A CRISPR/Cas12a Based Universal Lateral Flow Biosensor for the Sensitive and Specific Detection of African Swine-Fever Viruses in Whole Blood

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials, Chemicals, Reagents, and Methodology
2.2. Target DNA Design, Virus Collection, and DNA Extraction
2.3. PCR Amplification
2.4. Target Cleavage Assay
2.5. Construction of the LFB
2.6. ASFV Clinical Samples Preparation and Analysis
2.7. Statistical Analysis
3. Results
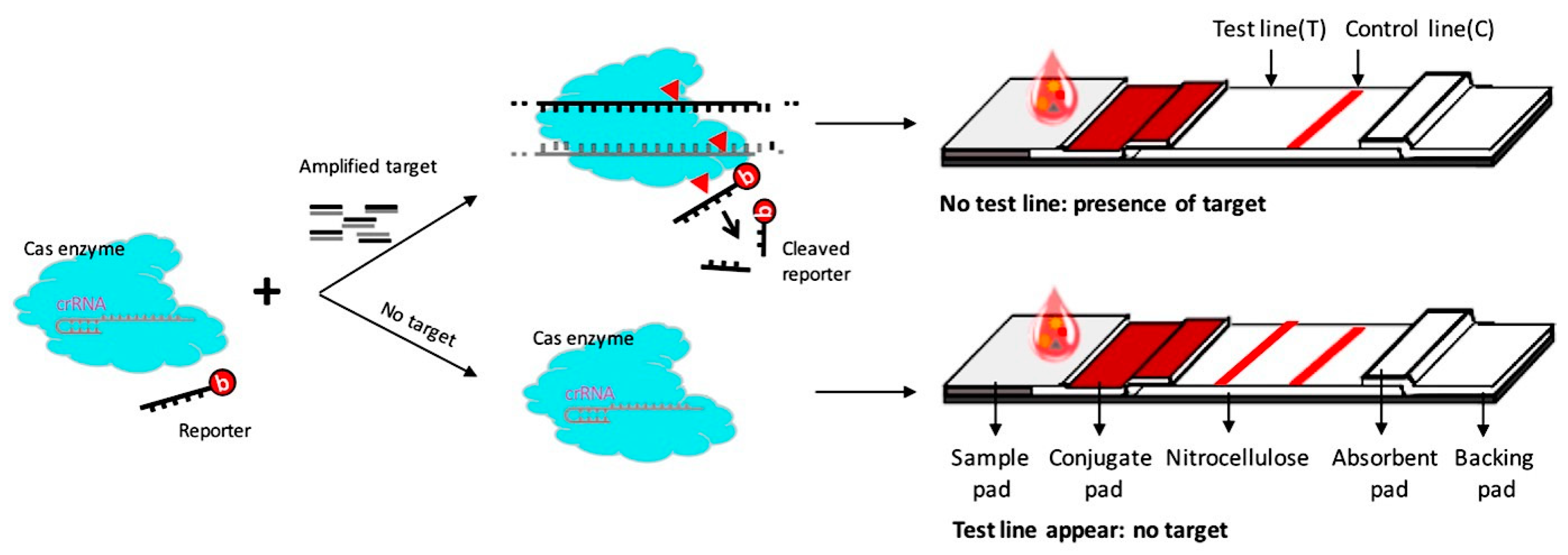
3.1. Overview of the CRISPR/Cas–LFB Detection System
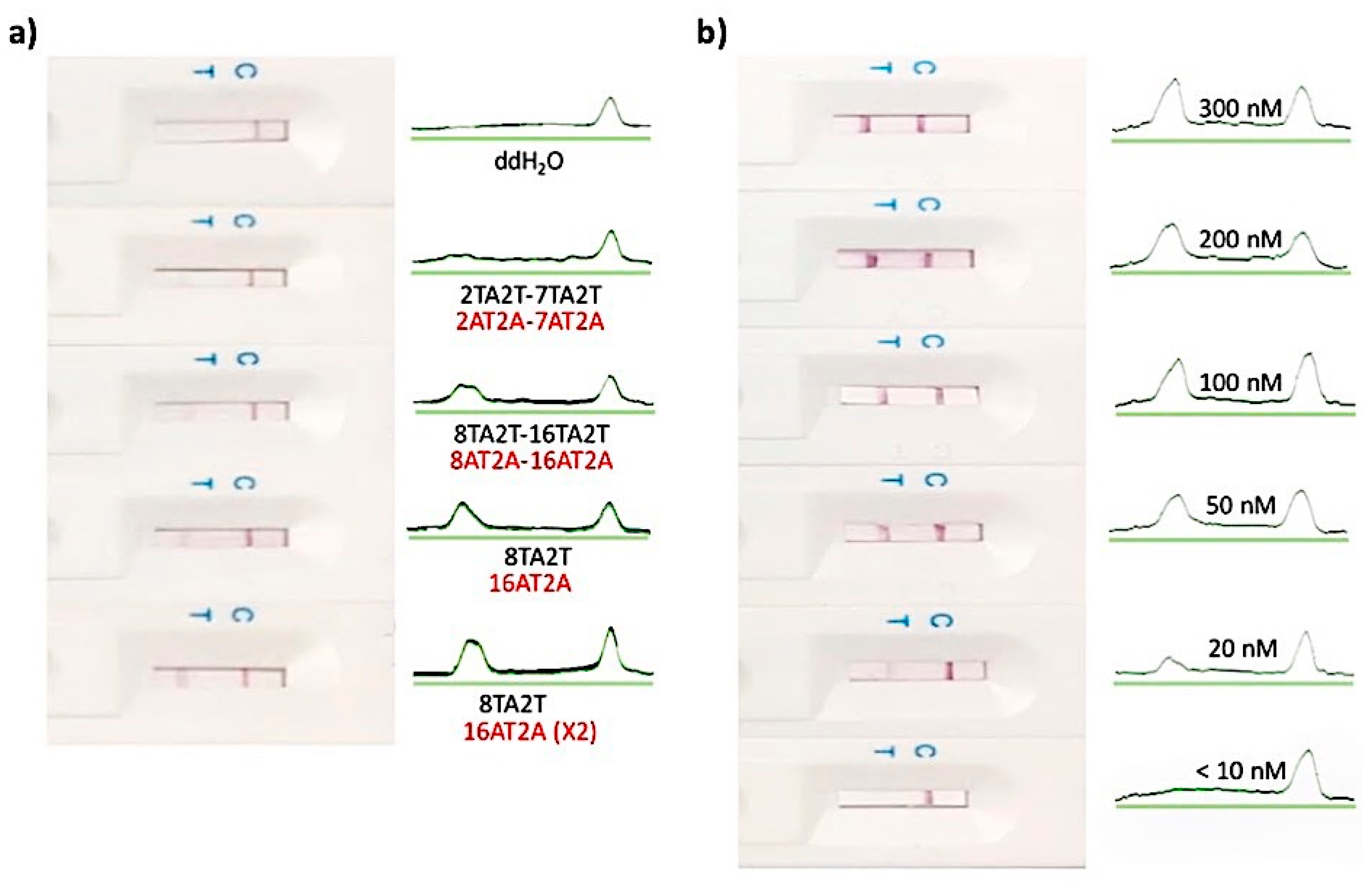
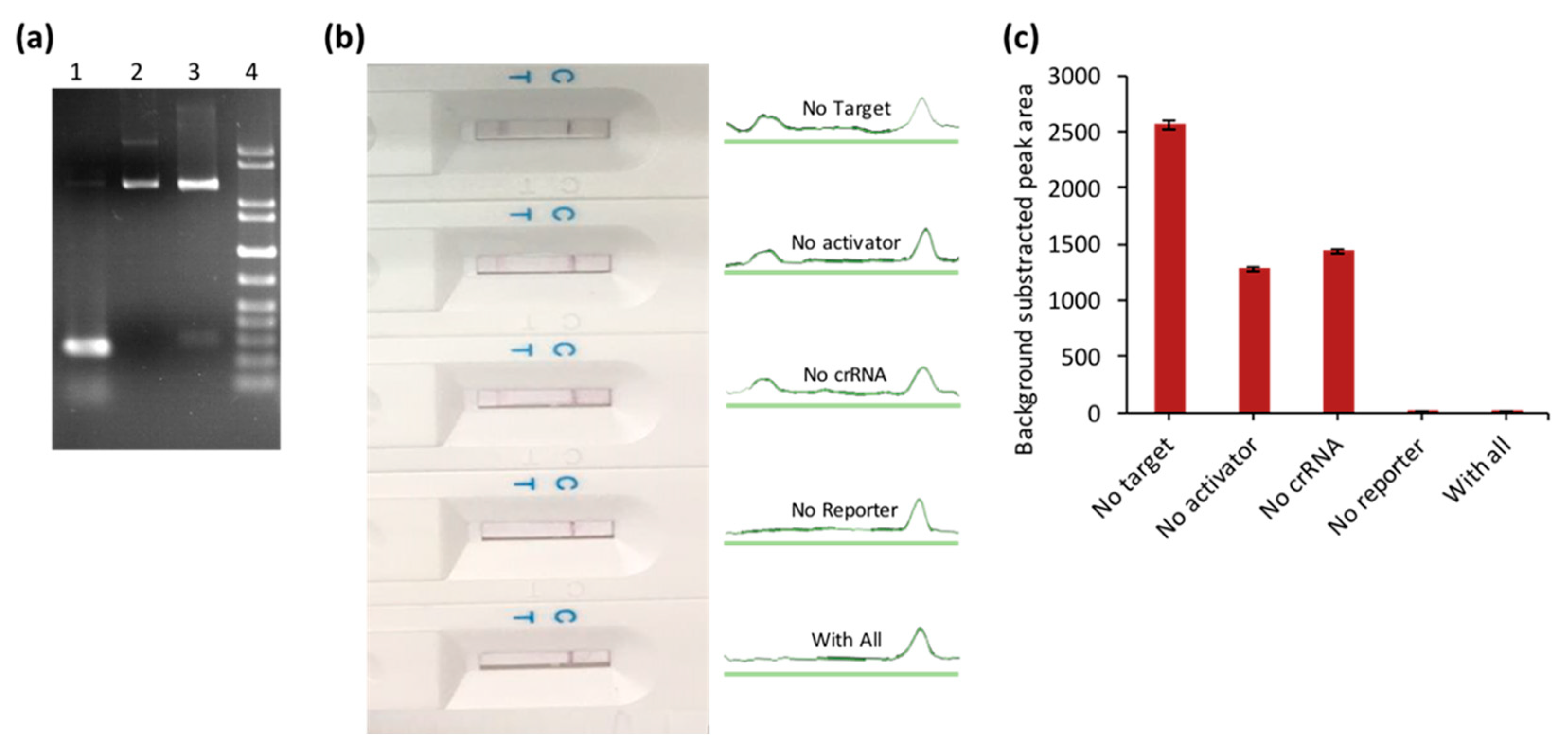
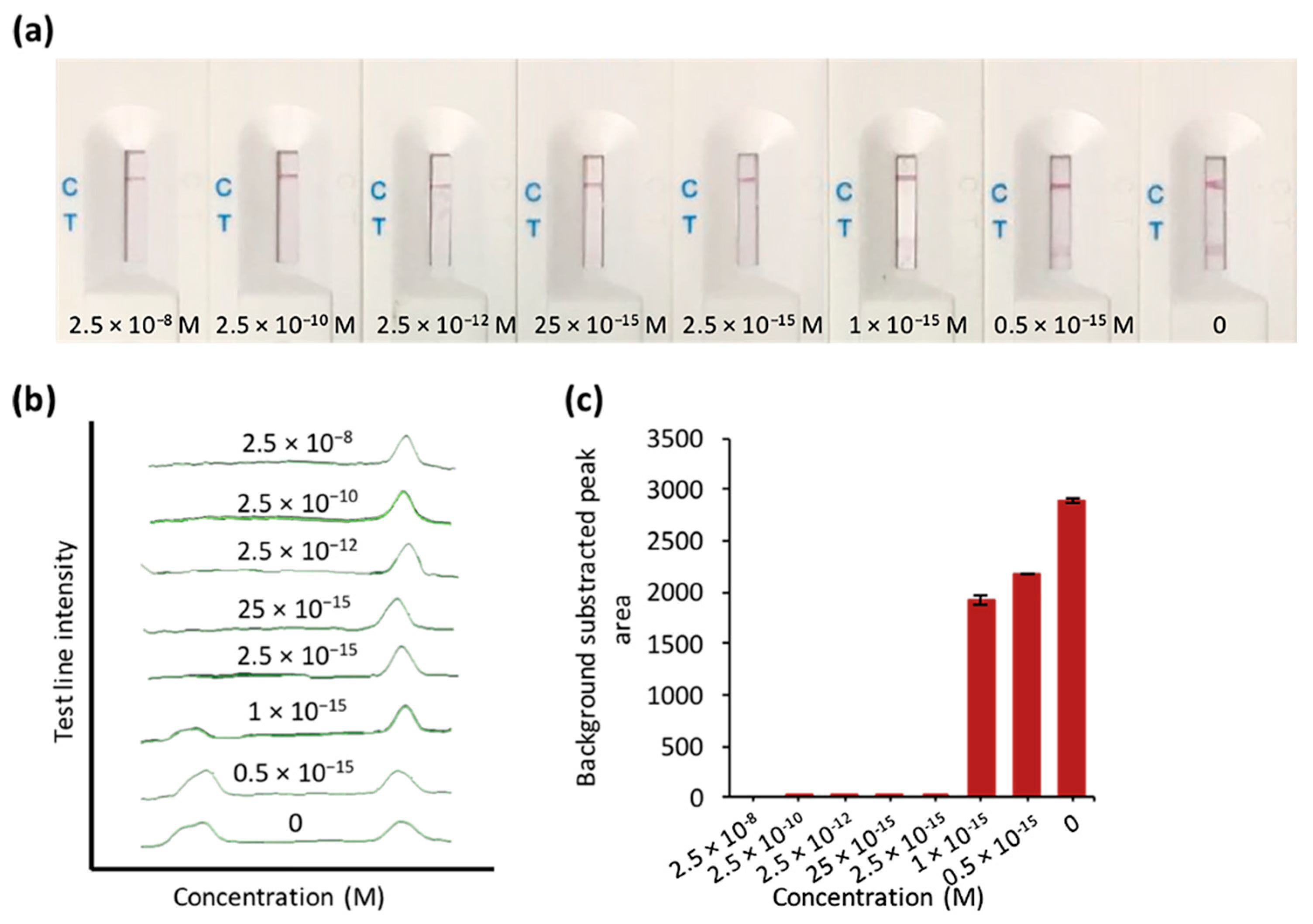
3.2. The Feasibility and Sensitivity Evaluation of CRISPR/Cas–LFB Assay Using Recombinant Plasmid
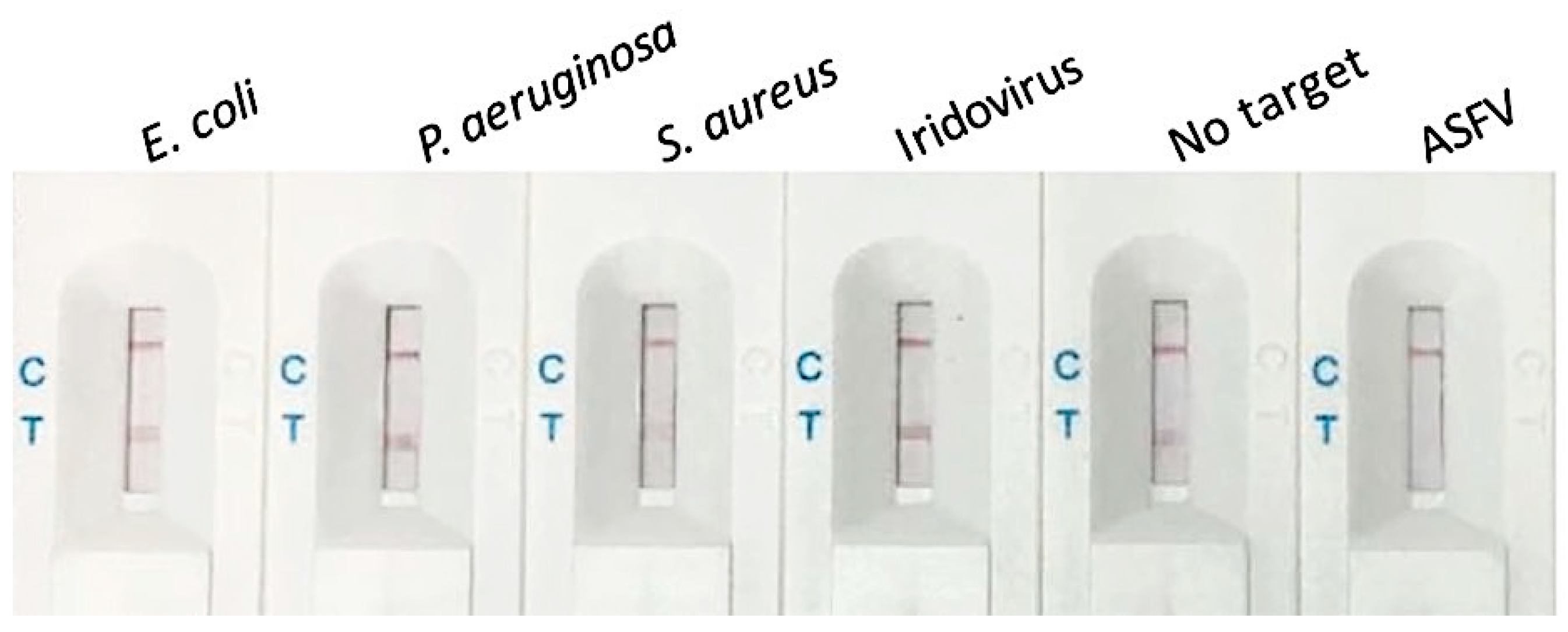
3.3. Selectivity of the Biosensor for ASFV Detection
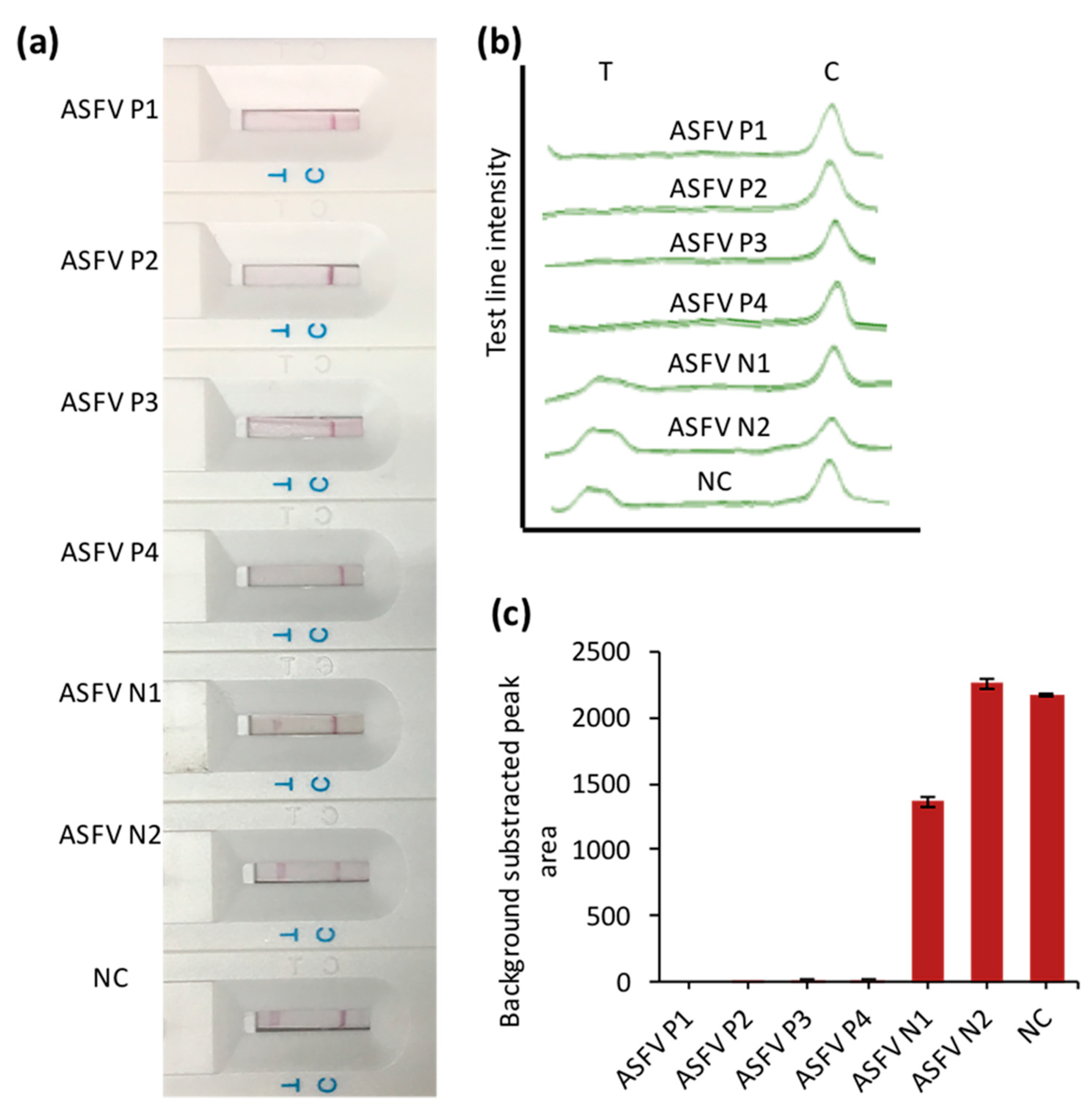
3.4. The Application of CRISPR/Cas–LFB Assay in Real Samples
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kleiboeker, S.B.; Scoles, G.A.; Burrage, T.G.; Sur, J. African swine fever virus replication in the midgut epithelium is required for infection of Ornithodoros ticks. J. Virol. 1999, 73, 8587–8598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, J.; Plowright, W.; Pierce, M.A. The epizootiology of African swine fever in Africa. Vet. Rec. 1969, 85, 668–674. [Google Scholar] [PubMed]
- Gomez-Villamandos, J.C.; Bautista, M.J.; Sanchez-Cordon, P.J.; Carrasco, L. Pathology of African swine fever: The role of monocyte-macrophage. Virus Res. 2013, 173, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, C.; Fernández-Pinero, J.; Pelayo, V.; Gazaev, I.; Markowska-Daniel, I.; Pridotkas, G.; Nieto, R.; Fernández-Pacheco, P.; Bokhan, S.; Nevolko, O.; et al. Genetic variation among African swine fever genotype II viruses, eastern and central Europe. Emerg. Infect Dis. 2014, 20, 1544–1547. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Fons, F.; Segalés, J.; Gortázar, C. A review of viral diseases of the European wild boar: Effects of population dynamics and reservoir rôle. Vet. J. 2008, 176, 158–169. [Google Scholar] [CrossRef]
- Gogin, A.; Gerasimov, V.; Malogolovkin, A.; Kolbasov, D. African swine fever in the North Caucasus region and the Russian Federation in years 2007–2012. Virus Res. 2013, 173, 198–203. [Google Scholar] [CrossRef]
- Śmietanka, K.; Woźniakowski, G.; Kozak, E.; Niemczuk, K.; Frączyk, M.; Bocian, Ł.; Kowalczyk, A.; Pejsak, Z. African Swine Fever Epidemic, Poland, 2014–2015. Emerg. Infect. Dis. 2016, 22, 1201–1207. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Vizcaino, J.M.; Mur, L.; Martinez-Lopez, B. African swine fever (ASF): Five years around Europe. Vet. Microbiol. 2013, 165, 45–50. [Google Scholar] [CrossRef]
- Liu, J.; Liu, B.; Shan, B.; Wei, S.; An, T.; Shen, G.; Chen, Z. Prevalence of African Swine Fever in China, 2018–2019. J. Med Virol. 2020, 92, 1023–1034. [Google Scholar] [CrossRef]
- Cubillos, C.; Gomez-Sebastian, S.; Moreno, N.; Nunez, M.C.; Mulumba-Mfumu, L.K.; Quembo, C.J.; Heath, L.; Etter, E.M.; Jori, F.; Escribano, J.M.; et al. African swine fever virus serodiagnosis: A general review with a focus on the analyses of African serum samples. Virus Res. 2013, 173, 159–167. [Google Scholar] [CrossRef]
- Agüero, M.; Fernández, J.; Romero, L.; Sánchez Mascaraque, C.; Arias, M.; Sánchez-Vizcaíno, J.M. Highly sensitive PCR assay for routine diagnosis of African swine fever virus in clinical samples. J. Clin. Microbiol. 2003, 41, 4431–4434. [Google Scholar] [CrossRef] [Green Version]
- Gallardo, C.; Nieto, R.; Soler, A.; Pelayo, V.; Fernández-Pinero, J.; Markowska-Daniel, I.; Pridotkas, G.; Nurmoja, I.; Granta, R.; Simón, A.; et al. Assessment of African Swine Fever Diagnostic Techniques as a Response to the Epidemic Outbreaks in Eastern European Union Countries: How To Improve Surveillance and Control Programs. J. Clin. Microbiol. 2015, 53, 2555–2565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sastre, P.; Gallardo, C.; Monedero, A.; Ruiz, T.; Arias, M.; Sanz, A.; Rueda, P. Development of a novel lateral flow assay for detection of African swine fever in blood. BMC Vet. Res. 2016, 12, 206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myhrvold, C.; Freije, C.A.; Gootenberg, J.S.; Abudayyeh, O.O.; Metsky, H.C.; Durbin, A.F.; Kellner, M.J.; Tan, A.L.; Paul, L.M.; Parham, L.A.; et al. Field deployable viral diagnostics using CRISPR-Cas13. Science 2018, 360, 444–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.Y.; Cheng, Q.X.; Wang, J.M.; Li, X.Y.; Zhang, Z.L.; Gao, S.; Cao, R.B.; Zhao, G.P.; Wang, J. CRISPR-Cas12a-assisted nucleic acid detection. Cell Discov. 2018, 4, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Li, S.; Wang, J.; Wang, G.; Zhao, G.; Wang, J. CRISPR-Cas12b-assisted nucleic acid detection platform for Nucleic Acid Detection and DNA Methylation Quantitation. ACS Synth. Biol. 2019, 10, 2228–2237. [Google Scholar] [CrossRef] [PubMed]
- Gootenberg, J.S.; Abudayyeh, O.O.; Kellner, M.J.; Joung, J.; Collins, J.J.; Zhang, F. Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science 2018, 360, 439–444. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.S.; Ma, E.; Harrington, L.B.; Da Costa, M.; Tian, X.; Palefsky, J.M.; Doudna, J.A. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science 2018, 360, 436–439. [Google Scholar] [CrossRef] [Green Version]
- Gootenberg, J.S.; Abudayyeh, O.O.; Lee, J.W.; Essletzbichler, P.; Dy, A.J.; Joung, J.; Verdine, V.; Donghia, N.; Daringer, N.M.; Freije, C.A.; et al. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 2017, 356, 438–442. [Google Scholar] [CrossRef] [Green Version]
- Harrington, L.B.; Burstein, D.; Chen, J.S.; Paez-Espino, D.; Ma, E.; Witte, I.P.; Cofsky, J.C.; Kyrpides, N.C.; Banfield, J.F.; Doudna, J.A. Programmed DNA destruction by miniature CRISPR-Cas14 enzymes. Science 2018, 362, 839–842. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Chen, S.; Guo, J.; Ma, X. Nanomaterial Labels in Lateral Flow Immunoassays for Point-of-Care-Testing. J. Mater. Sci. Technol. 2020, 60, 90–104. [Google Scholar] [CrossRef]
- Zeng, L.; Lie, P.; Fang, Z.; Xiao, Z. Lateral Flow Biosensors for the Detection of Nucleic Acid. In Nucleic Acid Detection: Methods and Protocols; Kolpashchikov, D.M., Gerasimova, Y.V., Eds.; Humana Press: Totowa, NJ, USA, 2013; pp. 161–167. [Google Scholar]
- Mukama, O.; Wu, J.; Li, Z.; Liang, Q.; Yi, Z.; Lu, X.; Liu, Y.; Liu, Y.; Hussain, M.; Makafe, G.G.; et al. An ultrasensitive and specific point-of-care CRISPR/Cas12 based lateral flow biosensor for the rapid detection of nucleic acids. Biosens. Bioelectron. 2020, 159, 112143. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Li, F.; Chen, Q.; Wu, J.; Duan, J.; Lei, X.; Zhang, Y.; Zhao, D.; Bu, Z.; Yin, H. Rapid detection of African swine fever virus using Cas12a-based portable paper diagnostics. Cell Discov. 2020, 6, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, J.; Lin, H.; Li, H.; Zhou, Y.; Liu, J.; Zhong, G.; Wu, L.; Jiang, W.; Du, H.; Yang, J.; et al. Cas12a-Based On-Site and Rapid Nucleic Acid Detection of African Swine Fever. Front. Microbiol. 2019, 10, 2830. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence (5′-3′) |
|---|---|
| Target ASFV DNA | TGTGAACAAAAGTTATGGGAAACCCGACCCCGAACCCACTTTGA GTCAAATCGAAGAAACACATTTGGTTCATTTTAATGCGCATTTT AAGCCTTATGTTCCAGTAGGGTTTGAATACAATAAAGTACGCCC GCATACGGGTACCCCCACCTTGGGAAACAAGCTTACCTTTGGTAT TCCCCAGTACGGAGACTTTTTCCATGATATGGTGGGCCACCATAT ATTGGGTGCATGTCATTCGTCCTGGCAGGATGCTCCGATTCAGGG CAC |
| PCR primers [11] | |
| Forward | AGTTATGGGAAACCCGACCC |
| Reverse | CCCTGAATCGGAGCATCCT |
| crRNA | UAAUUUCUACUAAGUGUAGAUATGCGCATTTTAAGCCTTATG TTC |
| Activator (24 bp) | TTTGGTTCATTTTAATGCGCATTTT |
| Reporter | Biotin-TTTTTTTTATT |
| Test-line probe (38 bases) | AATAAAAAAAAAAAAAAAAAATAAAAAAAAAAAAAAAA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, J.; Mukama, O.; Wu, W.; Li, Z.; Habimana, J.D.D.; Zhang, Y.; Zeng, R.; Nie, C.; Zeng, L. A CRISPR/Cas12a Based Universal Lateral Flow Biosensor for the Sensitive and Specific Detection of African Swine-Fever Viruses in Whole Blood. Biosensors 2020, 10, 203. https://0-doi-org.brum.beds.ac.uk/10.3390/bios10120203
Wu J, Mukama O, Wu W, Li Z, Habimana JDD, Zhang Y, Zeng R, Nie C, Zeng L. A CRISPR/Cas12a Based Universal Lateral Flow Biosensor for the Sensitive and Specific Detection of African Swine-Fever Viruses in Whole Blood. Biosensors. 2020; 10(12):203. https://0-doi-org.brum.beds.ac.uk/10.3390/bios10120203
Chicago/Turabian StyleWu, Jinghua, Omar Mukama, Wei Wu, Zhiyuan Li, Jean De Dieu Habimana, Yinghui Zhang, Rong Zeng, Chengrong Nie, and Lingwen Zeng. 2020. "A CRISPR/Cas12a Based Universal Lateral Flow Biosensor for the Sensitive and Specific Detection of African Swine-Fever Viruses in Whole Blood" Biosensors 10, no. 12: 203. https://0-doi-org.brum.beds.ac.uk/10.3390/bios10120203