
Cytokine-Mediated Inflammation in the Oral Cavity and Its Effect on Lipid Nanocarriers

 ,
,  , ,
, ,
Abstract
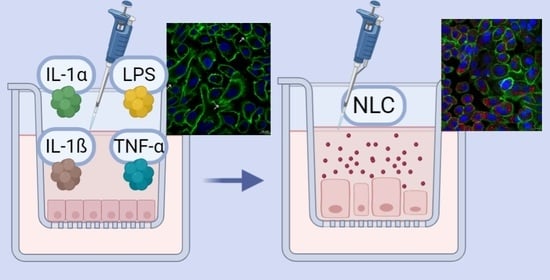
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Cell Culture
2.2.2. Toxicity Studies
2.2.3. Generation of Reactive Oxygen Species
2.2.4. Interleukin-8 Release
2.2.5. Determination of Cell Proliferation under Inflamed Conditions
2.2.6. Visualization of Phosphatidylserine Translocation
2.2.7. Visualization of Cell Morphological Changes
2.2.8. Visualization of Cell–Cell Contacts
2.2.9. Preparation of Nanostructured Lipid Carriers via High Pressure Homogenization
2.2.10. Particle Size and Zeta Potential
2.2.11. Interaction Studies of TR146 Cells with Nanostructured Lipid Carriers
2.2.12. Qualitative and Semi-Quantitative Nanostructured Lipid Carriers Uptake Studies
2.2.13. Determination of Active and Passive Transport Mechanisms
2.2.14. Statistical Analysis
3. Results
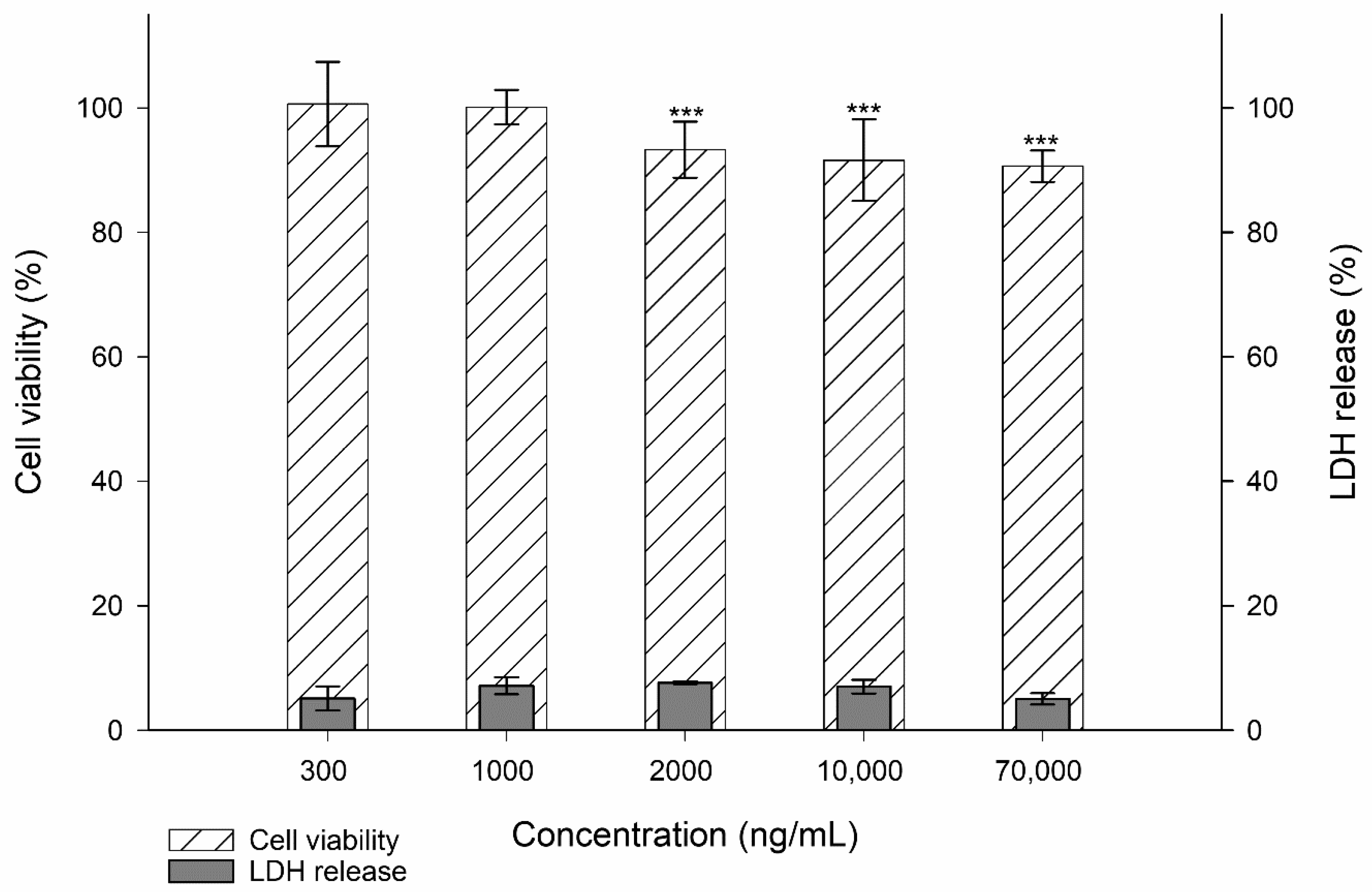
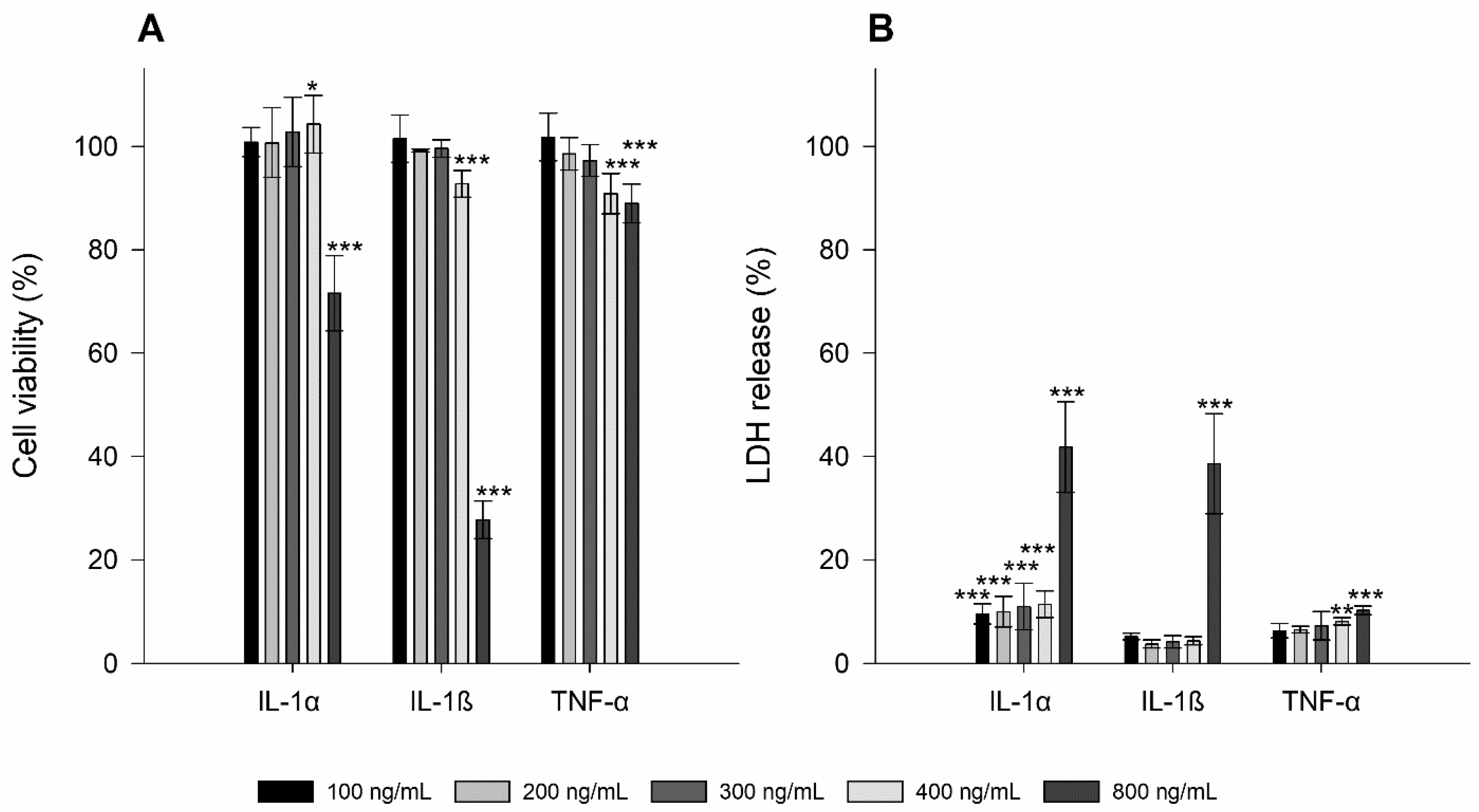
3.1. Toxicity Studies
3.2. Generation of Reactive Oxygen Species
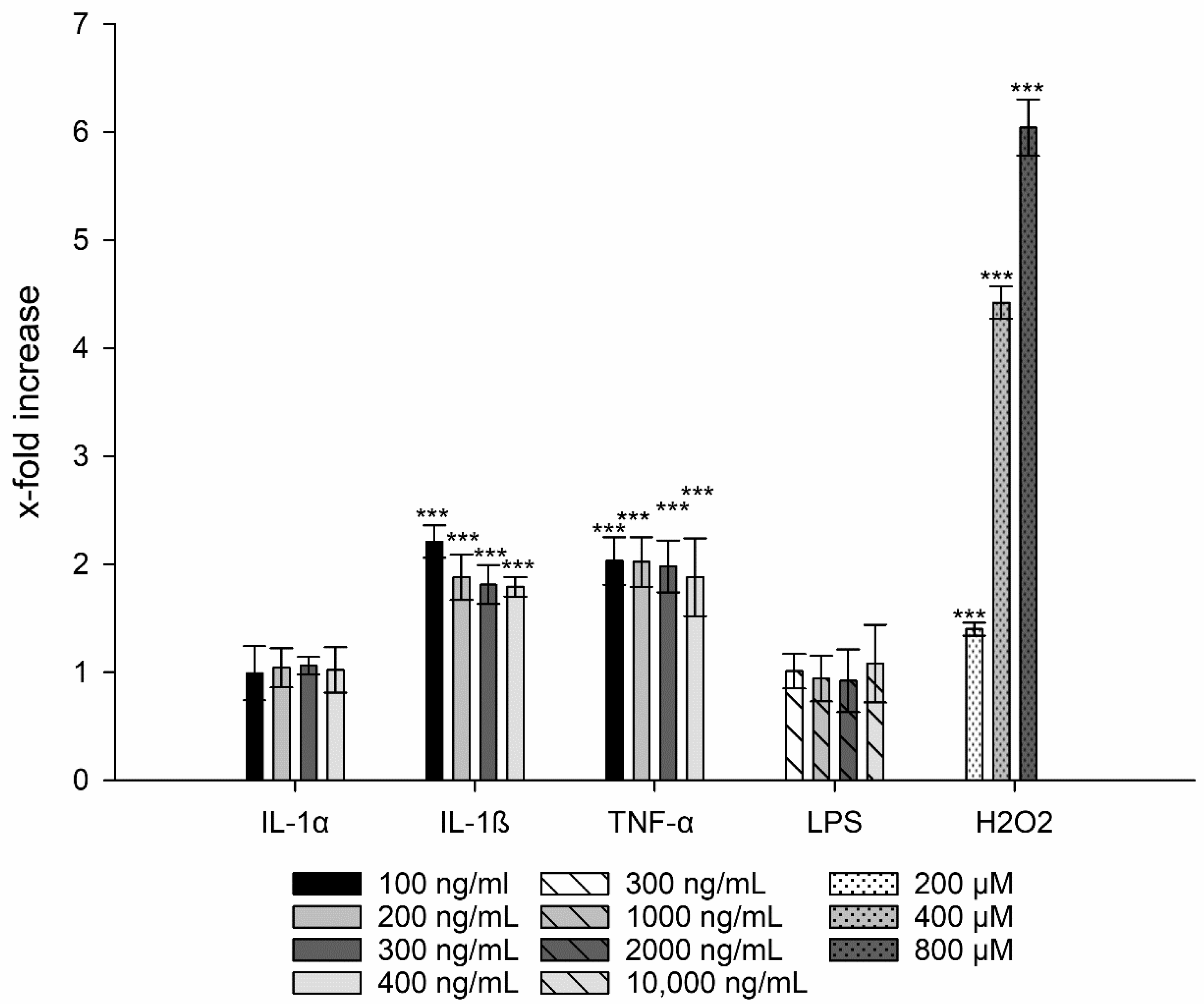
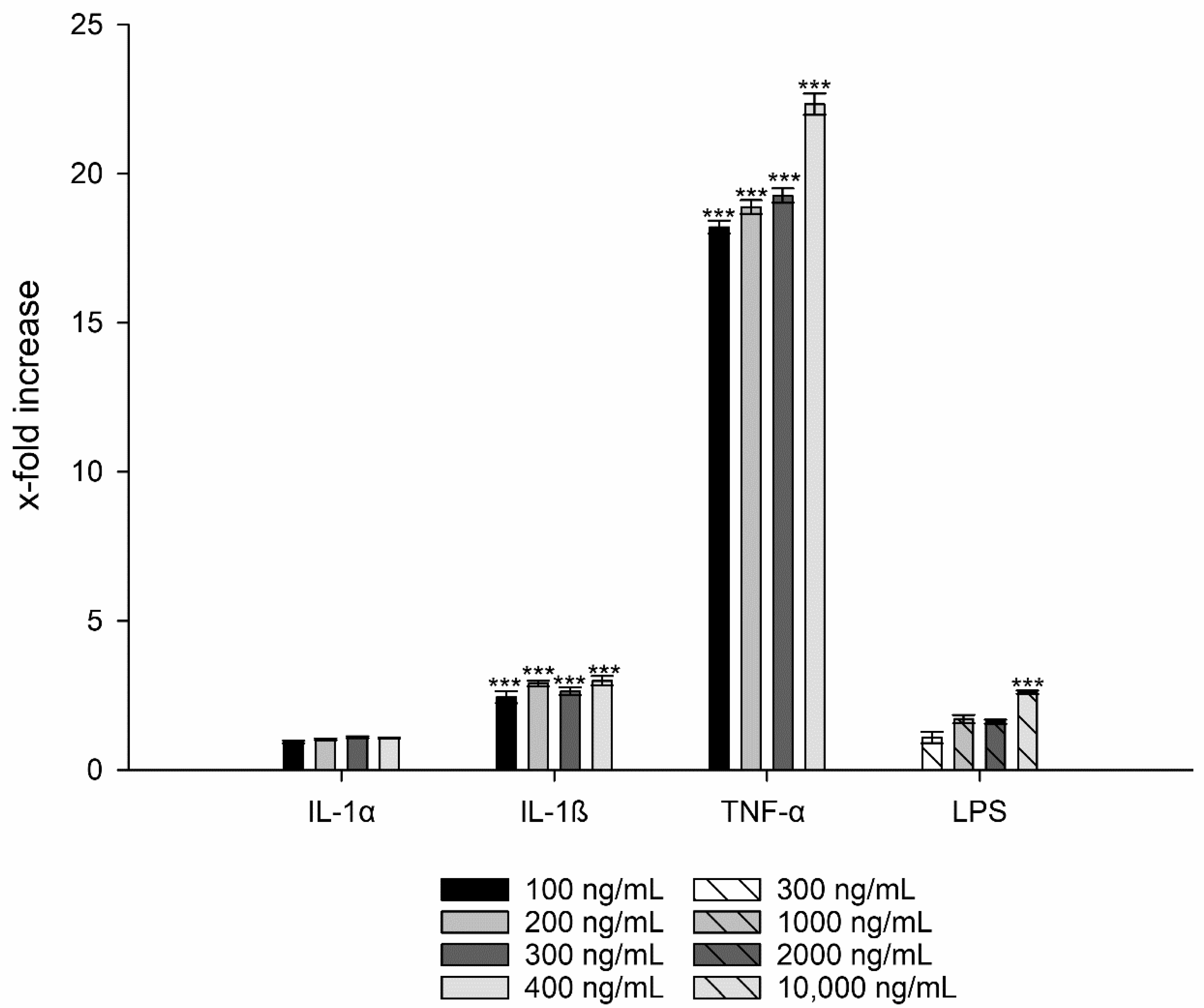
3.3. Interleukin-8 Release
3.4. Cell Proliferation under Inflamed Conditions
3.5. Visualization of Phosphatidylserine Translocation
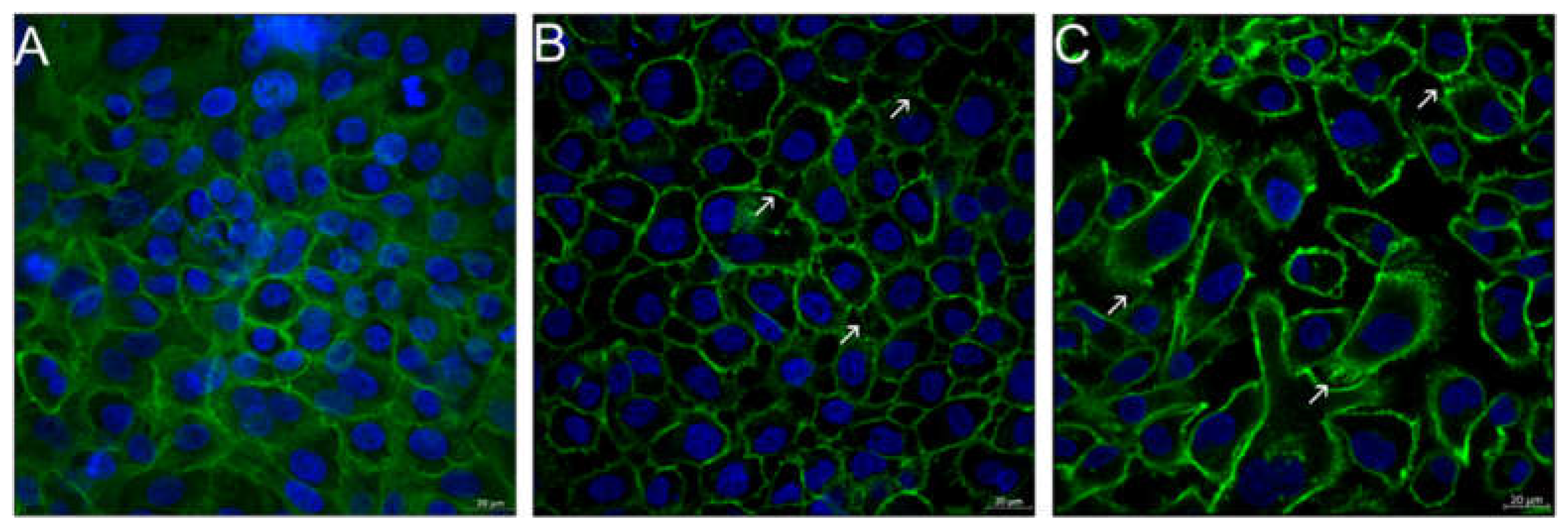
3.6. Visualization of Cell Morphological Changes
3.7. Loss of Cell–Cell Contacts (Desmosomes)
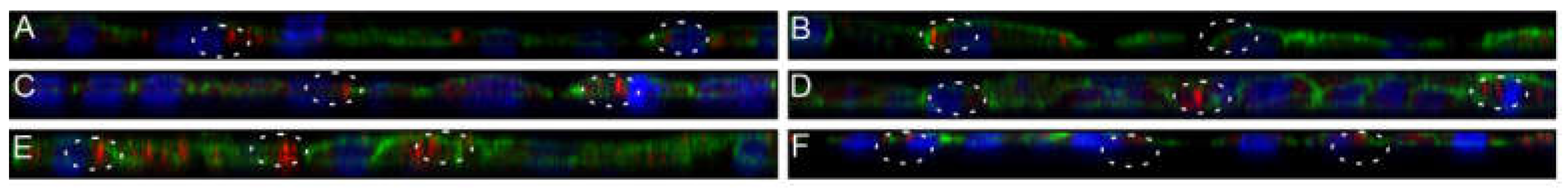
3.8. Nanostructured Lipid Carriers Interaction and Uptake Studies
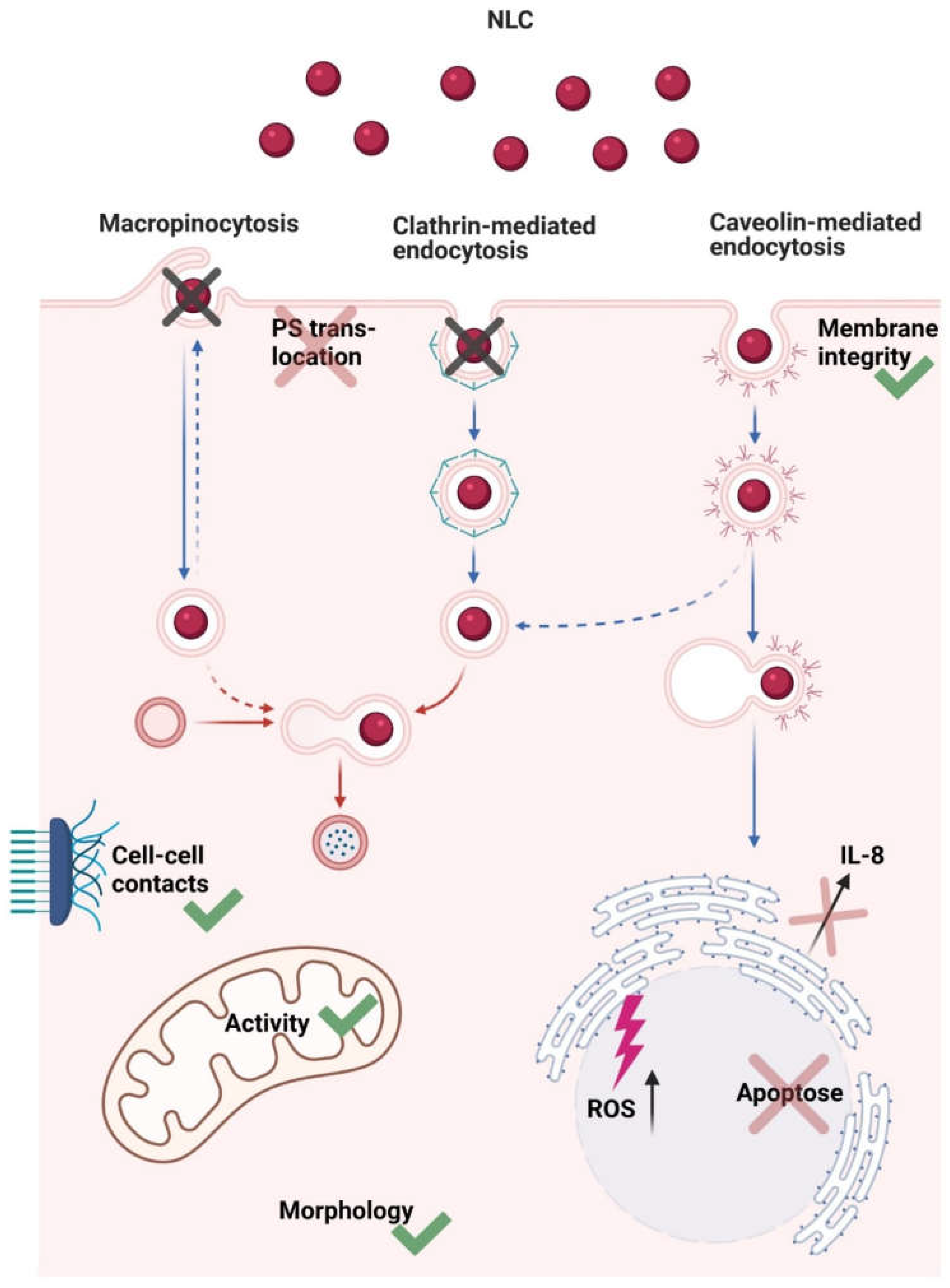
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization Oral Health. Available online: https://www.who.int/news-room/fact-sheets/detail/oral-health (accessed on 6 November 2019).
- Platform for Better Oral Health in Europe. The State of Oral Health in Europe. Available online: http://www.oralhealthplatform.eu/our-work/the-state-of-oral-health-in-europe/ (accessed on 6 November 2019).
- Hasturk, H.; Kantarci, A.; Van Dyke, T.E. Oral inflammatory diseases and systemic inflammation: Role of the macrophage. Front. Immunol. 2012, 3, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Politis, C.; Schoenaers, J.; Jacobs, R.; Agbaje, J.O. Wound Healing Problems in the Mouth. Front. Physiol. 2016, 7, 507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Oliveira, S.; Rosowski, E.E.; Huttenlocher, A. Neutrophil migration in infection and wound repair: Going forward in reverse. Nat. Rev. Immunol. 2016, 16, 378–391. [Google Scholar] [CrossRef] [PubMed]
- Andrews, C.; McLean, M.H.; Durum, S.K. Cytokine tuning of intestinal epithelial function. Front. Immunol. 2018, 9, 1270. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, S.; Uehara, A.; Tamai, R.; Takada, H. Innate immune responses in oral mucosa. J. Endotoxin Res. 2002, 8, 465–468. [Google Scholar] [CrossRef] [Green Version]
- Shnawa, I.M. Oral Epithelial Cytokines. Int. J. Vaccines Vaccin. 2016, 2. [Google Scholar] [CrossRef] [Green Version]
- Garlanda, C.; Dinarello, C.A.; Mantovani, A. The Interleukin-1 Family: Back to the Future. Immunity 2013, 39, 1003–1018. [Google Scholar] [CrossRef] [Green Version]
- Weber, A.; Wasiliew, P.; Kracht, M. Interleukin-1 (IL-1) pathway. Sci. Signal. 2010, 3. [Google Scholar] [CrossRef]
- Schmalz, G.; Schweikl, H.; Hiller, K.A. Release of prostaglandin E2, IL-6 and IL-8 from human oral epithelial culture models after exposure to compounds of dental materials. Eur. J. Oral Sci. 2000, 108, 442–448. [Google Scholar] [CrossRef]
- Gemmell, E.; Marshall, R.I.; Seymour, G.J. Cytokines and prostaglandins in immune homeostasis and tissue destruction in periodontal disease. Periodontol. 2000 1997, 14, 112–143. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, E.; Dittrich-Breiholz, O.; Holtmann, H.; Kracht, M. Multiple control of interleukin-8 gene expression. J. Leukoc. Biol. 2002, 72, 847–855. [Google Scholar] [CrossRef] [PubMed]
- Huguet-Casquero, A.; Xu, Y.; Gainza, E.; Pedraz, J.L.; Beloqui, A. Oral delivery of oleuropein-loaded lipid nanocarriers alleviates inflammation and oxidative stress in acute colitis. Int. J. Pharm. 2020, 586, 119515. [Google Scholar] [CrossRef] [PubMed]
- Pulito, C.; Cristaudo, A.; La Porta, C.; Zapperi, S.; Blandino, G.; Morrone, A.; Strano, S. Oral mucositis: The hidden side of cancer therapy. J. Exp. Clin. Cancer Res. 2020, 39, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Takenokuchi, M.; Kadoyama, K.; Yoshida, D.; Takaki, S.; Yamamoto, R.; Saigo, K.; Taniguchi, T. Evaluation of Absorbability of Macromolecular Substances in the Oral Mucosa and Skin using a Three-Dimensional Tissue Culture Model. Biol. Med. 2018, 10. [Google Scholar] [CrossRef]
- Yadev, N.P.; Murdoch, C.; Saville, S.P.; Thornhill, M.H. Evaluation of tissue engineered models of the oral mucosa to investigate oral candidiasis. Microb. Pathog. 2011, 50, 278–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bierbaumer, L.; Schwarze, U.Y.; Gruber, R.; Neuhaus, W. Cell culture models of oral mucosal barriers: A review with a focus on applications, culture conditions and barrier properties. Tissue Barriers 2018, 6, 1479568. [Google Scholar] [CrossRef]
- Rupniak, H.T.; Rowlatt, C.; Lane, E.B.; Steele, J.G.; Trejdosiewicz, L.K.; Hill, B.T.; Laskiewicz, B.; Povey, S. Characteristics of Four New Human Cell Lines Derived from Squamous Cell Carcinomas of the Head and Neck. JNCI J. Natl. Cancer Inst. 1985, 75, 621–635. [Google Scholar] [CrossRef]
- Jacobsen, J.; Nielsen, E.B.; Brondum-Nielsen, K.; Christensen, M.E.; Olin, H.-B.D.; Tommerup, N.; Rassing, M.R. Filter-grown TR146 cells as an in vitro model of human buccal epithelial permeability. Eur. J. Oral Sci. 1999, 107, 138–146. [Google Scholar] [CrossRef]
- Nielsen, H.M. In vitro models of the human buccal epithelium: The TR146 cell culture model and the porcine in vitro model. In Cell Culture Models of Biological Barriers: In Vitro Test Systems for Drug Absorption and Delivery; CRC Press: Boca Raton, FL, USA, 2002; p. 310. [Google Scholar]
- Teubl, B.J.; Absenger, M.; Fröhlich, E.; Leitinger, G.; Zimmer, A.; Roblegg, E. The oral cavity as a biological barrier system: Design of an advanced buccal in vitro permeability model. Eur. J. Pharm. Biopharm. 2013, 84, 386–393. [Google Scholar] [CrossRef]
- Schimpel, C.; Teubl, B.; Absenger, M.; Meindl, C.; Fröhlich, E.; Leitinger, G.; Zimmer, A.; Roblegg, E. Development of an Advanced Intestinal In Vitro Triple Culture Permeability Model to Study Transport of Nanoparticles. Mol. Pharm. 2014, 11, 808–818. [Google Scholar] [CrossRef]
- Lin, G.C.; Leitgeb, T.; Vladetic, A.; Friedl, H.P.; Rhodes, N.; Rossi, A.; Roblegg, E.; Neuhaus, W. Optimization of an oral mucosa in vitro model based on cell line TR146. Tissue Barriers 2020, 8, 1748459. [Google Scholar] [CrossRef] [Green Version]
- Teubl, B.J.; Meindl, C.; Eitzlmayr, A.; Zimmer, A.; Fröhlich, E.; Roblegg, E. In-vitro permeability of neutral polystyrene particles via buccal mucosa. Small 2013, 9, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Soomro, A.H.; Khan, E.; Noori, S.; Lone, M.A.; Syal, Z.; Sheikh, S. Assessment of cytokine release against oral mucosal cell line culture (tr146) stimulated by neutrophil elastase associated with behcet’s disease. Int. J. Dent. 2019, 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosokawa, Y.; Hosokawa, I.; Ozaki, K.; Matsuo, T. IL-27 Modulates Chemokine Production in TNF-α -Stimulated Human Oral Epithelial Cells. Cell. Physiol. Biochem. 2017, 43, 1198–1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sardaro, N.; della Vella, F.; Incalza, M.A.; Stasio, D.D.I.; Lucchese, A.; Contaldo, M.; Laudadio, C.; Petruzzi, M. Oxidative stress and oral mucosal diseases: An overview. In Vivo 2019, 33, 289–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Żukowski, P.; Maciejczyk, M.; Waszkiel, D. Sources of free radicals and oxidative stress in the oral cavity. Arch. Oral Biol. 2018, 92, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Petković, A.B.; Matić, S.M.; Stamatović, N.V.; Vojvodić, D.V.; Todorović, T.M.; Lazić, Z.R.; Kozomara, R.J. Proinflammatory cytokines (IL-1β and TNF-α) and chemokines (IL-8 and MIP-1α) as markers of peri-implant tissue condition. Int. J. Oral Maxillofac. Surg. 2010, 39, 478–485. [Google Scholar] [CrossRef]
- Oton-Leite, A.F.; Silva, G.B.L.; Morais, M.O.; Silva, T.A.; Leles, C.R.; Valadares, M.C.; Pinezi, J.C.D.; Batista, A.C.; Mendonça, E.F. Effect of low-level laser therapy on chemoradiotherapy-induced oral mucositis and salivary inflammatory mediators in head and neck cancer patients. Lasers Surg. Med. 2015, 47, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Basso, F.G.; Pansani, T.N.; Soares, D.G.; Scheffel, D.L.; Bagnato, V.S.; De Souza Costa, C.A.; Hebling, J. Biomodulation of Inflammatory Cytokines Related to Oral Mucositis by Low-Level Laser Therapy. Photochem. Photobiol. 2015, 91, 952–956. [Google Scholar] [CrossRef]
- Tetyczka, C.; Griesbacher, M.; Absenger-Novak, M.; Fröhlich, E.; Roblegg, E. Development of nanostructured lipid carriers for intraoral delivery of Domperidone. Int. J. Pharm. 2017, 526, 188–198. [Google Scholar] [CrossRef]
- Tetyczka, C.; Hodzic, A.; Kriechbaum, M.; Juraić, K.; Spirk, C.; Hartl, S.; Pritz, E.; Leitinger, G.; Roblegg, E. Comprehensive characterization of nanostructured lipid carriers using laboratory and synchrotron X-ray scattering and diffraction. Eur. J. Pharm. Biopharm. 2019, 139, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Lyklema, J. Electrokinetics after Smoluchowski. Colloids Surf. A Physicochem. Eng. Asp. 2003, 222, 5–14. [Google Scholar] [CrossRef]
- Krifka, S.; Spagnuolo, G.; Schmalz, G.; Schweikl, H. A review of adaptive mechanisms in cell responses towards oxidative stress caused by dental resin monomers. Biomaterials 2013, 34, 4555–4563. [Google Scholar] [CrossRef]
- Tobita, T.; Izumi, K.; Feinberg, S.E. Development of an in vitro model for radiation-induced effects on oral keratinocytes. Int. J. Oral Maxillofac. Surg. 2010, 39, 364–370. [Google Scholar] [CrossRef] [Green Version]
- Naumanen, P.; Lappalainen, P.; Hotulainen, P. Mechanisms of actin stress fibre assembly. J. Microsc. 2008, 231, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Tojkander, S.; Gateva, G.; Lappalainen, P. Actin stress fibers–assembly, dynamics and biological roles. J. Cell Sci. 2012, 125, 1855–1864. [Google Scholar] [CrossRef] [Green Version]
- Yuan, A.; Woo, S. Bin Adverse drug events in the oral cavity. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2015, 119, 35–47. [Google Scholar] [CrossRef] [Green Version]
- Spirk, C.; Hartl, S.; Pritz, E.; Gugatschka, M.; Kolb-Lenz, D.; Leitinger, G.; Roblegg, E. Comprehensive investigation of saliva replacement liquids for the treatment of xerostomia. Int. J. Pharm. 2019, 571, 118759. [Google Scholar] [CrossRef] [PubMed]
- Raber-Durlacher, J.E.; Elad, S.; Barasch, A. Oral mucositis. Oral Oncol. 2010, 46, 452–456. [Google Scholar] [CrossRef]
- Tetteh, E.K.; Morris, S. Evaluating the administration costs of biologic drugs: Development of a cost algorithm. Geogr. Nat. Resour. 2014, 4, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Balcão, V.M.; Vila, M.M.D.C. Structural and functional stabilization of protein entities: State-of-the-art. Adv. Drug Deliv. Rev. 2015, 93, 25–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winter, C.; Keimel, R.; Gugatschka, M.; Kolb, D.; Leitinger, G.; Roblegg, E. Investigation of changes in saliva in radiotherapy-induced head neck cancer patients. Int. J. Environ. Res. Public Health 2021, 18, 1629. [Google Scholar] [CrossRef] [PubMed]
- Naglik, J.R.; Moyes, D. Epithelial Cell Innate Response to Candida albicans. Adv. Dent. Res. 2011, 23, 50–55. [Google Scholar] [CrossRef] [Green Version]
- Weindl, G.; Wagener, J.; Schaller, M. Interaction of the mucosal barrier with accessory immune cells during fungal infection. Int. J. Med. Microbiol. 2011, 301, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Sorenson, B.S.; Khammanivong, A.; Guenther, B.D.; Ross, K.F.; Herzberg, M.C. IL-1 receptor regulates S100A8/A9-dependent keratinocyte resistance to bacterial invasion. Mucosal Immunol. 2012, 5, 66–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lalla, R.V.; Brennan, M.T.; Gordon, S.M.; Sonis, S.T.; Rosenthal, D.I.; Keefe, D.M. Oral Mucositis Due to High-Dose Chemotherapy and/or Head and Neck Radiation Therapy. J. Natl. Cancer Inst. Monogr. 2019, 2019. [Google Scholar] [CrossRef]
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem. Sci. 2010, 35, 505–513. [Google Scholar] [CrossRef] [Green Version]
- Kasahara, T.; Mukaida, N.; Yamashita, K.; Yagisawa, H.; Akahoshi, T.; Matsushima, K. IL-1 and TNF-α induction of IL-8 and monocyte chemotactic and activating factor (MCAF) mRNA expression in a human astrocytoma cell line. Immunology 1991, 74, 60–67. [Google Scholar]
- Bevers, E.M.; Comfurius, P.; Zwaal, R.F. Regulatory mechanisms in maintenance and modulation of transmembrane lipid asymmetry: Pathophysiological implications. Lupus 1996, 5, 480–487. [Google Scholar] [CrossRef]
- Ran, S.; Downes, A.; Thorpe, P.E. Increased Exposure of Anionic Phospholipids on the Surface of Tumor Blood Vessels. Cancer Res. 2002, 62, 6132–6140. [Google Scholar]
- Riedl, S.; Rinner, B.; Asslaber, M.; Schaider, H.; Walzer, S.; Novak, A.; Lohner, K.; Zweytick, D. In search of a novel target—Phosphatidylserine exposed by non-apoptotic tumor cells and metastases of malignancies with poor treatment efficacy. Biochim. Biophys. Acta Biomembr. 2011, 1808, 2638–2645. [Google Scholar] [CrossRef] [Green Version]
- Utsugi, T.; Schroit, A.J.; Connor, J.; Bucana, C.D.; Fidler, I.J. Elevated Expression of Phosphatidylserine in the Outer Membrane Leaflet of Human Tumor Cells and Recognition by Activated Human Blood Monocytes. Cancer Res. 1991, 51, 3062–3066. [Google Scholar]
- Zwaal, R.F.A.; Schroit, A.J. Pathophysiologic implications of membrane phospholipid asymmetry in blood cells. Blood 1997, 89, 1121–1132. [Google Scholar] [CrossRef]
- Burstyn-Cohen, T.; Maimon, A. TAM receptors, phosphatidylserine, inflammation, and cancer. Cell Commun. Signal. 2019, 17, 156. [Google Scholar] [CrossRef] [Green Version]
- Babiychuk, E.B.; Monastyrskaya, K.; Potez, S.; Draeger, A. Blebbing confers resistance against cell lysis. Cell Death Differ. 2011, 18, 80–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charras, G.T. A short history of blebbing. J. Microsc. 2008, 231, 466–478. [Google Scholar] [CrossRef] [PubMed]
- McGinley, E.L.; Fleming, G.J.P.; Moran, G.P. Development of a discriminatory biocompatibility testing model for non-precious dental casting alloys. Dent. Mater. 2011, 27, 1295–1306. [Google Scholar] [CrossRef] [PubMed]
- Jayatilake, J.A.M.S.; Samaranayake, Y.H.; Samaranayake, L.P. An ultrastructural and a cytochemical study of candidal invasion of reconstituted human oral epithelium. J. Oral Pathol. Med. 2005, 34, 240–246. [Google Scholar] [CrossRef]
- Nekrasova, O.; Green, K.J. Desmosome assembly and dynamics. Trends Cell Biol. 2013, 23, 537–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanna, A.N.; Berthiaume, L.G.; Kikuchi, Y.; Begg, D.; Bourgoin, S.; Brindley, D.N. Tumor Necrosis Factor-α Induces Stress Fiber Formation through Ceramide Production: Role of Sphingosine Kinase. Mol. Biol. Cell 2001, 12, 3618–3630. [Google Scholar] [CrossRef]
- Moharamzadeh, K.; Brook, I.M.; Van Noort, R.; Scutt, A.M.; Smith, K.G.; Thornhill, M.H. Development, optimization and characterization of a full-thickness tissue engineered human oral mucosal model for biological assessment of dental biomaterials. J. Mater. Sci. Mater. Med. 2008, 19, 1793–1801. [Google Scholar] [CrossRef] [PubMed]
- McHarg, S.; Hopkins, G.; Lim, L.; Garrod, D. Down-regulation of desmosomes in cultured cells: The roles of PKC, microtubules and lysosomal/proteasomal degradation. PLoS ONE 2014, 9, e108570. [Google Scholar] [CrossRef] [PubMed]
- Raiko, L.; Siljamäki, E.; Mahoney, M.G.; Putaala, H.; Suominen, E.; Peltonen, J.; Peltonen, S. Hailey-Hailey disease and tight junctions: Claudins 1 and 4 are regulated by ATP2C1 gene encoding Ca(2+)/Mn(2+) ATPase SPCA1 in cultured keratinocytes. Exp. Dermatol. 2012, 21, 586–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishii, K.; Green, K.J. Cadherin function: Breaking the barrier. Curr. Biol. 2001, 11, R569–R572. [Google Scholar] [CrossRef] [Green Version]
- Koga, H.; Tsuruta, D.; Ohyama, B.; Ishii, N.; Hamada, T.; Ohata, C.; Furumura, M.; Hashimoto, T. Desmoglein 3, its pathogenecity and a possibility for therapeutic target in pemphigus vulgaris. Expert Opin. Ther. Targets 2013, 17, 293–306. [Google Scholar] [CrossRef] [PubMed]
- Al-Sadi, R.; Guo, S.; Ye, D.; Ma, T.Y. TNF-α modulation of intestinal epithelial tight junction barrier is regulated by ERK1/2 activation of Elk-1. Am. J. Pathol. 2013, 183, 1871–1884. [Google Scholar] [CrossRef] [Green Version]
- Ma, T.Y.; Iwamoto, G.K.; Hoa, N.T.; Akotia, V.; Pedram, A.; Boivin, M.A.; Said, H.M. TNF-α-induced increase in intestinal epithelial tight junction permeability requires NF-κB activation. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 286. [Google Scholar] [CrossRef] [Green Version]
- Marchiando, A.M.; Shen, L.; Vallen Graham, W.; Weber, C.R.; Schwarz, B.T.; Austin, J.R.; Raleigh, D.R.; Guan, Y.; Watson, A.J.M.; Montrose, M.H.; et al. Caveolin-1-dependent occludin endocytosis is required for TNF-induced tight junction regulation in vivo. J. Cell Biol. 2010, 189, 111–126. [Google Scholar] [CrossRef] [Green Version]
- Neurath, M.F. Cytokines in inflammatory bowel disease. Nat. Rev. Immunol. 2014, 14, 329–342. [Google Scholar] [CrossRef]
- Grabinger, T.; Bode, K.J.; Demgenski, J.; Seitz, C.; Delgado, M.E.; Kostadinova, F.; Reinhold, C.; Etemadi, N.; Wilhelm, S.; Schweinlin, M.; et al. Inhibitor of Apoptosis Protein-1 Regulates Tumor Necrosis Factor–Mediated Destruction of Intestinal Epithelial Cells. Gastroenterology 2017, 152, 867–879. [Google Scholar] [CrossRef] [Green Version]
- McLean, M.H.; Neurath, M.F.; Durum, S.K. Targeting interleukins for the treatment of inflammatory bowel disease—What lies beyond anti-TNF therapy? Inflamm. Bowel Dis. 2014, 20, 389–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teubl, B.J.; Schimpel, C.; Leitinger, G.; Bauer, B.; Fröhlich, E.; Zimmer, A.; Roblegg, E. Interactions between nano-TiO2 and the oral cavity: Impact of nanomaterial surface hydrophilicity/hydrophobicity. J. Hazard. Mater. 2015, 286, 298–305. [Google Scholar] [CrossRef]
- Strachan, J.B.; Dyett, B.P.; Nasa, Z.; Valery, C.; Conn, C.E. Toxicity and cellular uptake of lipid nanoparticles of different structure and composition. J. Colloid Interface Sci. 2020, 576, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Voigt, J.; Christensen, J.; Shastri, V.P. Differential uptake of nanoparticles by endothelial cells through polyelectrolytes with affinity for caveolae. Proc. Natl. Acad. Sci. USA 2014, 111, 2942–2947. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Chen, M.; Su, Z.; Sun, M.; Ping, Q. Size-exclusive effect of nanostructured lipid carriers on oral drug delivery. Int. J. Pharm. 2016, 511, 524–537. [Google Scholar] [CrossRef] [PubMed]
- Beloqui, A.; Solinís, M.Á.; Gascón, A.R.; del Pozo-Rodríguez, A.; des Rieux, A.; Préat, V. Mechanism of transport of saquinavir-loaded nanostructured lipid carriers across the intestinal barrier. J. Control. Release 2013, 166, 115–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fröhlich, E.; Roblegg, E. Oral uptake of nanoparticles: Human relevance and the role of in vitro systems. Arch. Toxicol. 2016, 90, 2297–2314. [Google Scholar] [CrossRef] [PubMed]
- Bae, G.D.; Park, E.Y.; Kim, K.; Jang, S.E.; Jun, H.S.; Oh, Y.S. Upregulation of caveolin-1 and its colocalization with cytokine receptors contributes to beta cell apoptosis. Sci. Rep. 2019, 9, 16785. [Google Scholar] [CrossRef] [PubMed]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tetyczka, C.; Hartl, S.; Jeitler, R.; Absenger-Novak, M.; Meindl, C.; Fröhlich, E.; Riedl, S.; Zweytick, D.; Roblegg, E. Cytokine-Mediated Inflammation in the Oral Cavity and Its Effect on Lipid Nanocarriers. Nanomaterials 2021, 11, 1330. https://0-doi-org.brum.beds.ac.uk/10.3390/nano11051330
Tetyczka C, Hartl S, Jeitler R, Absenger-Novak M, Meindl C, Fröhlich E, Riedl S, Zweytick D, Roblegg E. Cytokine-Mediated Inflammation in the Oral Cavity and Its Effect on Lipid Nanocarriers. Nanomaterials. 2021; 11(5):1330. https://0-doi-org.brum.beds.ac.uk/10.3390/nano11051330
Chicago/Turabian StyleTetyczka, Carolin, Sonja Hartl, Ramona Jeitler, Markus Absenger-Novak, Claudia Meindl, Eleonore Fröhlich, Sabrina Riedl, Dagmar Zweytick, and Eva Roblegg. 2021. "Cytokine-Mediated Inflammation in the Oral Cavity and Its Effect on Lipid Nanocarriers" Nanomaterials 11, no. 5: 1330. https://0-doi-org.brum.beds.ac.uk/10.3390/nano11051330