1. Introduction
Tissue engineering is gaining increasing attention in oral bone surgery in relation to the need for new bone substitute materials.
Considering the high variability and complex architecture of bone defects in the oral and maxillofacial district, oral bone surgery requires tailorable, customizable, adaptable, and shapeable biomaterials for bone regeneration. These biomaterials should positively interact with mineralizing/remodeling cells, must be biocompatible and bioactive, easily insertable, and possibly requiring minimally invasive surgical procedures [
1,
2].
Different bone defects frequently occur in the oral and maxillofacial area. These defects may be caused by periradicular/periapical lesions and periapical abscesses, odontogenic cysts, peri-implantitis, periodontitis, bone trauma, bone atrophies and tumors, and are responsible for tooth loss, occlusal dysfunctions, and functional alterations.
A novel approach in tissue engineering is represented by the design of innovative green and biologically derived materials to combine good biological properties with eco-friendly productive processes.
In this context, the development of eco-sustainable materials produced by using biological/organic wastes (i.e., obtained from controlled fermentation of agro-wastes) or natural components represents an environmental-responsive approach of the new industrial science [
3,
4].
The use of organic wastes represents an ethical and valuable approach to reduce environmental pollution. Moreover, green chemistry does not entail the generation of toxic or harmful wastes during the production processes [
3,
4].
Polymers are widely used to design materials for tissue engineering and possess attractive properties for bone regeneration. Natural polymers, including peptides (gelatin and collagen), natural poly-esters (polyhydroxyalkanoates, poly(β-hydroxybutyrate and poly(β-hydroxybutyrate-co-β-hydroxyvalerate)) and polysaccharides (alginates i.e., mannuronate/guluronate-based copolymers, cellulose, chitin, hyaluronic acid, pectin and starch), have been used to prepare sponges [
5], membranes [
6], and recently porous scaffolds [
7,
8] as well for tissue engineering. Various polysaccharides form hydrogels through physical or chemical cross-linking, and many of them possess environmentally responsive properties. Hydrogel refers to cross-linked water-swollen polymer networks forming a porous structure with high water entrapment property and leaching capability.
Polysaccharide-based hydrogels have proven ideal for biomedical and pharmaceutical applications due to their intrinsic biocompatibility, degradability, environment sensitivity to pH, temperature, and possibility to incorporate specific biomolecules [
9,
10].
Porous hydrogels allow for the easier transport of mineralizing cells and nutrients into the biomaterials, ensuring a suitable environment for new bone tissue regeneration [
11,
12]. Natural polysaccharides are interesting components adopted as part of innovative approaches and strategies to fabricate biodegradable, biocompatible, and green matrices for regenerative medicine as they have demonstrated biocompatibility, no hydrophobic behavior, and biodegradation in non-toxic components.
Seaweed-derived (algae-based) natural polysaccharides, consisting of linear copolymers of D-mannuronic acid and L-guluronic acid units, showed the ability to retain water and gelling, forming biocompatible and stable hydrogels with adjustable porosity and low immunogenicity and biodegradability in physiological conditions. Gelation occurs in the presence of cross-linking divalent ions (as calcium and magnesium) and the mechanical properties of hydrogels increase with increasing concentrations of divalent ions [
13]. In addition to the chemical cross-linking gelation, hydrogel preparation can be achieved using other ecologically sustainable non-toxic techniques, such as freeze casting [
5,
6] or phase separation and ultrasonic cavitation [
14].
Mannuronic/guluronic acids-based polymers have been used to produce through the freeze casting method either 3D highly porous sponges/scaffolds with vasculogenic properties on human embryonic stem cells [
5] or membranes having mechanical and physical properties adequate for skin tissue engineering [
6].
Hydrogels, with their lack of native ligand sites suitable for mammalian cells attachment [
13], showed in vivo uncontrolled degradation rates and low mechanical stiffness [
15]. Hydrogels can be doped with magnesium phosphate or reinforced with polymer fibers to improve their mechanical properties and to provide a favorable substrate for cells [
16] or can be combined with polypeptides, such as collagen and/or gelatin, to mimic the human extracellular matrix and provide sites for cell attachment and colonization [
10].
Gelatin is a heterogeneous mixture of water-soluble peptides produced by the partial hydrolysis of collagen extracted from the skin, bones, and connective tissues of animals. Gelatin is biocompatible and biodegradable, exhibits low antigenicity, does not produce harmful by-products upon enzymatic degradation, and is available at low cost [
10]. Gelatin has been recently used to produce 3D-printed scaffolds with high porosity, promising mechanical properties and no cytotoxic effects on human dermal fibroblasts [
8].
The association of mannuronic/guluronic acids-based polymers and gelatin has been used in some biomedical applications, such as drug delivery systems [
17], skin tissue engineering [
18], and cartilage tissue engineering [
4,
19]. However, they do not appear suitable for applications in bone tissue engineering due to low mechanical properties, non-controllable resorbability, and difficulties inherent to reaching high porosity values [
20].
Hydraulic calcium silicates (CaSi) demonstrated to be effective fillers in bone regeneration procedures in dentistry [
21] in relation to their biointeractivity [
22,
23,
24], apatite forming ability [
22,
23,
25,
26,
27], biocompatibility, and capability to induce the differentiation of several mineralizing cells, such as human bone marrow stromal cells [
28,
29], orofacial bone mesenchymal stem cells [
30], cementoblasts [
31], and oral derived periapical cyst mesenchymal stem cells [
32].
The combination of calcium phosphates such as dicalcium phosphate dihydrate (DCPD) to CaSi materials was demonstrated to enhance their biological properties [
30,
33,
34], and apatite-forming ability [
33,
35,
36].
The novelty of the present study was to design and produce seaweed-based green mineral-filled hydrogels, constituted of copolymers of sodium D-mannuronate and L-guluronate units, gelatin, and nanoparticles of CaSi and DCPD, conceived for the regeneration of non-load bearing oral bone defects and to evaluate their chemical-physical and biological (osteogenic and angiogenic commitment of stem cells) properties. Similar formulations have never been reported.
2. Materials and Methods
2.1. Mineral-Filled Hydrogels Preparation
The hydrogels were designed and prepared in the Laboratory of Green Biomaterials at the Dental School of the University of Bologna.
Poly(sodium D-mannuronate-co-L-guluronate) powder (Sigma Aldrich, St. Louis, MO, USA), gelatin from bovine skin powder (Sigma Aldrich, St. Louis, MO, USA), and calcium chloride (Sigma Aldrich, St. Louis, MO, USA) were used.
Dicalcium phosphate dihydrate (DCPD; CaHPO
4·2H
2O, Sigma-Aldrich, Steinheim, Germany), wollastonite, and CaSi powder (Aalborg, Denmark)—composed of dicalcium silicate, tricalcium silicate, tricalcium aluminate, and calcium sulfate and prepared by melt-quenching technique followed by grinding using agate ball milling procedures (particle size less than 5 microns) [
22,
35,
37]—were used as bioactive biointeractive mineral fillers.
Poly(sodium D-mannuronate-co-L-guluronate) and gelatin components were present in a 1:1 weight ratio and formed the organic hydrogel phase Polysaccharide/Polypeptide (PP).
PP was filled with various amounts of mineral powders and different PP-x:y formulations were prepared, where x and y represent the weight % of CaSi and DCPD, respectively. Three formulations were studied: PP-16:16, PP-33:22, PP-31:31.
Bioactive mineral powders were added in 50 mL of demineralized water and kept at 60 °C and 700 rpm for 30 min under vigorous stirring, to achieve a homogeneous dispersion. Then, gelatin (polypeptide) powder was slowly added to the dispersion and maintained at 60 °C and 1200 rpm for 90 min. Finally, the copolymer of D-mannuronate/L-guluronate sodium salts (polysaccharide) was added to the dispersion and vigorously stirred at 60 °C and 1500 rpm for 5 min (
Figure 1a). The slurry mixture was then poured on paraffin-covered Petri dishes (diameter 60 mm, height 10 mm), or a polydimethylsiloxane cylindrical mold (diameter 10 mm, height 60 mm) and cooled at 0–4 °C for at least 2 days (
Figure 1b,c). The hydrogel solidification through chemical cross-linking was obtained by the calcium ions released from CaSi and DCPD mineral fillers.
Hydrogel without mineral fillers (used as control, PP-CTRL) was prepared as follows: 1g CaCl2 was dissolved in 50 mL of demineralized water and kept at 60 °C and 700 rpm for 20 min under vigorous stirring to achieve a homogeneous dispersion. Gelatin powder was then added and stirred at 60 °C and 1000 rpm for 60 min. Finally, the copolymer of D-mannuronate/L-guluronate sodium salts powder was added to the dispersion (gelatin to copolymer weight ratio 1:1) and vigorously stirred at 1500 rpm. The slurry mixture was poured inside paraffin-covered Petri dishes (diameter 60 mm, height 10 mm) or into polydimethylsiloxane-based cylindrical molds (diameter 10 mm, height 60 mm) and cooled at 0–4 °C for at least 2 days.
2.2. Calcium Release and Alkalizing Activity (pH of Soaking Water)
Cylindrical molds (10 ± 0.1 mm diameter and 10 ± 0.1 mm thick; n = 10 for each composition) were immersed in 10 mL of deionized water inside polypropylene-sealed containers and stored at 37 °C. The soaking water was collected and replaced at six-time endpoints (3 h and 1, 3, 7, 14, and 28 days). The collected water was analyzed for pH and calcium ions using a potentiometric method under magnetic stirring at room temperature (25 °C). The pH was measured using a selective temperature-compensated electrode (Sen Tix Sur WTW, Weilheim, Germany) connected to a multi-parameter laboratory meter (inoLab 750 WTW, Weilheim, Germany) previously calibrated with standard solutions. The amount of calcium ions was measured using a calcium probe (calcium ion electrode, Eutech instruments Pte Ltd., Singapore City, Singapore) after the addition of 2% of ionic strength adjuster (ISA) 4 mol/L KCl (WTW, Weilheim, Germany).
2.3. Solubility, Porosity, Water Sorption
Cubic samples (10 ± 0.1 mm long × 10 ± 0.1 mm wide × 10 ± 0.1 mm thick; n = 8 for each composition) were weighed to determine the initial mass (I) and the mass immersed in 20 mL of distilled water at 37 °C for 24 h (S). Then, the specimens were removed from the water, the excess water from the surface of each sample was removed using a moistened filter paper (20 mL of distilled water dropped on a 9 cm wide 12.5-cm-long glass plate covered by a filter paper), and the saturated mass (M) was recorded.
Finally, the samples were dried at 37 °C until the weight was stable, and the final dry mass (D) was recorded.
Open pores volume (V
OP = M − D, in cm
3), impervious portion volume (V
IP = D − S, in cm
3), and apparent porosity (P = [(M − D)/V] × 100) were calculated following the Archimedes principle (and ASTM C266-88) [
24,
33,
38].
Water sorption (WS = [(M − D)/D] × 100) and solubility (S = [(I − D)/D] × 100) were calculated as a percentage of the original weight. Each weight measurement was repeated three times using an analytical balance (Bel Engineering series M, Monza, Italy) and determined to the nearest 0.001 g. Mean values of the measures were reported.
2.4. Setting Time
Cylindrical samples (10 ± 0.1 mm diameter and 10 ± 0.1mm thick;
n = 3 for each composition) were placed at 37 °C and 95% relative humidity. The initial setting time was measured by evaluating the absence of indentation caused by a Gilmore needle (ASTM C 226-07 Standard test method for time of setting of cement paste by Gillmore needles) [
22,
24,
33,
38] at room temperature (25 °C). The Gilmore needle to evaluate the initial setting time weighed 113.4 g and was 2.12 mm in diameter.
2.5. Radiopacity
Cylindrical samples (10.0 mm diameter, 1.0 mm thick;
n = 3 per group) were radiographed using a radiographic unit with a reference aluminum step wedge (60 mm long, 10 mm wide thickness varying from 2 to 6 mm in 1 mm-increments) following ISO 9917-2007 [
24,
38,
39]. The target-film distance was approximately 30 cm with the sample at 3 cm from the surface of the radiographic tube, 0.13 s exposure at 70 KVp, and 8 mA. The film was processed and scanned. The radiographic density (color intensity) data were converted into aluminum step-wedge equivalent thickness (mmAl) using the software Image J (NIH software, Bethesda, MD, USA) [
24,
38,
39].
2.6. Surface Micromorphology and Apatite Nucleation in Hank Balanced Salt Solution
The apatite forming ability was evaluated following ISO 23317 (implants for surgery—in vitro evaluation for apatite-forming ability of implant materials). Hank Balanced Salt Solution (HBSS, Cambrex Bioscience, Verviers, Belgium) having a composition of inorganic ions similar to human blood plasma was used as simulated body fluid [
23,
24,
33,
40,
41]. HBSS composition was: K
+ 5.8 mM, Ca
2+ 1.27 mM, HCO
3− 4.17 mM, SO
42− 0.81 mM, Mg
2+ 0.81 mM, Na
+ 141.6 mM, H
2PO
4− 0.44 mM, HPO
42− 0.336 mM, and Cl
− 144.7 mM.
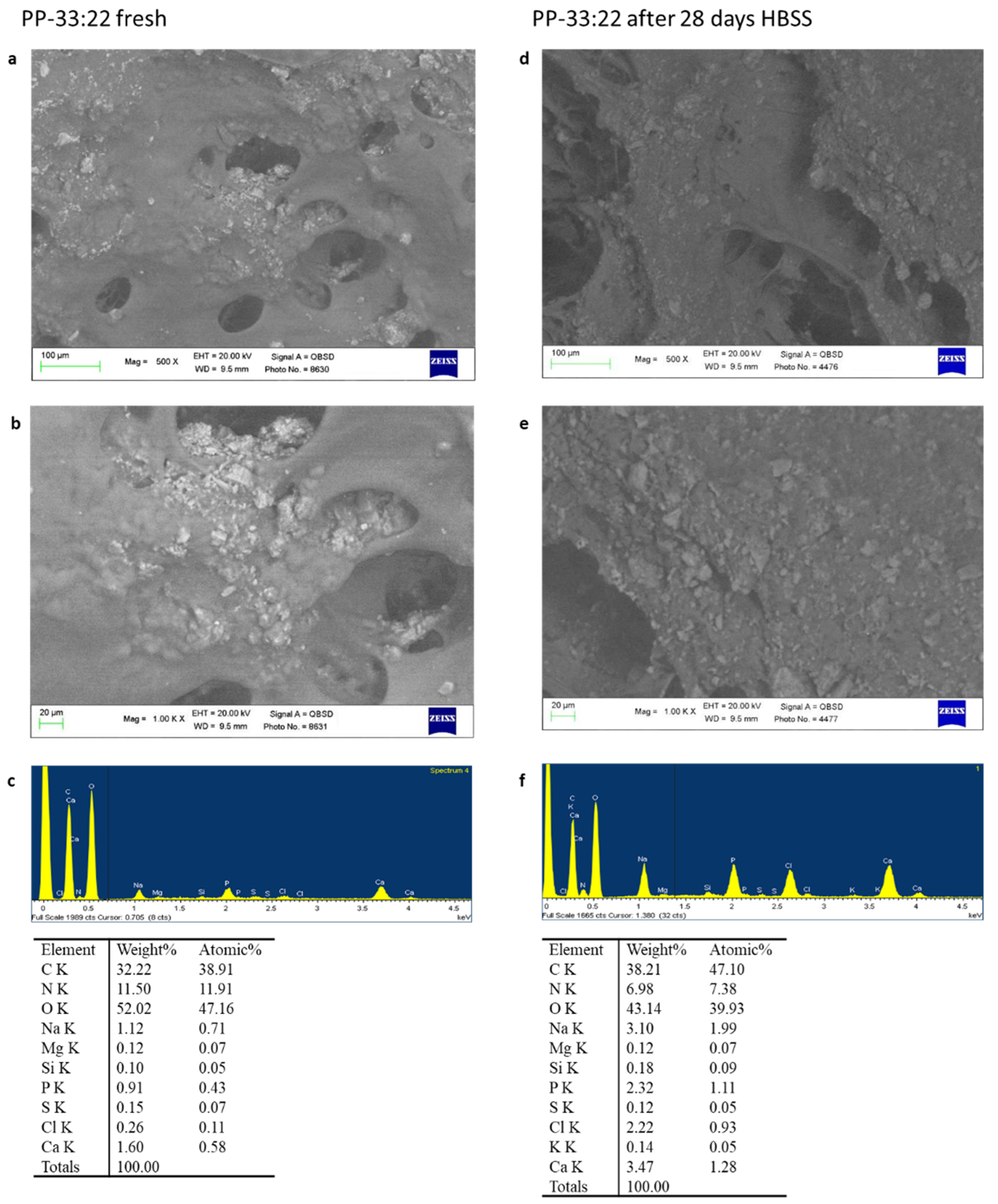
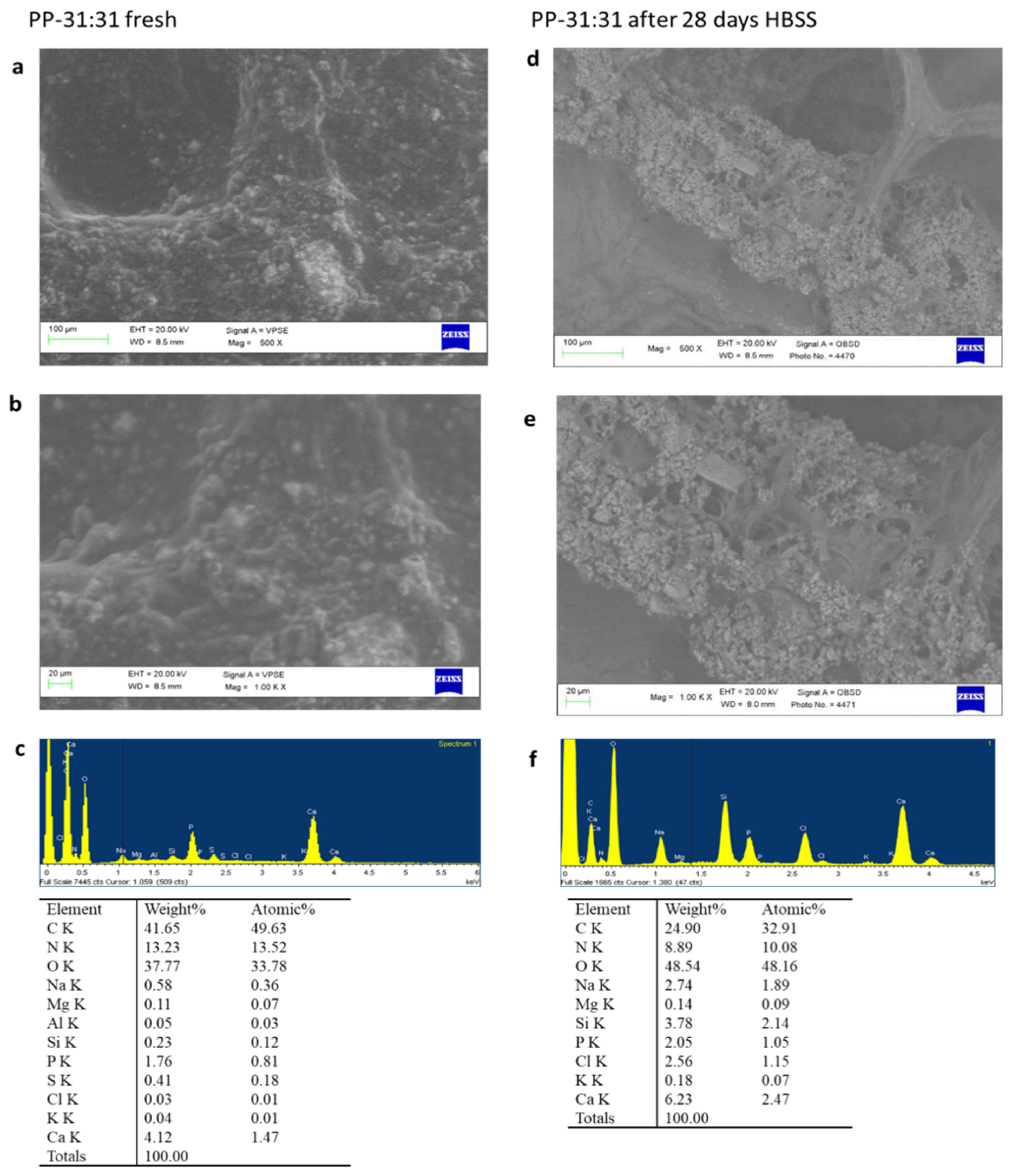
Set fresh samples and samples aged 28 days in HBSS were analysed by ESEM-EDX, FT-Raman, and micro-Raman.
An environmental scanning electron microscope (ESEM, Zeiss EVO 50; Carl Zeiss, Oberkochen, Germany) connected to a secondary electron detector for energy dispersive X-ray analysis (EDX; Oxford INCA 350 EDS, Abingdon, UK) using computer-controlled software (Inca Energy Version 18, Abingdon, UK) was used. Specimens were placed directly on the ESEM stub and examined uncoated in wet conditions at low vacuum (100 Pascal). EDX microchemical analysis was carried out in random areas of 50 × 50 µm to evaluate the relative element content. Elemental microanalysis (weight % and atomic %) with the ZAF correction method was performed in full frame to analyze entire areas [
23,
24,
33,
40,
41].
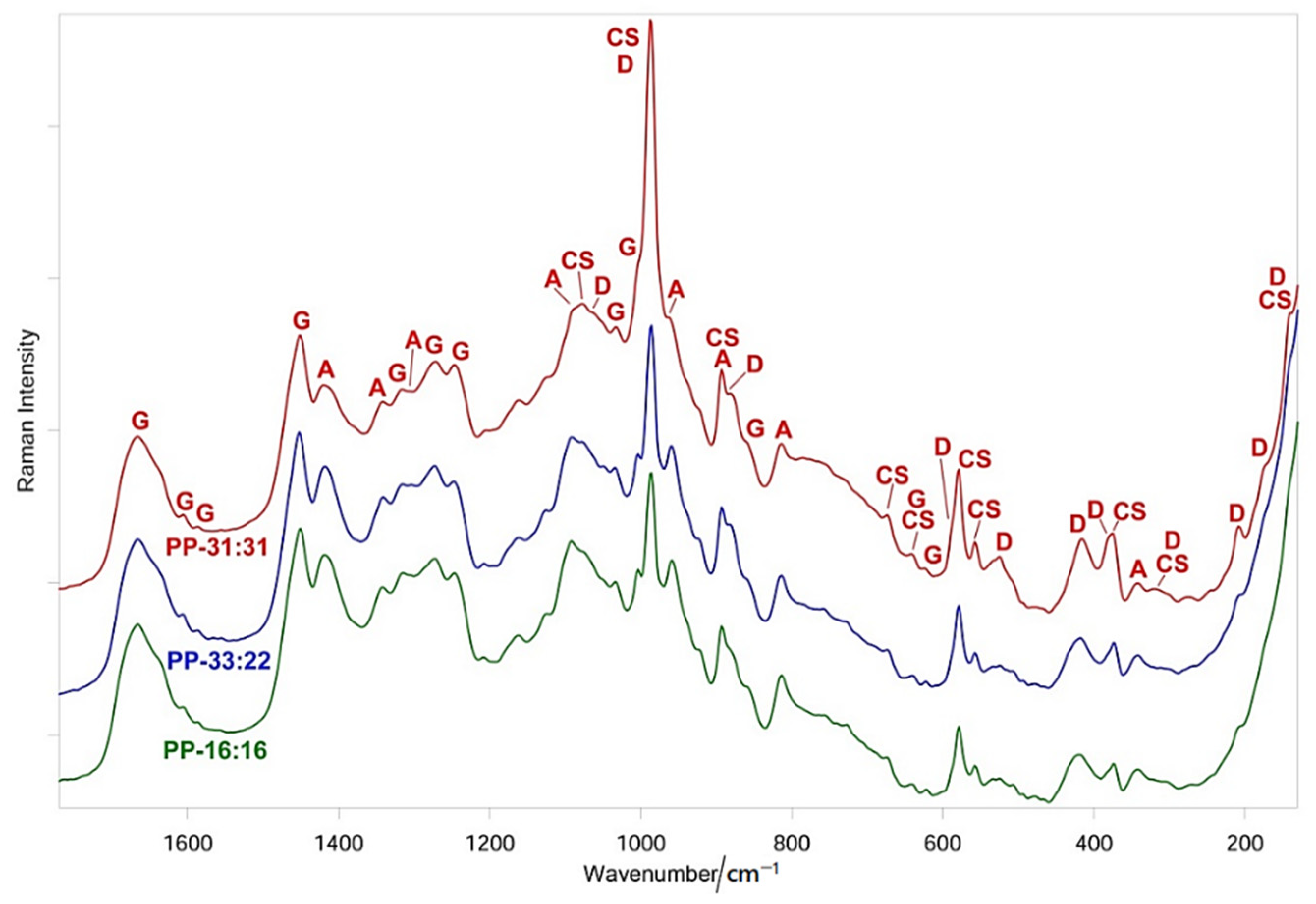
Spectroscopic vibrational techniques (FT-Raman and micro-Raman) were used to characterize the mineral fillers, polymeric matrices, and mineral-filled hydrogels before (i.e., fresh samples) and after apatite nucleation tests.
FT-Raman spectra were recorded by using a Bruker MultiRam FT-Raman spectrometer equipped with a cooled Ge-diode detector (Bruker Optik GmbH, Ettlingen, Germany). The excitation source was a Nd3+-YAG laser (1064 nm) in the backscattering (180°) configuration. The focused laser beam diameter was about 100 µm, the spectral resolution 4 cm−1, and the laser power for the sample was about 120 mW. Three-five Raman spectra were non-destructively recorded on three-five different positions of each composite sample and averaged.
Micro-Raman spectra were obtained by using an NRS-2000C Jasco spectrometer (Jasco Inc., Easton, MD, USA) with a microscope of 100× magnification. All the spectra were recorded in back-scattering conditions with 5 cm−1 spectral resolution by using the 532 nm green diode-pumped solid-state laser driver (RgBLase LLC, Fremont, CA, USA) with a power of about 20 mW. A 160 K cooled digital charge-coupled device (Spec-10: 100B, Roper Scientific Inc., Sarasota, FL, USA) was used as a detector. Due to the poor quality of the micro-Raman spectra of the fresh samples, this technique was only used to investigate the presence of a mineral deposition on the hydrogels, in addition to FT-Raman spectroscopy. This technique was also used to characterize the fresh samples and to disclose possible interactions between the phases through the comparative analysis of the spectra.
2.7. Cell Tests
2.7.1. Hydrogel Disks Sterilization
Cylindrical samples (10.0 mm diameter, 1.0 mm thick) were immersed in 250 mL of 70% ethanol solution for 20 min, then rinsed for 3 consecutive times with 5 mL of distilled water using a syringe [
42].
2.7.2. Cells Test: Direct Contact and Extract Test
Human mesenchymal stem cells (MSCs) derived from vascular wall were harvested in accordance with Local Ethics Committee Approval (protocol APP-13-01). The isolation process and stemness properties are detailed elsewhere [
43]. Human MSCs were plated at the density of 5.5 × 10
4 cells/well in 12-well plates and cultured in Dulbecco’s Modified Eagle’s Medium (DMEM, Euroclone, Milan, Italy) with 10% Fetal Bovine Serum (FBS, Euroclone, Milan, Italy) in an incubator at 37 °C and 5% CO
2 to allow cell adhesion and monolayer formation.
Hydrogels disks (PP-16:16, PP-33:22, PP-31:31) were placed into the wells in contact with MSCs and cultured for 3, 7, and 14 days. The culture medium was changed every 3 days. MSCs cultured without hydrogels were used as control. These experiments were used to evaluate the effects of hydrogels on vasculogenic and osteogenic MSCs differentiation.
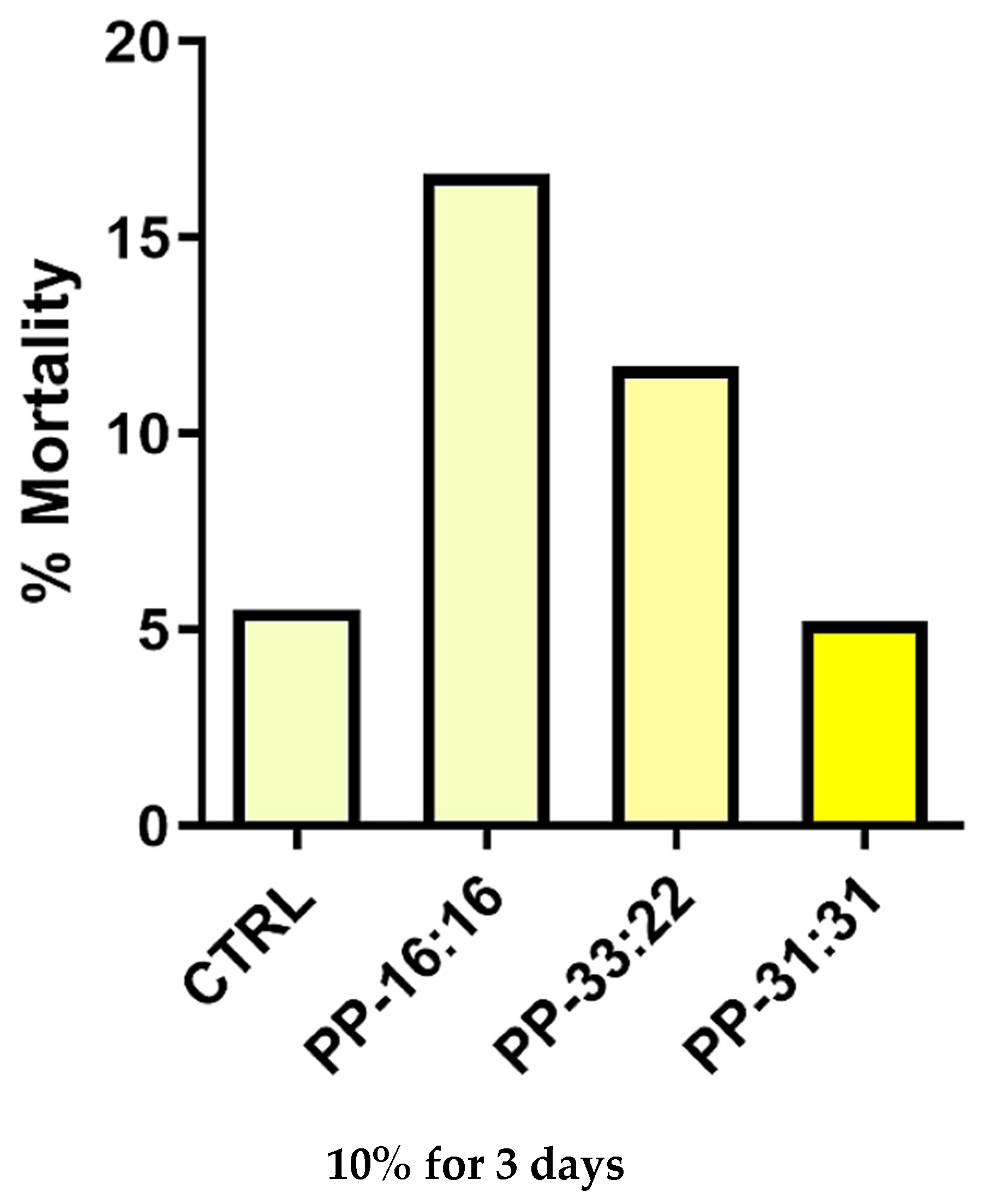
Additional experiments using the extracts from each hydrogel formulation were performed to evaluate the biocompatibility (cell mortality) and MSCs commitment (expression of vasculogenic and osteogenic markers). ISO 10993-5:2009 (clause 4.2.3.2: Extraction conditions) was used to prepare the extracts by using a 37 °C extraction temperature instead of 50 °C to simulate the clinical conditions. Extracts were obtained by immersion of hydrogel disks (10.0 mm diameter, 1.0 mm thick) into 5 mL of sterile water for 72 h followed by filtration before their addition to culture medium. MSCs were incubated for 3 days in DMEM with 10% FBS and 10% of extract from each hydrogel formulation, and cell mortality was assessed in each experimental condition. Briefly, MSCs were detached, centrifuged, and manually counted. The mortality was calculated as the ratio between the death cells number and the total cells number × 100, and values were expressed as percentages.
2.7.3. CD31, ALP and OCN Gene Expression
Total RNA was extracted from MSCs placed in contact with different hydrogel formulations using PureZOL (Bio-Rad, Hercules, CA, USA) according to the manufacturer’s instructions. Reverse transcription was performed from 0.25 ng RNA through iScript cDNA Synthesis Kit (Bio-Rad, Hercules, CA, USA). Real-time PCR was carried out using SsoAdvanced™ Universal SYBR
® Green Supermix (Bio-Rad Hercules, CA, USA) and amplified through CFX Connect™ Real-Time PCR Detection System (Bio-Rad Hercules, CA, USA). The forward and reverse primers sequences (Sigma-Merck, Milan, Italy) are listed in
Table 1. Glyceraldehyde 3-phosphate dehydrogenase (
GAPDH) was used as housekeeping gene. The expression of target genes was normalized on
GAPDH and analyzed using the 2^
−ΔΔCt relative quantification methods. Results were expressed as fold changes relative to the CTRL group.
The expression of CD31 (a specific marker of vascular differentiation), Alkaline phosphatase (ALP), and Osteocalcin (OCN) (both markers of osteogenic differentiation) were analyzed.
2.8. Statistical Analysis
Statistical analysis of the chemical physical tests was performed using Sigmaplot 12 (Systat, Chicago, IL, USA). Calcium release and alkalizing activity were analyzed using two-way ANOVA followed by RM Student–Newman–Keuls test (p < 0.05). Different letters represent statistically significant differences (p < 0.05) in the same line (capital letters) or in the same column (small letters). A one-way ANOVA followed by RM Student–Newman–Keuls test (p < 0.05) was used to analyse statistically significant differences of the materials for solubility, porosity, water absorption, radiopacity, and setting time.
Statistical analysis of cell tests were carried out using GraphPad Prism software (San Diego, Ca, USA). The differences between experimental groups were evaluated using two-way ANOVA followed by Dunnett’s multiple comparison test. Results from three independent experiments are reported as mean ± standard deviation (SD). p values < 0.05 were considered statistically significant.
4. Discussion
The occurrence of bone defects in oral maxilla or mandibula is a frequent clinical condition. The healing of bony deficit, caused by infections (periradicular, abscesses, periimplantitis), periodontitis, cysts, insufficient peri-implant bone, osteoporosis, trauma, tumor excision, and bone necrosis, requires treatments favouring bone regeneration.
There are increasing attention and grants allocated to “green”, i.e., eco-sustainable chemistry and technology. In this context, the use of natural bio-derived compounds (possibly abundant in nature) having high biocompatibility and biological interactivity to design materials for bone regeneration represents a futuristic ecological approach.
Mannuronic/guluronic acid-based polysaccharides can be synthetized from marine seaweed comprised in
Phaeophyceae class. Commercial polysaccarydes are produced mainly from wild brown algae of
Ascophyllum, Durvillaea, Ecklonia, Laminaria, Lessonia, Macrocystis, Saccharina, and
Sargassum genera. Approximately 1500–2000 species of brown algae are present worldwide [
48] and are abundant in several world areas, including the Mediterranean, North East Atlantic (North Sea and Baltic Areas), North America, and South East Asia [
48].
The coastal invasion of brown algae is a critical worldwide environmental phenomenon reported in Ireland, the Mediterranean Sea, and the Caribbean Sea [
49,
50]. The harvesting of algae from eutrophic coasts, where the proliferation of invading seaweeds is favoured by water nutrients, represents an attractive green approach adopting a new ecological concept whereby invasive algae are removed from coastal areas for the production of useful algae-based biomaterials (circular chemistry concept).
Mannuronic/guluronic acid-based polysaccharides possess several biological advantages when compared to other synthetic polymers, including hydrophilicity, absence of toxicity, and low immunogenicity [
51]. The drawbacks of these polysaccharides, such as low mechanical properties, uncontrolled degradation, and lack of adhesion ligands for mammalian cells, may be avoided/surpassed through their combination with other polymers and/or fillers. This is the strategy that we used to project the grafts of the present study.
Therefore, the design of bio-based composite scaffolds, constituted by different natural polymers having various biological effects favouring bone regeneration processes, represents a very attractive strategy.
Recently, experimental bio-based stiff/solid polymers containing collagen, chitosan, carrageenan, or alginate, in combination with calcium compounds as calcium chloride, calcium phosphate, calcium sulphate, hydroxyapatite, or glass have been conceived and tested [
52,
53,
54].
Scaffolds composed of fibrin, mannuronic/guluronic acid, and calcium phosphate were demonstrated to be non-cytotoxic, biodegradable, biointeractive, and were able to induce pro-osteogenic and angiogenic differentiation on mammalian macrophages [
52]. Porous scaffolds made of mannuronic/guluronic acid, carrageenan, and calcium silicate showed fibroblasts biocompatibility and the ability to nucleate apatite [
53]. Hydroxyapatite/chitosan and mannuronic/guluronic acid-based scaffolds loaded with antibacterial molecules were biocompatible, released biointeractive calcium ions, and induced the osteogenic differentiation of bone marrow stem cells [
54].
The association of mannuronic/guluronic acids with gelatine hydrogel revealed biocompatibility and absence of cytotoxicity for skin tissue engineering [
55] or wound healing application [
56]. To date, no similar hydrogels have been proposed for clinical application in bone tissue engineering, due to their lack of suitable physical-chemical and biointeractive properties.
In the present study, innovative green mineral-filled hydrogels have been developed starting from bio-derived sources through low-temperature, non-toxic, and low time-consuming procedures. The graft biomaterials have been conceived to be placed in non-load-bearing bone sites or in alveolar bone sites during healing, with the purpose to support bone tissue regeneration. Clinical studies and conventional oral rehabilitation protocols indicate that functional loading can be postponed for several months, waiting for new bone formation [
57].
The impact of inorganic fillers addition in hydrogels has been widely reported in literature [
58,
59,
60]. Inorganic mineral fillers, such as calcium phosphates, calcium silicates, and calcium carbonates, provide activating biointeractive signals by releasing biologically active ions [
61,
62]. Moreover, the fillers anisotropy in the presence of electrical stimulation may be useful to guide the resident cell growth, proliferation, and attachment [
58,
59,
60]. The use of inorganic fillers, such as hydroxyapatite or silicate nanoparticles, may provide additional properties to the soft and flat hydrogel surface, imparting stiffness strength [
58,
59,
60]. The inclusion of specific inorganic fillers (such as silver) in hydrogels may confer antibacterial properties [
63,
64].
In our study, the rationale for the addition of high percentages of CaSi and DCPD (up to 62% by weight) minerals is therefore related to the need to confer consistence to the hydrogel and to improve the biological properties through biointeractive and biologically active ion-releasing fillers [
40,
41].
CaSi are widely used in oral surgery and endodontics for their positive bio-interaction with bone tissues (i.e., periapical bone) [
21,
65,
66]. When in contact with body fluids, CaSi can create a microenvironment favorable for apatite nucleation, through the formation of a hydrated silica rich layer, the release of calcium ions, and the increase of local pH [
23,
29,
36]. The leached calcium ions have and epigenetic effect on mineralizing cells [
67,
68] and silicon possesses angiogenic properties [
69,
70].
A calcium phosphate component has also been included to provide further positive epigenetic chemical signals to cells involved in regenerative processes of bone. In addition, the association of calcium phosphate compounds and CaSi bioactive material improves and accelerates the apatite nucleation [
35]. DCPD provides an additional phosphate source early after contact with watery fluids for its progressive dissolution [
33,
35]. DCPD is the most soluble calcium orthophosphate salt at pH > 8.2 due to the presence of structural water [
71] and converts into apatite after contact with water or fluids.
In the present study, the designed hydrogels can crosslink through divalent ions (i.e calcium ions) rapidly released from CaSi and calcium phosphate. All mineral-filled hydrogels released high calcium ions and showed alkalizing ability during the 28 days test period. The ion release values particularly increased during the first three days and remained stable at all the tested endpoints (seven, 14, and 28 and days).
It has been reported that soluble molecular signals, such as calcium ions, trigger the cascade of cell differentiation into osteoblast lineage [
29] and create the conditions for a bone bonding structure (apatite coating layer) useful in bone defects [
34]. Released calcium ions specifically modulate osteopontin,
OCN, and
ALP in mineralizing cells. The occurrence of alkaline pH increased the activity/growth of osteoblasts and other mineralizing cells [
72].
In addition, silicon from CaSi likely provided an additional positive stimulus for mesenchymal stem cell differentiation into vascular lineage [
73]. SiOH silanol groups may be exposed by CaSi and increase the nucleation of apatite [
74].
Our study analysed the gene expression of markers for osteogenesis (ALP and OCN) and vascularization (CD31) by MSCs cultured in contact with mineral-filled hydrogels and with hydrogels extracts.
ALP and
OCN are markers of initial and late osteogenic differentiation.
ALP expression increases in cells at alkaline pH during the first stages of mineral phase formation, while
OCN expression indicates calcified tissue formation.
OCN binds hydroxyapatite crystals into extracellular calcified matrix [
71].
CD31, also known as platelet endothelial cell adhesion molecule 1 (PECAM-1), is a sensitive and specific marker for vascular differentiation [
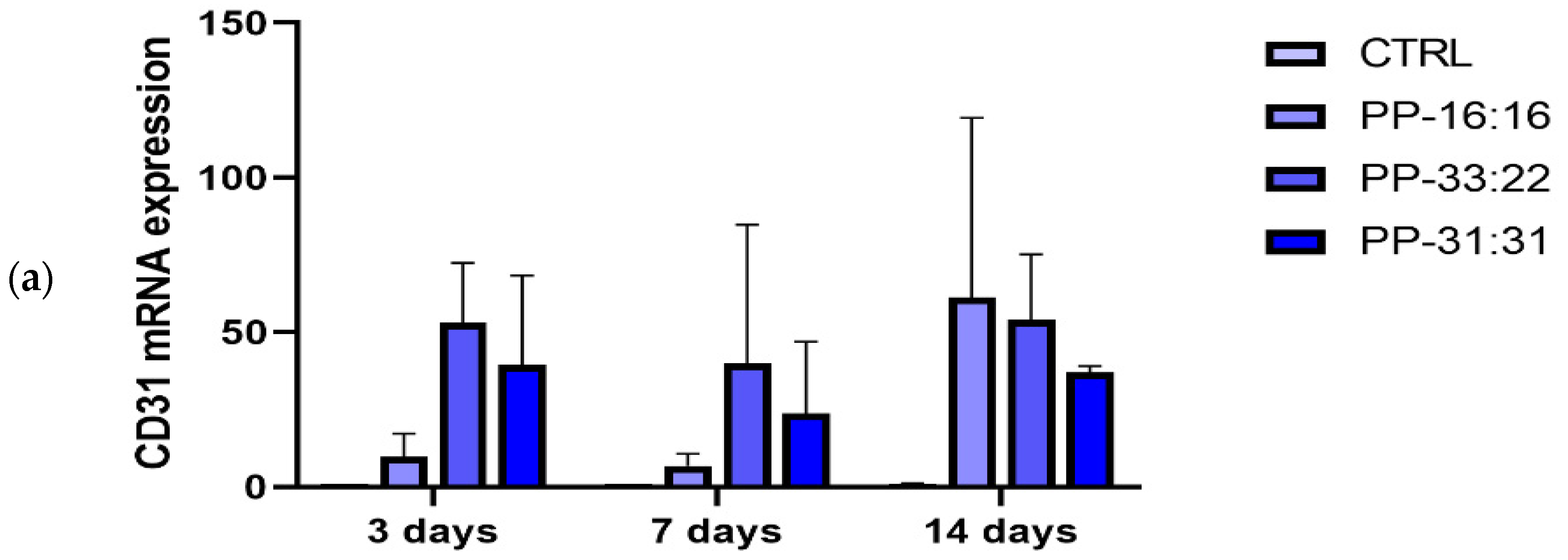
75]. In our study, the gene expression of MSCs cultured in contact with mineral-filled hydrogels showed that the addition of increasing percentages of CaSi and DCPD favored the expression of the pro-angiogenic marker
CD31 (
Figure 9a), with limited osteogenic marker expression (
ALP and
OCN,
Figure 9b,c). The expression of this gene was high (over 40-fold increase at 14 days for all the formulations), although non-statistically significant due to the high standard deviation.
Differently, MSCs cultured with hydrogels extracts induced an expression of the osteogenic markers (
Figure 10b,c) and not of the angiogenic marker. This could be attributed to the stable calcium ions released by the mineral-filled formulations during the first days immersion, as evident in
Table 2.
It should be clarified that the differences in gene expression between the two experimental conditions (MSCs in contact with the mineral-filled hydrogels and MSCs in presence of 10% extracts) can be likely related to a lower concentration of biologically active ions in 10%.
Neoangiogenesis is a necessary prerequisite for bone regeneration, new bone tissue development, and remodeling processes. Oral bone is highly vascularized. An ideal biomaterial should favor vascular endothelial cells attachment, proliferation, and vascular network formation, should allow the nutrients influx and the later mineralizing cell colonization [
76,
77].
Recent studies [
32,
78,
79] showed the expression of osteogenic and proangiogenic genes by MSCs in contact with CaSi and DCPD in polymer-based scaffolds. However, it has been reported that materials with low mineral filling showed a negligible induction of
OCN expression by MSCs derived from human periapical cyst after 21 days of culture [
32] and a moderate expression of
OCN from human adipose derived MSCs after 14 days [
79]. In addition, a moderate pro-angiogenic stimulus on vascular wall mesenchymal stem cells was originated by materials with low mineral filling [
79].
In this study, a reduction of MSCs mortality in presence of hydrogel extracts was observed when higher amounts of mineral fillers were used. Similar and low percentages of dead cells, comparable to the CTRL, was obtained when cells were cultured in the presence of PP-31:31, the formulation with the highest amount of CaSi and DCPD. A previous study on alginate-gelatine scaffold extracts showed that high extracellular calcium ions lead to an improvement of cell viability [
80], in agreement with our cell mortality tests. Extracellular calcium ions have an impact on cell behaviour, viability, and proliferation [
22,
28,
29,
30,
31,
33,
40,
41].
In the present study, the results of cell mortality indicate and confirm that the biologically active ions released from mineral-filled hydrogels with the highest percentage of CaSi-DCPD (i.e., PP:31-31) lead to a reduction of cell mortality and to an improvement of biological properties of the materials (increased gene expression). This effect may also be related to both the hydrophilic behavior of the bio-based hydrogel and to the mineral layer nucleation in the formulation with higher mineral fillers inclusion.
Raman spectroscopy showed that the different phases of mineral-filled hydrogels interact between each other. In particular, conformationally sensitive bands of gelatin (i.e., Amide I and III) underwent shifts and changes in relative intensities, suggesting that the inorganic fillers induced structural rearrangements of the organic phase. Wavenumber shifts were detected also in the bands of the former components.
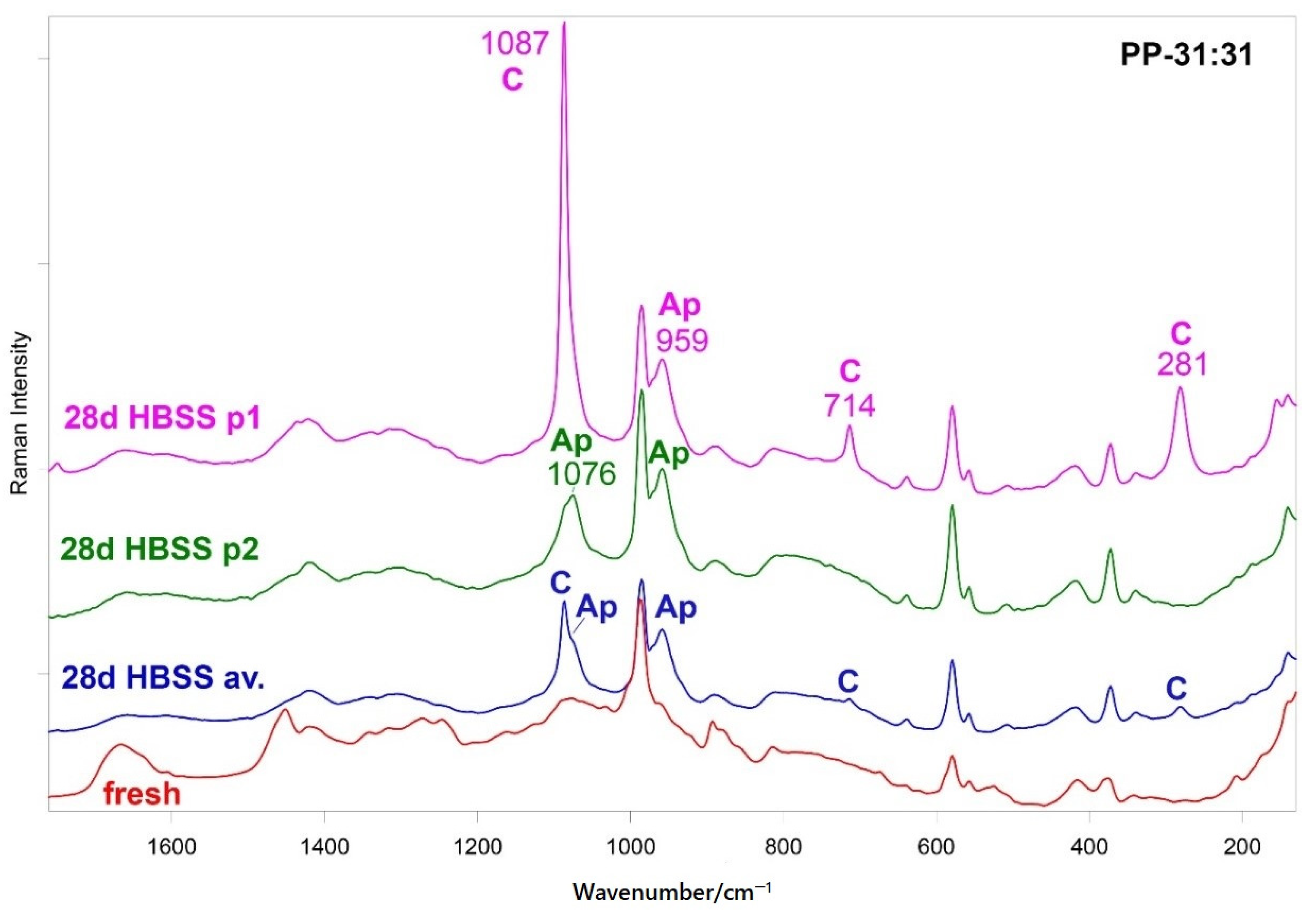
Raman spectroscopy disclosed B-type carbonated apatite (and calcite) nucleation only on the hydrogel containing the highest number of inorganic fillers, i.e., PP-31:31 (
Figure 6 and
Figure S4), while such phases were not detected on the other mineral-filled hydrogels. The Raman spectra showed that upon mineral deposition the gelatin component underwent conformational rearrangements towards a more unordered state, as revealed by the wavenumber downshift of Amide I and the strengthening of the 1272 cm
−1 band. At the same time, the COO
− groups belonging to mannuronate/guluronate (and likely also to aspartate and glutamate residues of gelatin) complexed calcium, as revealed by the strengthening of the COO
− stretching modes and by the appearance of the band at 1436 cm
−1 (
Figure 6 and
Figure 7), assigned to calcium alginate [
43].
Raman spectroscopy showed that no calcium ion saturation was attained in the CTRL hydrogel, on the basis of the relatively low shift of the COO
− symmetric stretching (from 1414 to 1419 cm
−1, see
Table S1) and breathing modes of the glycosidic ring (from 1096 to 1095 cm
−1, see
Table S1). For calcium ions saturated hydrogels shift by 20 and 10 cm
−1 have been reported for the former and the latter modes, respectively [
44,
47].
Our mineral-filled hydrogels showed attractive physical properties when compared with PP-CTRL. High porosity and water sorption likely related to each other and to calcium release were observed, in particular for hydrogels filled with high amounts of CaSi and DCPD (PP-33:22 and PP-31:31) (
Table 4).
The formulation with no mineral filler (designed as control) revealed low porosity and water sorption, low ions release and demonstrated complete dissolution after 3–7 days immersion in simulated body fluids.
We underline that high porosity is required for vascularization and neoangiogenesis processes in order to allow the uptake of nutrients and growth factors from peripheral blood vessels followed by cells colonization and proliferation and new blood vessel formation into biomaterial structure [
81].
ESEM analysis revealed marked surface modifications of the mineral-filled hydrogels when immersed in HBSS. This may be related to polymer degradation, mineral granules exposition, and apatite nucleation, all indexes of a dynamic modification of the mineral-filled hydrogel structure over time during the contact with body fluid. Indeed, a shift to a more porous surface was observed after 28 days.
All formulations revealed pore sizes over 100 μm after immersion in HBSS, markedly larger than that of fresh hydrogels. PP-16:16, the formulation with a lower percentage of fillers, had very limited pores when compared to PP-31:31 (the formulation with the highest percentage of fillers).
Pore size is important to allow different biological functions. Pores within 20 microns allow cell–extracellular matrix interactions and nutrients/metabolites transport. Pores larger than 100 μm are useful for new vessel formation and new bone formation [
82].
The chemical-physical and biological data obtained in the present study support the multifaceted/multimodal action of the designed mineral-filled hydrogels for oral bone defects. Such biomaterials (mainly PP-31:31) can act as vasculogenesis promoters and osteogenesis inducers through the direct stimulation of MSCs by their biointeractive properties and the release of biologically active signals.


 ,
, 













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}