells by fluorescence microscopy. J. Cell Biol. 1981, 88, 526–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baracca, A.; Sgarbi, G.; Solaini, G.; Lenaz, G. Rhodamine 123 as a probe of mitochondrial membrane potential: Evaluation of proton flux through F0 during ATP synthesis. Biochim. Biophys. Acta (Bba)—Bioenerg. 2003, 1606, 137–146. [Google Scholar] [CrossRef] [Green Version]
- Gokerkucuk, E.B.; Tramier, M.; Bertolin, G. Imaging Mitochondrial Functions: From Fluorescent Dyes to Genetically-Encoded Sensors. Genes 2020, 11, 125. [Google Scholar] [CrossRef] [Green Version]
- Georgieva, M.; Vasileva, B.; Speranza, G.; Wang, D.; Stoyanov, K.; Draganova-Filipova, M.; Zagorchev, P.; Sarafian, V.; Miloshev, G.; Krasteva, N. Amination of Graphene Oxide Leads to Increased Cytotoxicity in Hepatocellular Carcinoma Cells. Int. J. Mol. Sci. 2020, 21, 2427. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- De Both, N.J.; Vermey, M.; Dinjens, W.N.; Bosman, F.T. A comparative evaluation of various invasion assays testing colon carcinoma cell lines. Br. J. Cancer 1999, 81, 934–941. [Google Scholar] [CrossRef] [Green Version]
- Crowley, L.C.; Scott, A.P.; Marfell, B.J.; Boughaba, J.A.; Chojnowski, G.; Waterhouse, N.J. Measuring Cell Death by Propidium Iodide Uptake and Flow Cytometry. Cold Spring Harb. Protoc. 2016, 2016. [Google Scholar] [CrossRef]
- Lammel, T.; Boisseaux, P.; Fernández-Cruz, M.-L.; Navas, J.M. Internalization and cytotoxicity of graphene oxide and carboxyl graphene nanoplatelets in the human hepatocellular carcinoma cell line Hep G2. Part. Fibre Toxicol 2013, 10, 27. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Wei, K.; Zhang, W.; Bai, Y.; Sun, Y.; Gu, J. Graphene Oxide Quantum Dots Incorporated into a Thin Film Nanocomposite Membrane with High Flux and Antifouling Properties for Low-Pressure Nanofiltration. ACS Appl. Mater. Interfaces 2017, 9, 11082–11094. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Z.; Wang, J.; Li, J.; Lin, Y. Graphene and graphene oxide: Biofunctionalization and applications in biotechnology. Trends Biotechnol. 2011, 29, 205–212. [Google Scholar] [CrossRef]
- Ghosh, S.; Chatterjee, K. Poly(Ethylene Glycol) Functionalized Graphene Oxide in Tissue Engineering: A Review on Recent Advances. Int. J. Nanomed. 2020, 15, 5991–6006. [Google Scholar] [CrossRef]
- Luo, N.; Weber, J.K.; Wang, S.; Luan, B.; Yue, H.; Xi, X.; Du, J.; Yang, Z.; Wei, W.; Zhou, R.; et al. PEGylated graphene oxide elicits strong immunological responses despite surface passivation. Nat. Commun. 2017, 8, 14537. [Google Scholar] [CrossRef] [Green Version]
- Peng, J.; Gong, P.; Li, S.; Kong, F.; Ge, X.; Wang, B.; Guo, L.; Liu, Z.; You, J. A smart bioresponsive nanosystem with dual-modal imaging for drug visual loading and targeted delivery. Chem. Eng. J. 2020, 391, 123619. [Google Scholar] [CrossRef]
- Huang, R.-X.; Zhou, P.-K. DNA damage response signaling pathways and targets for radiotherapy sensitization in cancer. Signal. Transduct. Target. 2020, 5, 60. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Deng, Y.; Meng, X.; Chang, H.; Ling, C.; Li, D.; Wang, Q.; Lu, T.; Yang, Y.; Song, G.; et al. Genotoxicity evaluation of silica nanoparticles in murine: A systematic review and meta-analysis. Toxicol. Mech. Methods 2021, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Georgieva, M.; Zagorchev, P.; Miloshev, G. Random, double- and single-strand DNA breaks can be differentiated in the method of Comet assay by the shape of the comet image. Electrophoresis 2015, 36, 2553–2560. [Google Scholar] [CrossRef]
- Kianmehr, M.; Amiri, M.; Ebrahimzadeh-Bideskan, A.; Hajavi, J. DNA damage assessment in the lymphocytes of construction painters by comet assay. Toxicol. Ind. Health 2016, 32, 1902–1909. [Google Scholar] [CrossRef]
- Gurcan, C.; Taheri, H.; Bianco, A.; Delogu, L.G.; Yilmazer, A. A closer look at the genotoxicity of graphene based materials. J. Phys. Mater. 2019, 3, 014007. [Google Scholar] [CrossRef]
- Hinzmann, M.; Jaworski, S.; Kutwin, M.; Jagiełło, J.; Koziński, R.; Wierzbicki, M.; Grodzik, M.; Lipińska, L.; Sawosz, E.; Chwalibog, A. Nanoparticles containing allotropes of carbon have genotoxic effects on glioblastoma multiforme cells. Int. J. Nanomed. 2014, 9, 2409–2417. [Google Scholar] [CrossRef] [Green Version]
- De Marzi, L.; Ottaviano, L.; Perrozzi, F.; Nardone, M.; Santucci, S.; De Lapuente, J.; Borras, M.; Treossi, E.; Palermo, V.; Poma, A. Flake size-dependent cyto and genotoxic evaluation of graphene oxide on in vitro A549, CaCO2 and vero cell lines. J. Biol. Regul. Homeost. Agents 2014, 28, 281–289. [Google Scholar] [PubMed]
- Akhavan, O.; Ghaderi, E.; Emamy, H. Nontoxic concentrations of PEGylated graphene nanoribbons for selective cancer cell imaging and photothermal therapy. J. Mater. Chem. 2012, 22, 20626–20633. [Google Scholar] [CrossRef]
- Wu, K.; Zhou, Q.; Ouyang, S. Direct and Indirect Genotoxicity of Graphene Family Nanomaterials on DNA—A Review. Nanomaterials 2021, 11, 2889. [Google Scholar] [CrossRef]
- Ronot, X.; Benel, L.; Adolphe, M.; Mounolou, J.C. Mitochondrial analysis in living cells: The use of rhodamine 123 and flow cytometry. Biol. Cell 1986, 57, 1–7. [Google Scholar] [CrossRef]
- Gatei, M.; Scott, S.P.; Filippovitch, I.; Soronika, N.; Lavin, M.F.; Weber, B.; Khanna, K.K. Role for ATM in DNA damage-induced phosphorylation of BRCA1. Cancer Res. 2000, 60, 3299–3304. [Google Scholar]
- Choi, M.; Kipps, T.; Kurzrock, R. ATM Mutations in Cancer: Therapeutic Implications. Mol. Cancer Ther. 2016, 15, 1781–1791. [Google Scholar] [CrossRef] [Green Version]
- Xiong, H.; Zhang, J. Expression and clinical significance of ATM and PUMA gene in patients with colorectal cancer. Oncol. Lett. 2017, 14, 7825–7828. [Google Scholar] [CrossRef]
- Ahlskog, J.K.; Larsen, B.D.; Achanta, K.; Sørensen, C.S. ATM/ATR-mediated phosphorylation of PALB2 promotes RAD51 function. EMBO Rep. 2016, 17, 671–681. [Google Scholar] [CrossRef] [Green Version]
- Son, M.Y.; Hasty, P. Homologous recombination defects and how they affect replication fork maintenance. AIMS Genet. 2019, 5, 192–211. [Google Scholar] [CrossRef]
- Zhao, W.; Steinfeld, J.B.; Liang, F.; Chen, X.; Maranon, D.G.; Jian Ma, C.; Kwon, Y.; Rao, T.; Wang, W.; Sheng, C.; et al. BRCA1-BARD1 promotes RAD51-mediated homologous DNA pairing. Nature 2017, 550, 360–365. [Google Scholar] [CrossRef]
- Zhang, J. The role of BRCA1 in homologous recombination repair in response to replication stress: Significance in tumorigenesis and cancer therapy. Cell Biosci. 2013, 3, 11. [Google Scholar] [CrossRef] [Green Version]
- Nordén, B.; Takahashi, M. Understanding Rad51 function is a prerequisite for progress in cancer research. QRB Discov. 2020, 1, e9. [Google Scholar] [CrossRef]
- Sage, J.M.; Gildemeister, O.S.; Knight, K.L. Discovery of a novel function for human Rad51: Maintenance of the mitochondrial genome. J. Biol. Chem. 2010, 285, 18984–18990. [Google Scholar] [CrossRef] [Green Version]
- Qiao, G.B.; Wu, Y.L.; Yang, X.N.; Zhong, W.Z.; Xie, D.; Guan, X.Y.; Fischer, D.; Kolberg, H.C.; Kruger, S.; Stuerzbecher, H.W. High-level expression of Rad51 is an independent prognostic marker of survival in non-small-cell lung cancer patients. Br. J. Cancer 2005, 93, 137–143. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wang, W.-Y.; Xiao, J.-H.; Xu, F.; Liao, D.-Y.; Xie, L.; Wang, J.; Luo, F. Overexpression of Rad51 Predicts Poor Prognosis in Colorectal Cancer: Our Experience with 54 Patients. PLoS ONE 2017, 12, e0167868. [Google Scholar] [CrossRef] [Green Version]
- Grundy, M.K.; Buckanovich, R.J.; Bernstein, K.A. Regulation and pharmacological targeting of RAD51 in cancer. Nar. Cancer 2020, 2, zcaa024. [Google Scholar] [CrossRef] [PubMed]
- Li, X.L.; Zhou, J.; Chen, Z.R.; Chng, W.J. P53 mutations in colorectal cancer—Molecular pathogenesis and pharmacological reactivation. World J. Gastroenterol. 2015, 21, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Iacopetta, B. TP53 mutation in colorectal cancer. Hum. Mutat. 2003, 21, 271–276. [Google Scholar] [CrossRef]
- Banin, S.; Moyal, L.; Shieh, S.; Taya, Y.; Anderson, C.W.; Chessa, L.; Smorodinsky, N.I.; Prives, C.; Reiss, Y.; Shiloh, Y.; et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 1998, 281, 1674–1677. [Google Scholar] [CrossRef] [PubMed]
- Nakano, K.; Vousden, K.H. PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell 2001, 7, 683–694. [Google Scholar] [CrossRef]
- Shan, Z.; Liu, Q.; Li, Y.; Wu, J.; Sun, D.; Gao, Z. PUMA decreases the growth of prostate cancer PC-3 cells independent of p53. Oncol. Lett. 2017, 13, 1885–1890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreis, N.-N.; Louwen, F.; Yuan, J. The Multifaceted p21 (Cip1/Waf1/CDKN1A) in Cell Differentiation, Migration and Cancer Therapy. Cancers 2019, 11, 1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javelaud, D.; Besançon, F. CDKN1A (cyclin-dependent kinase inhibitor 1A). Atlas Genet. Cytogenet. Oncol. Haematol. 2001, 5, 170–171. [Google Scholar] [CrossRef]
- Jin, Y.H.; Yoo, K.J.; Lee, Y.H.; Lee, S.K. Caspase 3-mediated cleavage of p21WAF1/CIP1 associated with the cyclin A-cyclin-dependent kinase 2 complex is a prerequisite for apoptosis in SK-HEP-1 cells. J. Biol. Chem. 2000, 275, 30256–30263. [Google Scholar] [CrossRef] [Green Version]
- Bedelbaeva, K.; Snyder, A.; Gourevitch, D.; Clark, L.; Zhang, X.-M.; Leferovich, J.; Cheverud, J.M.; Lieberman, P.; Heber-Katz, E. Lack of p21 expression links cell cycle control and appendage regeneration in mice. Proc. Natl. Acad. Sci. USA 2010, 107, 5845–5850. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Wettersten, H.I.; Park, S.-H.; Weiss, R.H. Small-molecule inhibitors of p21 as novel therapeutics for chemotherapy-resistant kidney cancer. Future Med. Chem. 2013, 5, 991–994. [Google Scholar] [CrossRef] [Green Version]
- Chen, A.; Huang, X.; Xue, Z.; Cao, D.; Huang, K.; Chen, J.; Pan, Y.; Gao, Y. The Role of p21 in Apoptosis, Proliferation, Cell Cycle Arrest, and Antioxidant Activity in UVB-Irradiated Human HaCaT Keratinocytes. Med. Sci. Monit. Basic Res. 2015, 21, 86–95. [Google Scholar] [CrossRef] [Green Version]
- Nikitaki, Z.; Hellweg, C.E.; Georgakilas, A.G.; Ravanat, J.L. Stress-induced DNA damage biomarkers: Applications and limitations. Front. Chem. 2015, 3, 35. [Google Scholar] [CrossRef] [Green Version]
- Georgakilas, A.G.; Martin, O.A.; Bonner, W.M. p21: A Two-Faced Genome Guardian. Trends Mol. Med. 2017, 23, 310–319. [Google Scholar] [CrossRef]
- Zhu, Q.; Xiao, S.; Hua, Z.; Yang, D.; Hu, M.; Zhu, Y.T.; Zhong, H. Near Infrared (NIR) Light Therapy of Eye Diseases: A Review. Int. J. Med. Sci. 2021, 18, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Hansen, L.T.; Helleday, T.; Sorensen, C.S.; Spang-Thomsen, M. A putative role for p21 (Waf1/Cip1) in regulation of RAD51 in response to etoposide and ionizing radiation treatment of small cell lung cancer cells. Cancer Res. 2004, 64, 612. [Google Scholar]
- Raderschall, E.; Bazarov, A.; Cao, J.; Lurz, R.; Smith, A.; Mann, W.; Ropers, H.H.; Sedivy, J.M.; Golub, E.I.; Fritz, E.; et al. Formation of higher-order nuclear Rad51 structures is functionally linked to p21 expression and protection from DNA damage-induced apoptosis. J. Cell Sci. 2002, 115, 153–164. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence 5′–3′ |
|---|---|
| Hs_GAPDH_For | ACCAGGTGGTCTCCTCTGACTTCAA |
| Hs_GAPDH_Rev | ACCCTGTTGCTGTAGCCAAATTCG |
| Mmus_GAPDH_For | CGACTTCAACAGCAACTCCCA |
| Mmus_GAPDH_Rev | AGCCGTATTCATTGTCATACCAGG |
| Hs_ATM_For | TGCTGTGAGAAAACCATGGAAGTGA |
| Hs_ATM_Rev | TCCGGCCTCTGCTGTAAATACAAAG |
| Mmus_ATM_For | AGGTGTCTTCAGAAGGTGCTGTG |
| Mmus_ATM_Rev | CCTCTACAATGGTCAGCAGGGT |
| Hs_TP53_For | AACAGCTTTGAGGTGCGTGTTTGTG |
| Hs_TP53_Rev | AGAGGAGCTGGTGTTGTTGGGCA |
| Mmus_TP53_For | GGAGAGTATTTCACCCTCAAGATCC |
| Mmus_TP53_Rev | AGACTCCTCTGTAGCATGGGC |
| HsBBC3_For (PUMA) | TACGAGCGGCGGAGACAAG |
| HsBBC3_Rev (PUMA) | GGTAAGGGCAGGAGTCCCAT |
| Mmus_BBC3_For (PUMA) | TACGAGCGGCGGAGACAA |
| Mmus_BBC3_Rev (PUMA) | GCTCCAGGATCCCTGGGTAA |
| Hs_CDKN1a_For | AGAGGAAGACCATGTGGACCTGTCA |
| Hs_CDKN1a_Rev | AGAAATCTGTCATGCTGGTCTGCC |
| Mmus_CDKN1a_For | ATCTCAGGGCCGAAAACGGA |
| Mmus_CDKN1a_Rev | TCTTGCAGAAGACCAATCTGCG |
| Hs_Rad51_For | TCAAGCATCAGCCATGATGGTAGAA |
| Hs_Rad51_Rev | AGAAACCTGGCCAAGTGCATCTG |
| Mmus_Rad51_For | CCCAAGTAGATGGAGCAGCCA |
| Mmus_Rad51_Rev | TTTCTCAGGTACAGCCTGGTGG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krasteva, N.; Staneva, D.; Vasileva, B.; Miloshev, G.; Georgieva, M. Bioactivity of PEGylated Graphene Oxide Nanoparticles Combined with Near-Infrared Laser Irradiation Studied in Colorectal Carcinoma Cells. Nanomaterials 2021, 11, 3061. https://0-doi-org.brum.beds.ac.uk/10.3390/nano11113061
Krasteva N, Staneva D, Vasileva B, Miloshev G, Georgieva M. Bioactivity of PEGylated Graphene Oxide Nanoparticles Combined with Near-Infrared Laser Irradiation Studied in Colorectal Carcinoma Cells. Nanomaterials. 2021; 11(11):3061. https://0-doi-org.brum.beds.ac.uk/10.3390/nano11113061
Chicago/Turabian StyleKrasteva, Natalia, Dessislava Staneva, Bela Vasileva, George Miloshev, and Milena Georgieva. 2021. "Bioactivity of PEGylated Graphene Oxide Nanoparticles Combined with Near-Infrared Laser Irradiation Studied in Colorectal Carcinoma Cells" Nanomaterials 11, no. 11: 3061. https://0-doi-org.brum.beds.ac.uk/10.3390/nano11113061