
Controlled Metal–Support Interactions in Au/CeO2–Mg(OH)2 Catalysts Activating the Direct Oxidative Esterification of Methacrolein with Methanol to Methyl Methacrylate

 , , , , and
, , , , and
Abstract
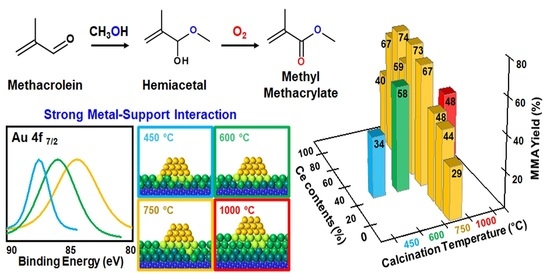
:
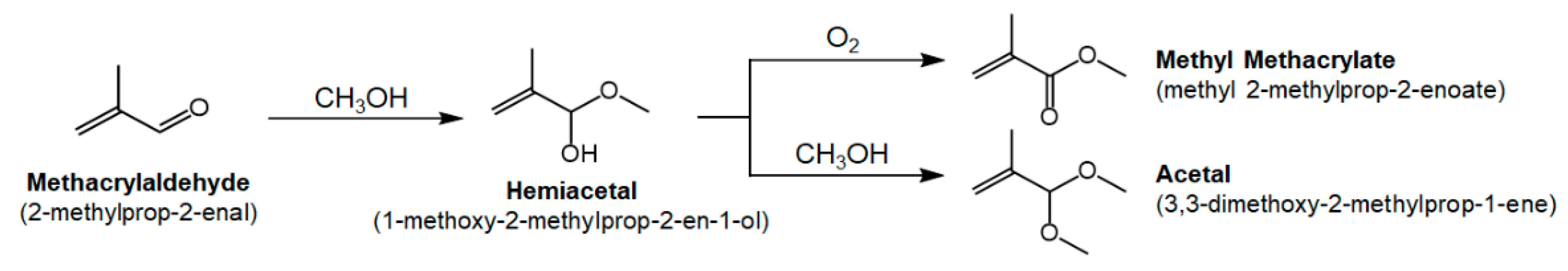
1. Introduction
2. Materials and Methods
2.1. Preparation of Materials
2.2. Characterization of Materials
2.3. Reaction Studies
3. Results and Discussion
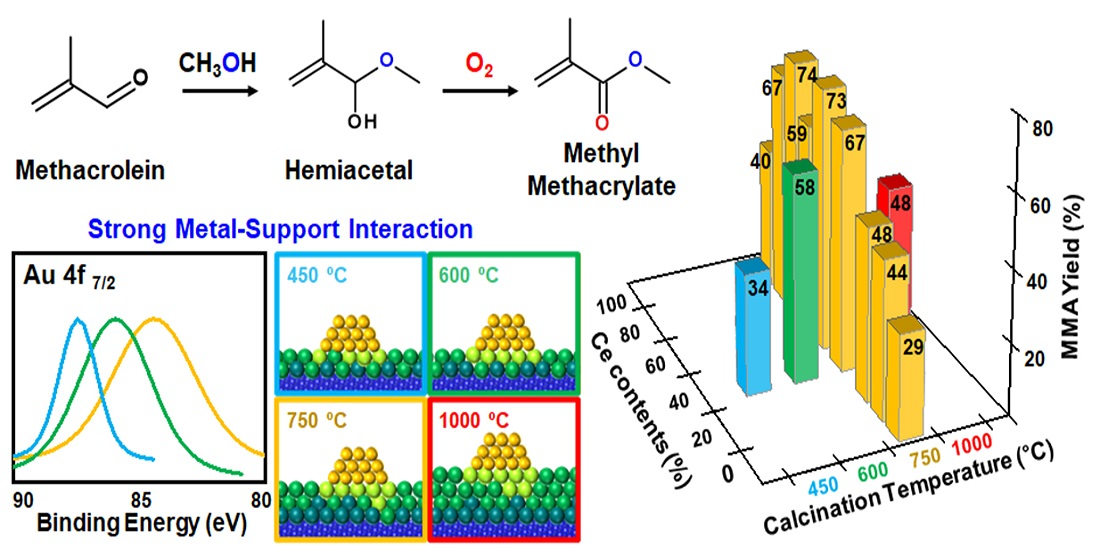
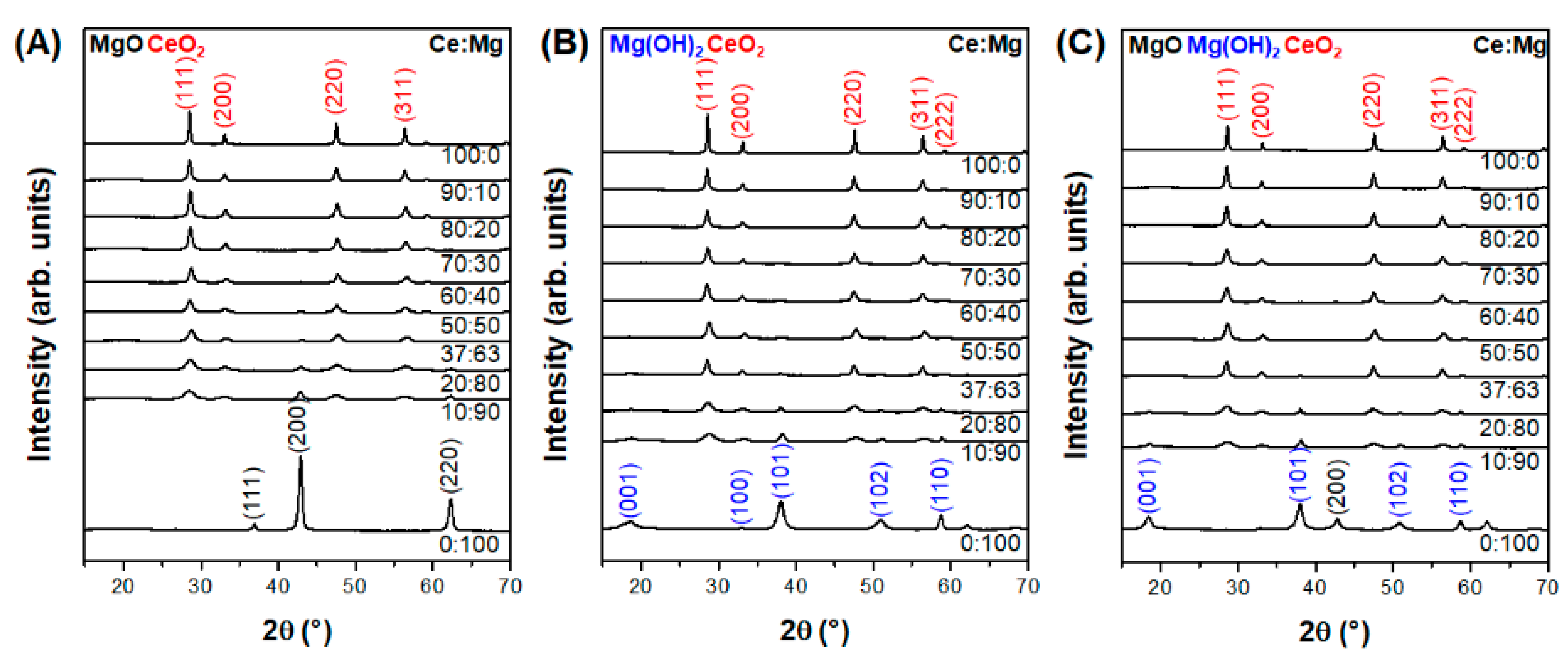
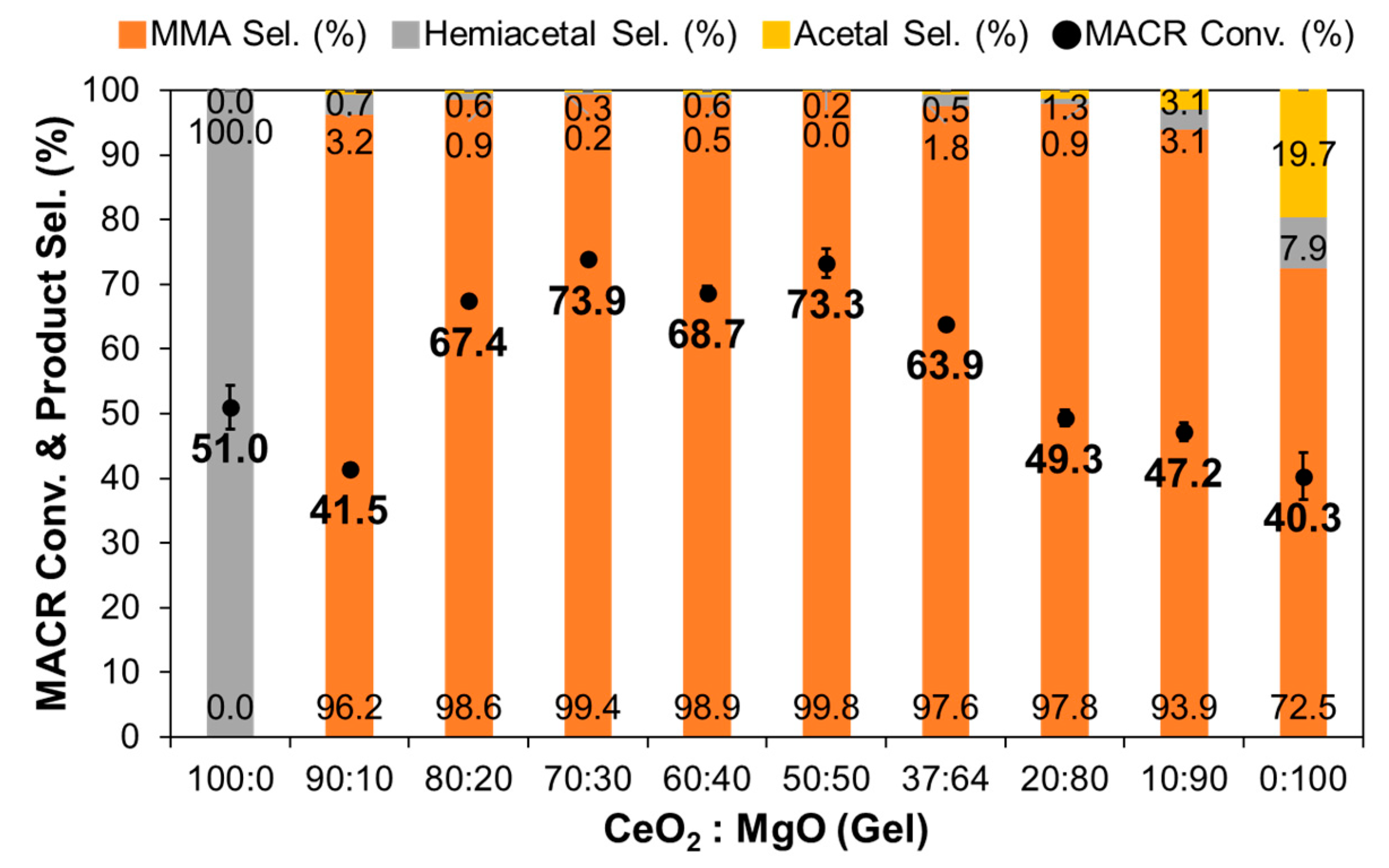
3.1. Effect of Composition in CeO2–Mg(OH)2 Supporting Au Nanoparticles
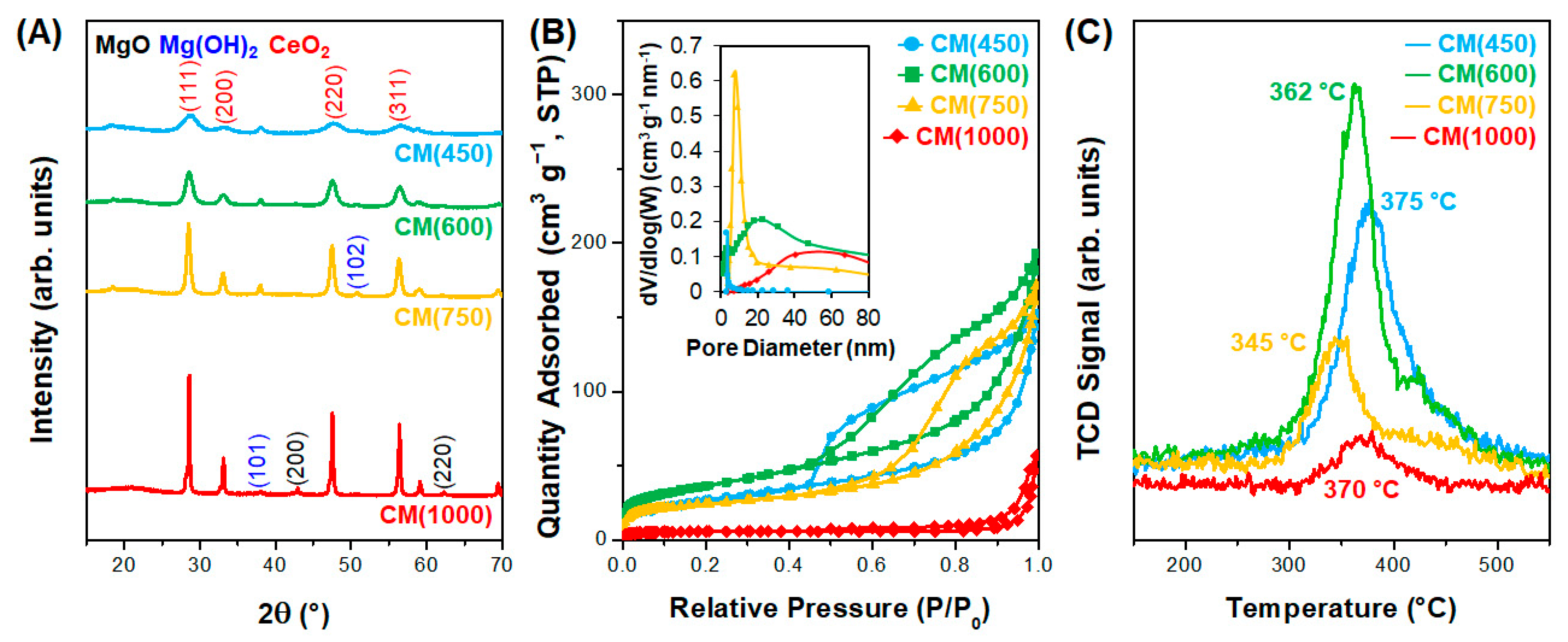
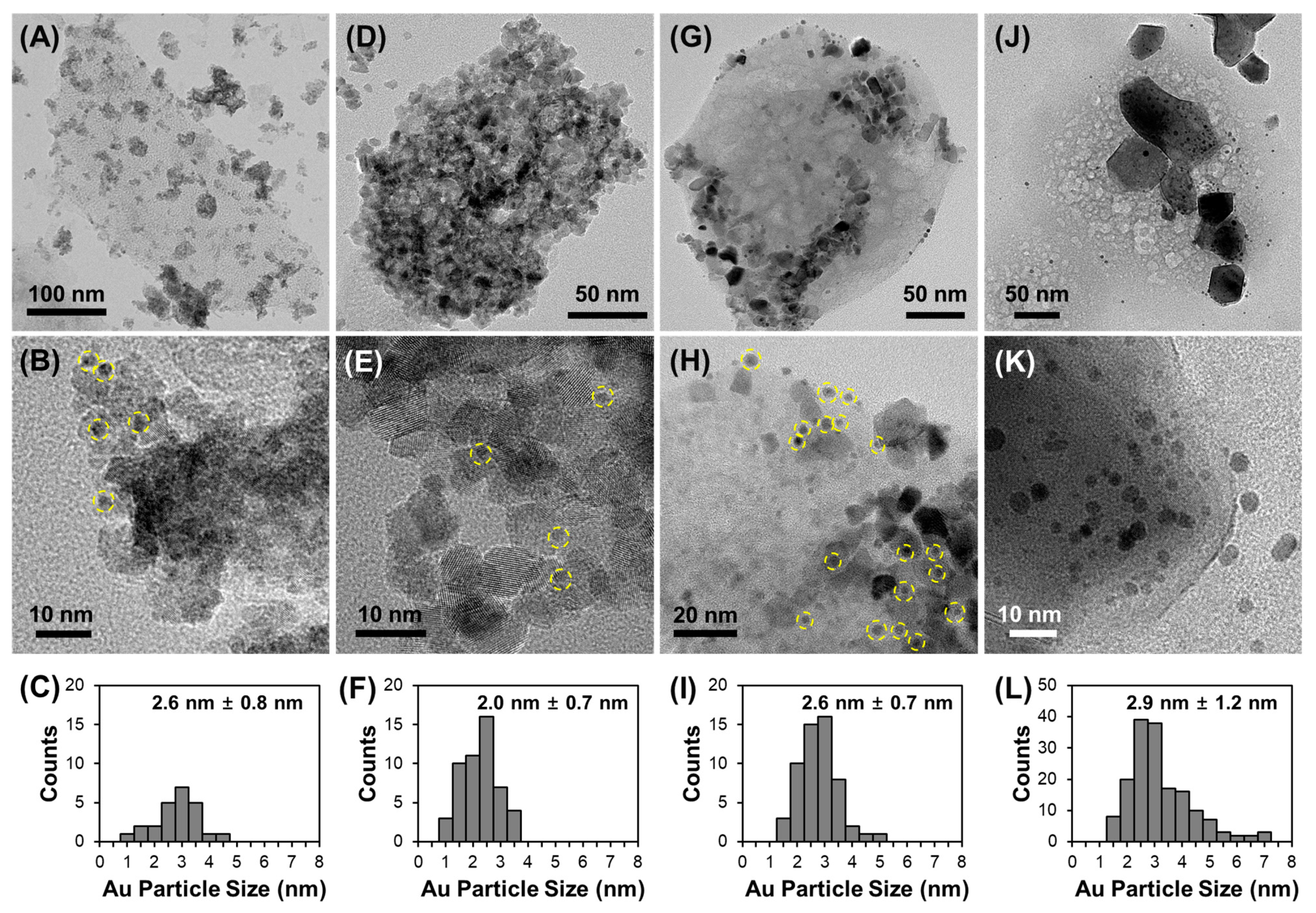
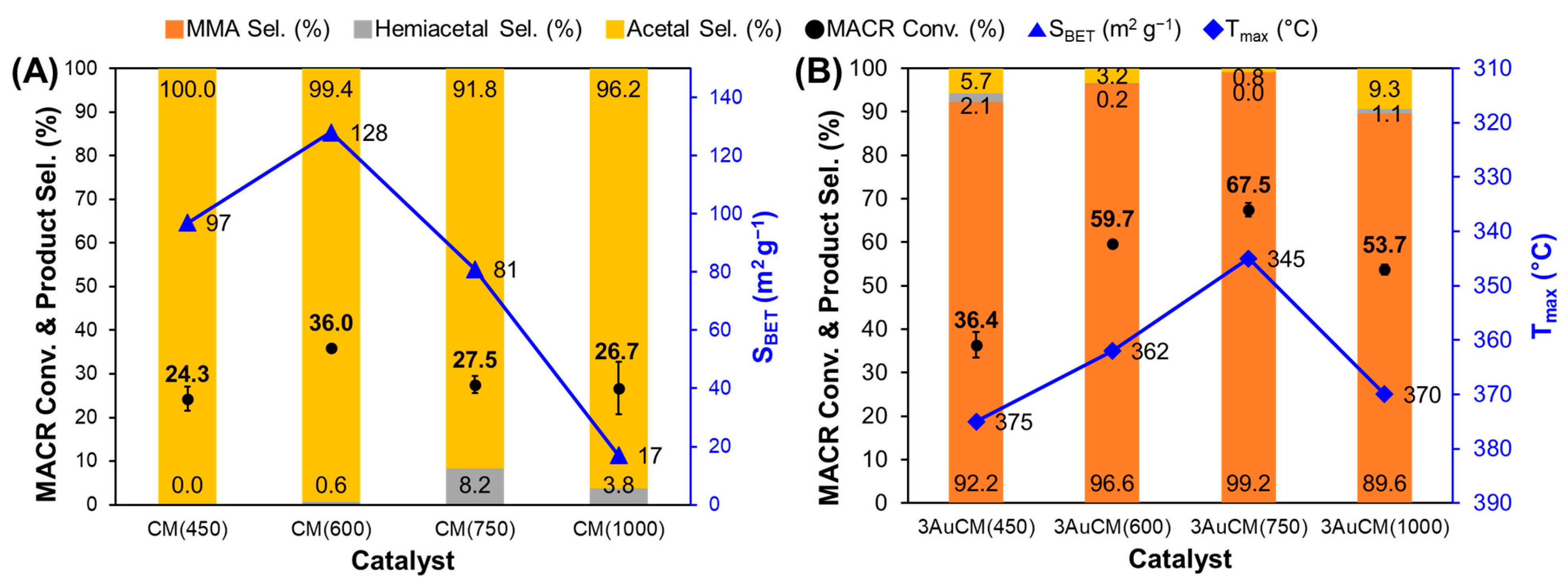
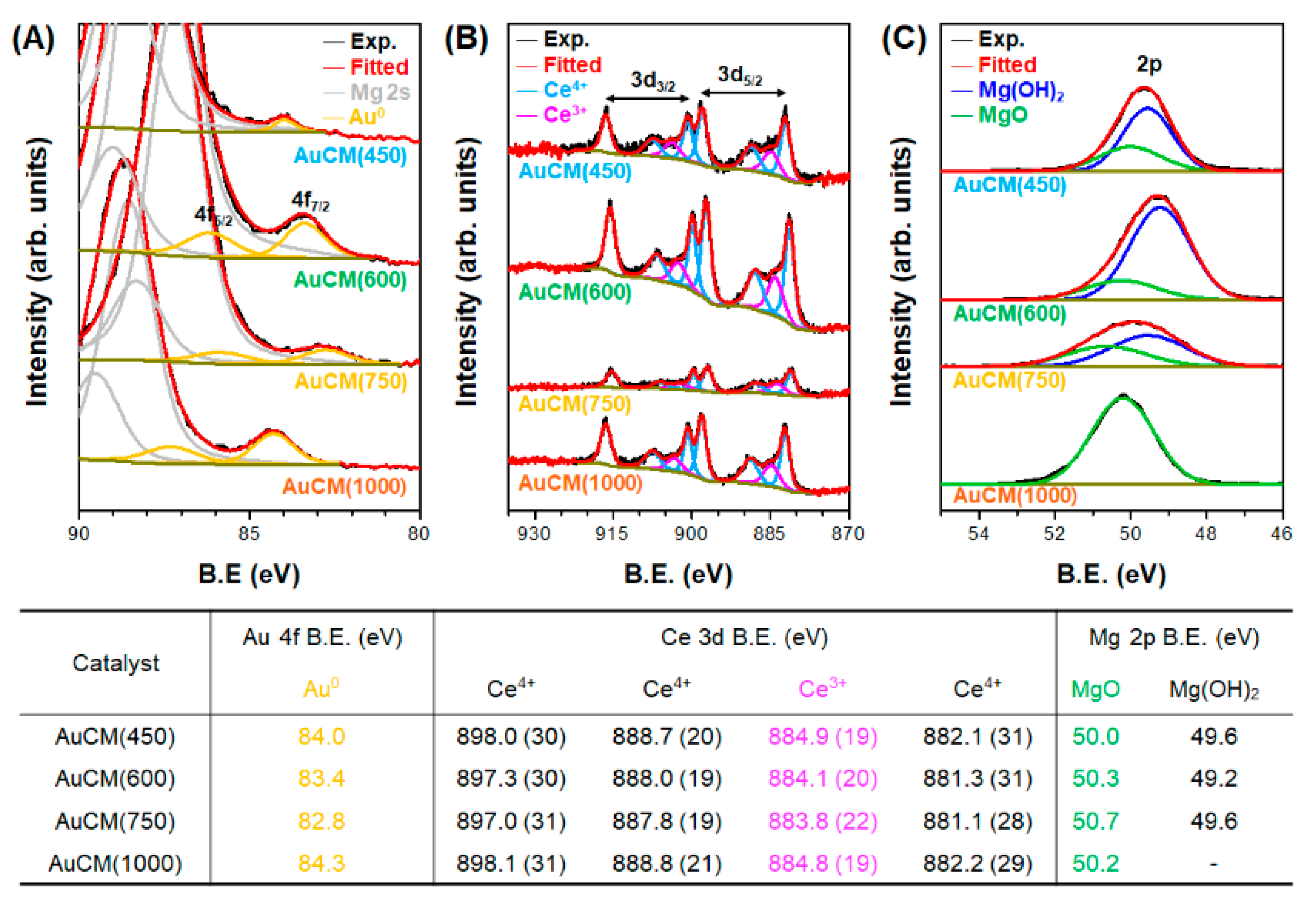
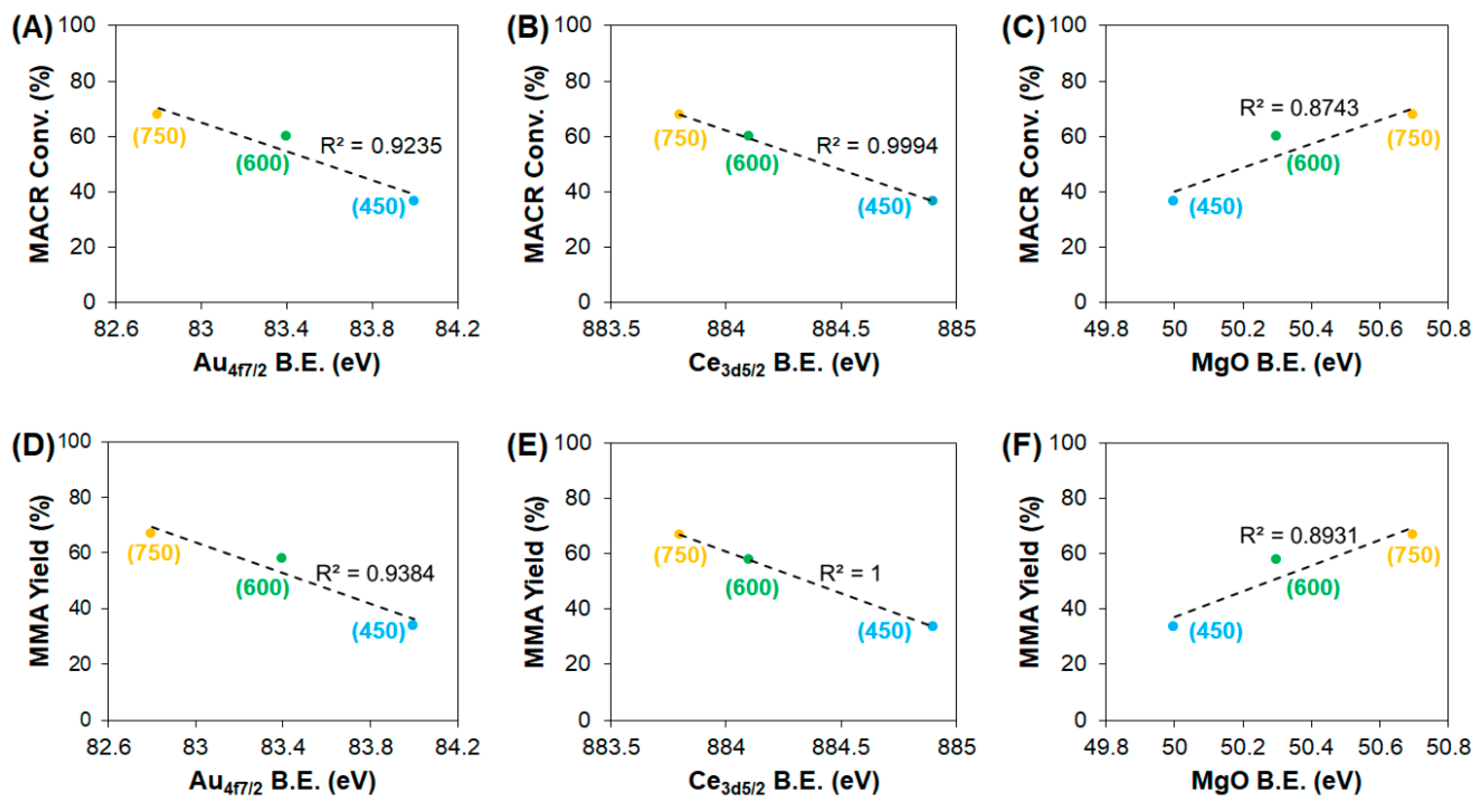
3.2. Effect of Support Crystallinities in CeO2–Mg(OH)2 Supporting Au Nanoparticles
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Zhang, H.; Liu, G.; Shi, L.; Ye, J. Single-Atom Catalysts: Emerging Multifunctional Materials in Heterogeneous Catalysis. Adv. Energy Mater. 2018, 8, 1701343. [Google Scholar] [CrossRef]
- Jagadeesan, D. Multifunctional nanocatalysts for tandem reactions: A leap toward sustainability. Appl. Catal. A 2016, 511, 59–77. [Google Scholar] [CrossRef]
- Felpin, F.X.; Fouquet, E. Heterogeneous Multifunctional Catalysts for Tandem Processes: An Approach toward Sustainability. Chem. Sus. Chem. 2008, 1, 718–724. [Google Scholar] [CrossRef]
- Tang, Z.; Fiorilli, S.L.; Heeres, H.J.; Pescarmona, P.P. Multifunctional Heterogeneous Catalysts for the Selective Conversion of Glycerol into Methyl Lactate. ACS Sustain. Chem. Eng. 2018, 6, 10923–10933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.; Zhang, S.; Zhang, J.; Yan, R. Modified extra-large mesoporous silica supported Au–Ni as a highly efficient catalyst for oxidative coupling of aldehydes with methanol. RSC Adv. 2014, 4, 58769–58772. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, X.; Ding, G.; Zheng, H.; Li, X.; Li, Y.; Zhu, Y. One-step Conversion of Fructose to Furfuryl Alcohol in a Continuous Fixed-bed Reactor: The Important Role of Supports. Chem. Cat. Chem. 2019, 11, 2118–2125. [Google Scholar] [CrossRef]
- Lim, S.; Kwon, S.; Kim, N.; Na, K. A Multifunctional Au/CeO2–Mg(OH)2 Catalyst for One-Pot Aerobic Oxidative Esterification of Aldehydes with Alcohols to Alkyl Esters. Nanomaterials 2021, 11, 1536. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.Y.; Wang, A.; Zhang, T.; Mou, C.Y. Catalysis by gold: New insights into the support effect. Nano Today 2013, 8, 403–416. [Google Scholar] [CrossRef]
- Gao, T.; Chen, J.; Fang, W.; Cao, Q.; Su, W.; Dumeignil, F. Ru/MnXCe1OY catalysts with enhanced oxygen mobility and strong metal-support interaction: Exceptional performances in 5-hydroxymethylfurfural base-free aerobic oxidation. J. Catal. 2018, 368, 53–68. [Google Scholar] [CrossRef]
- Chen, J.Z.; Gao, J.; Probus, P.R.; Liu, W.; Wu, X.; Wegener, E.C.; Kropf, A.J.; Zemlyanov, D.; Zhang, G.; Yang, X.; et al. The effect of strong metal–support interaction (SMSI) on Pt–Ti/SiO2 and Pt–Nb/SiO2 catalysts for propane dehydrogenation. Catal. Sci. Technol. 2020, 10, 5973–5982. [Google Scholar] [CrossRef]
- Chen, J.C.; Talpade, A.; Canning, G.A.; Probus, P.R.; Ribeiro, F.H.; Datye, A.K.; Miller, J.T. Strong metal-support interaction (SMSI) of Pt/CeO2 and its effect on propane dehydrogenation. Catal. Today 2021, 371, 4–10. [Google Scholar] [CrossRef]
- Kim, Y.; Song, Y.; Choi, Y.; Jeong, K.; Park, J.H.; Ko, K.C.; Na, K. Catalytic Consequences of Supported Pd Catalysts on Dehydrogenative H2 Evolution from 2-[(n-Methylcyclohexyl)methyl]piperidine as the Liquid Organic Hydrogen Carrier. ACS Sustain. Chem. Eng. 2021, 9, 809–821. [Google Scholar] [CrossRef]
- Shanmugapriya, S.; Zhu, P.; Yan, C.; Asiri, A.M.; Zhang, X.; Selvan, R.K. Multifunctional High-Performance Electrocatalytic Properties of Nb2O5 Incorporated Carbon Nanofibers as Pt Support Catalyst. Adv. Mater. Interfaces 2019, 6, 1900565. [Google Scholar] [CrossRef]
- Sankar, M.; He, Q.; Engel, R.V.; Sainna, M.A.; Logsdail, A.J.; Roldan, A.; Willock, D.J.; Agarwal, N.; Kiely, C.J.; Hutchings, G.J. Role of the Support in Gold-Containing Nanoparticles as Heterogeneous Catalysts. Chem. Rev. 2020, 120, 3890–3938. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Chen, L.; Bao, Y.; Zhang, Y.; Wang, J.; Fu, M.; Wu, J.; Ye, D. The Applications of Morphology Controlled ZnO in Catalysis. Catalysts 2016, 6, 188. [Google Scholar] [CrossRef] [Green Version]
- Xiong, J.; Mei, X.; Liu, J.; Wei, Y.; Zhao, Z.; Xie, Z.; Li, J. Efficiently multifunctional catalysts of 3D ordered meso-macroporous Ce0.3Zr0.7O2-supported PdAu@CeO2 core-shell nanoparticles for soot oxidation: Synergetic effect of Pd-Au-CeO2 ternary components. Appl. Catal. B 2019, 251, 247–260. [Google Scholar] [CrossRef]
- Yang, Y.; Ren, Z.; Zhou, S.; Wei, M. Perspectives on Multifunctional Catalysts Derived from Layered Double Hydroxides toward Upgrading Reactions of Biomass Resources. ACS Catal. 2021, 11, 6440–6454. [Google Scholar] [CrossRef]
- Han, K.; Xu, S.; Wang, Y.; Wang, S.; Zhao, L.; Kambonde, J.; Yu, H.; Shi, W.F. Confining Ni and ceria in silica shell as synergistic multifunctional catalyst for methane dry reforming reaction. J. Power Sources 2021, 506, 230232. [Google Scholar] [CrossRef]
- Wang, B.; Liu, F.; Guan, W.; Wang, A.; Zhang, T. Promoting the Effect of Au on the Selective Hydrogenolysis of Glycerol to 1,3-Propanediol over the Pt/WOx/Al2O3 Catalyst. ACS Sustain. Chem. Eng. 2021, 9, 5705–5715. [Google Scholar] [CrossRef]
- Tian, Y.; Li, Y.; Zheng, Y.; Wang, M.; Zuo, C.; Huang, H.; Yin, D.; Fu, Z.; Tan, J.; Zhou, Z. Nano-Au/MCeOx Catalysts for the Direct Oxidative Esterification of Methylacrolein to Methyl Esters. Ind. Eng. Chem. Res. 2019, 58, 19397–19405. [Google Scholar] [CrossRef]
- Diao, Y.; Yan, R.; Zhang, S.; Yang, P.; Li, Z.; Wang, L.; Dong, H. Effects of Pb and Mg doping in Al2O3-supported Pd catalyst on direct oxidative esterification of aldehydes with alcohols to esters. J. Mol. Catal. A Chem. 2009, 303, 35–42. [Google Scholar] [CrossRef]
- Wang, B.; Ran, W.; Sun, W.; Wang, K. Direct Oxidative Esterification of Aldehyde with Alcohol to Ester over Pd/Styrene-Divinyl Benzene Copolymer Catalyst. Ind. Eng. Chem. Res. 2012, 51, 3932–3938. [Google Scholar] [CrossRef]
- Wan, X.; Deng, W.; Zhang, Q.; Wang, Y. Magnesia-supported gold nanoparticles as efficient catalysts for oxidative esterification of aldehydes or alcohols with methanol to methyl esters. Catal. Today 2014, 233, 147–154. [Google Scholar] [CrossRef]
- Zuo, C.; Tian, Y.; Zheng, Y.; Wang, L.; Fu, Z.; Jiao, T.; Wang, M.; Huang, H.; Li, Y. One step oxidative esterification of methacrolein with methanol over Au-CeO2/γ-Al2O3 catalysts. Catal. Commun. 2019, 124, 51–55. [Google Scholar] [CrossRef]
- Li, Y.; Yun, T.; Yanxia, Z.; Tingting, G.; Zhongjun, F.; Tiantian, J.; Ming, W.; Haofei, H.; Cuncun, Z. Direct oxidation esterification of methacrolein with methanol: Oxygen vacancy promotion of Zr-doped Au/CeO2 nanorods. Can. J. Chem. Eng. 2020, 98, 767–774. [Google Scholar] [CrossRef]
- Qi, M.; Wu, X.; Wang, L.; Song, Y.; Diao, Y. The effect of the bimetallic Pd-Pb structures on direct oxidative esterification of methacrolein with methanol. Mol. Catal. 2021, 510, 111714. [Google Scholar] [CrossRef]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of Gases in Multimolecular Layers. J. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Barrett, E.P.; Joyner, L.G.; Halenda, P.P. The Determination of Pore Volume and Area Distributions in Porous Substances. I. Computations from Nitrogen Isotherms. J. Chem. Soc. 1951, 73, 373–380. [Google Scholar] [CrossRef]
- Gulati, U.; Rajesh, U.C.; Rawat, D.S.; Zaleski, J.M. Development of magnesium oxide–silver hybrid nanocatalysts for synergistic carbon dioxide activation to afford esters and heterocycles at ambient pressure. Green Chem. 2020, 22, 3170. [Google Scholar] [CrossRef]
- Bayeh, A.W.; Lin, G.Y.; Chang, Y.C.; Kabtamu, D.M.; Chen, G.C.; Chen, H.Y.; Wang, K.C.; Wang, Y.M.; Chiang, T.C.; Huang, H.C.; et al. Oxygen-Vacancy-Rich Cubic CeO2 Nanowires as Catalysts for Vanadium Redox Flow Batteries. ACS Sustain. Chem. Eng. 2020, 8, 16757–16765. [Google Scholar] [CrossRef]
- Winburn, R.S.; Lerach, S.L.; Jarabek, B.R.; Wisdom, M.A.; Grier, D.G.; McCarthy, G.J. Quantitative XRD Analysis of Coal Combustion By-Products by the Rietveld Method. Testing with Standard Mixtures. Adv. X-Ray Anal. 2000, 42, 387–396. [Google Scholar]
- Chrysochoou, M.; Dermatas, D. Application of the Rietveld method to assess chromium(VI) speciation in chromite ore processing residue. J. Hazard. Mater. 2007, 141, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Barrio, L.; Agnoli, S.; Senanayake, S.D.; Evans, J.; Kubacka, A.; Estrella, M.; Hanson, J.C.; Martinez-Arias, A.; Fernandez-Garcia, M.; et al. High Activity of Ce1xNixO2y for H2 Production through Ethanol Steam Reforming: Tuning Catalytic Performance through Metal–Oxide Interactions. Angew. Chem. Int. Ed. 2010, 49, 9680–9684. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Li, Z.; Ma, H.; Gao, Z. Surface composition and catalytic activity of La-Fe mixed oxides for methane oxidation. Appl. Surf. Sci. 2015, 351, 709–714. [Google Scholar] [CrossRef]
- Mironyuk, I.F.; Gun’ko, V.M.; Povazhnyak, M.O.; Zarko, V.I.; Chelyadin, V.M.; Leboda, R.; Skubiszewska-Zieba, J.; Janusz, W. Magnesia formed on calcination of Mg(OH)2 prepared from natural bischofite. Appl. Surf. Sci. 2006, 252, 4071–4082. [Google Scholar] [CrossRef]
- Li, Y.; Zheng, Y.; Wang, L.; Fu, Z. Oxidative Esterification of Methacrolein to Methyl Methacrylate over Supported Gold Catalysts Prepared by Colloid Deposition. Chem. Cat. Chem. 2017, 9, 1960–1968. [Google Scholar] [CrossRef]
- Ali, B.; Yusup, S.; Quitain, A.T.; Alnarabiji, M.S.; Kamil, R.N.M.; Kida, T. Synthesis of novel graphene oxide/bentonite bi-functional heterogeneous catalyst for one-pot esterification and transesterification reactions. Energy Convers. Manag. 2018, 171, 1801–1812. [Google Scholar] [CrossRef]
- Kuljiraseth, J.; Wangriya, A.; Malones, J.M.C.; Klysubun, W.; Jitkarnka, S. Synthesis and characterization of AMO LDH-derived mixed oxides with various Mg/Al ratios as acid–basic catalysts for esterification of benzoic acid with 2-ethylhexanol. Appl. Catal. B 2019, 243, 415–427. [Google Scholar] [CrossRef]
- Paul, B.; Khatun, R.; Sharma, S.K.; Adak, S.; Singh, G.; Das, D.; Siddiqui, N.; Bhandari, S.; Joshi, V.; Sasaki, T.; et al. Fabrication of Au Nanoparticles Supported on One-Dimensional La2O3 Nanorods for Selective Esterification of Methacrolein to Methyl Methacrylate with Molecular Oxygen. ACS Sustain. Chem. Eng. 2019, 7, 3982–3994. [Google Scholar] [CrossRef]
- Zhou, D.; Chen, X.; Wei, X.; Tang, L.; Liang, J.; Wang, S.; Wang, L. Insights into the synergetic mechanism of basic active site and oxygen vacancy on thermally activated MOFs for the colophony esterification: Experiments and DFT calculations. Ind. Crops Prod. 2021, 166, 113486. [Google Scholar] [CrossRef]
- Pal, R.; Wang, L.M.; Pei, Y.; Wang, L.S.; Zeng, X.C. Unraveling the Mechanisms of O2 Activation by Size-Selected Gold Clusters: Transition from Superoxo to Peroxo Chemisorption. J. Am. Chem. Soc. 2012, 134, 9438–9445. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Yamaguchi, T.; Matsushita, K.; Iitsuka, C.; Miura, J.; Akaogi, T.; Ishida, H. Aerobic Oxidative Esterification of Aldehydes with Alcohols by Gold−Nickel Oxide Nanoparticle Catalysts with a Core−Shell Structure. ACS Catal. 2013, 3, 1845–1849. [Google Scholar] [CrossRef]
- Li, Y.; Wang, L.; Yan, R.; Han, J.; Zhang, S. Gold nanoparticles supported on Ce–Zr oxides for the oxidative esterification of aldehydes to esters. Catal. Sci. Technol. 2015, 5, 3682. [Google Scholar] [CrossRef]
- Kim, M.; Su, Y.; Fukuoka, A.; Hensen, E.J.M.; Nakajima, K. Aerobic Oxidation of 5-(Hydroxymethyl)furfural Cyclic Acetal Enables Selective Furan-2,5-dicarboxylic Acid Formation with CeO2-Supported Gold Catalyst. Angew. Chem. Int. Ed. 2018, 57, 8235–8239. [Google Scholar] [CrossRef]
- Taketoshi, A.; Ishida, T.; Murayama, T.; Honma, T.; Haruta, M. Oxidative esterification of aliphatic aldehydes and alcohols with ethanol over gold nanoparticle catalysts in batch and continuous flow reactors. Appl. Catal. A 2019, 585, 117169. [Google Scholar] [CrossRef]
- Fujita, T.; Ishida, T.; Shibamoto, K.; Honma, T.; Ohashi, H.; Murayama, T.; Haruta, M. CO Oxidation over Au/ZnO: Unprecedented Change of the Reaction Mechanism at Low Temperature Caused by a Different O2 Activation Process. ACS Catal. 2019, 9, 8364–8372. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ce:Mg Ratio | MACR Conv. (%) | MMA Sel. (%) | Hemiacetal Sel. (%) | Acetal Sel. (%) | YMMA (%) [b] | |||
|---|---|---|---|---|---|---|---|---|
| Gel | Rietveld | |||||||
| Ce | Mg | CeO2 | MgO | |||||
| 100 | 0 | 100 | 0 | 51.0 | 0.0 | 100.0 | 0.0 | 0.0 |
| 90 | 10 | 98 | 2 | 41.5 | 96.2 | 3.2 | 0.7 | 39.9 |
| 80 | 20 | 97 | 3 | 67.4 | 98.6 | 0.9 | 0.6 | 66.5 |
| 70 | 30 | 92 | 8 | 73.9 | 99.4 | 0.2 | 0.3 | 73.5 |
| 60 | 40 | 85 | 16 | 59.9 | 98.8 | 0.0 | 1.2 | 59.2 |
| 50 | 50 | 79 | 21 | 73.3 | 99.8 | 0.0 | 0.2 | 73.2 |
| 37 | 63 | 73 | 27 | 67.5 | 99.2 | 0.0 | 0.8 | 67.0 |
| 20 | 80 | 48 | 52 | 49.3 | 97.8 | 0.9 | 1.3 | 48.2 |
| 10 | 90 | 26 | 74 | 47.2 | 93.9 | 3.1 | 3.1 | 43.3 |
| 0 | 100 | 0 | 100 | 40.3 | 72.5 | 7.9 | 19.7 | 29.2 |
| Catalyst | Au Content (wt.%) [b] | Au Size (nm) [c] | Au Dispersion (%) [d] | SBET (m2 g−1) [e] | Dp (nm) [f] | Vtot (cm3 g−1) [g] | BT (mmol g−1) [h] | Tmax (°C) [i] | MACR Conv. (%) | MMA Sel. (%) | Hemiacetal Sel. (%) | Acetal Sel. (%) | YMMA (%) [j] | TON [k] | STY [l] |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AuCM (450) | 2.7 | 2.6 | 45.0 | 97 | 3.3 | 0.23 | 0.32 | 375 | 36.4 | 92.2 | 2.1 | 5.7 | 33.6 | 413 | 381 |
| AuCM (600) | 2.7 | 2.0 | 58.4 | 128 | 5.4 | 0.28 | 0.45 | 362 | 59.7 | 96.6 | 0.2 | 3.2 | 57.7 | 264 | 255 |
| AuCM (750) | 2.5 | 2.6 | 44.4 | 81 | 7.4 | 0.25 | 0.24 | 345 | 67.5 | 99.2 | 0.0 | 0.8 | 67.0 | 521 | 517 |
| AuCM (1000) | 1.6 | 2.9 | 40.3 | 17 | 40.3 | 0.07 | 0.07 | 370 | 53.7 | 89.6 | 1.1 | 9.2 | 48.1 | 714 | 640 |
| Catalyst | Quantity (g) | Methanol/MACR | Temp. (°C) | Pressure (MPa) | Time (h) | MACR Conv. (%) | MMA Sel. (%) | TON | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| AuCM(750) | 1.5 | 5/1 | 80 | 0.9 | 1 | 67.5 | 99.2 | 521 | This work |
| Au–CeO2/ɤ–Al2O3 | 0.5 | 20/1 | 70 | 0.2 | 2 | 97 | 90 | - | [24] |
| Au/CexZryOz | 0.5 | 20/1 | 80 | 0.3 | 2 | 99 | 74 | 577 | [25] |
| Pd3Pb | 2.5 | 8/1 | 80 | 0.3 | 2 | 89 | 79 | - | [26] |
| Au/ZnO | 0.5 | 30/1 | 70 | 0.2 | 2 | 99.9 | 85.9 | - | [36] |
| Au/La2O3 | 0.1 | 8/1 | 70 | 0.2 | 2 | 89 | 98 | 1136 | [39] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, N.; Lim, S.; Kwon, S.; Choi, Y.; Lee, J.-W.; Na, K. Controlled Metal–Support Interactions in Au/CeO2–Mg(OH)2 Catalysts Activating the Direct Oxidative Esterification of Methacrolein with Methanol to Methyl Methacrylate. Nanomaterials 2021, 11, 3146. https://0-doi-org.brum.beds.ac.uk/10.3390/nano11113146
Kim N, Lim S, Kwon S, Choi Y, Lee J-W, Na K. Controlled Metal–Support Interactions in Au/CeO2–Mg(OH)2 Catalysts Activating the Direct Oxidative Esterification of Methacrolein with Methanol to Methyl Methacrylate. Nanomaterials. 2021; 11(11):3146. https://0-doi-org.brum.beds.ac.uk/10.3390/nano11113146
Chicago/Turabian StyleKim, Nagyeong, Seulgi Lim, Seungdon Kwon, Yuyeol Choi, Ji-Woong Lee, and Kyungsu Na. 2021. "Controlled Metal–Support Interactions in Au/CeO2–Mg(OH)2 Catalysts Activating the Direct Oxidative Esterification of Methacrolein with Methanol to Methyl Methacrylate" Nanomaterials 11, no. 11: 3146. https://0-doi-org.brum.beds.ac.uk/10.3390/nano11113146