
Genomic Insights into the Distribution and Phylogeny of Glycopeptide Resistance Determinants within the Actinobacteria Phylum

 , and
, and
Abstract
:1. Introduction
2. Results
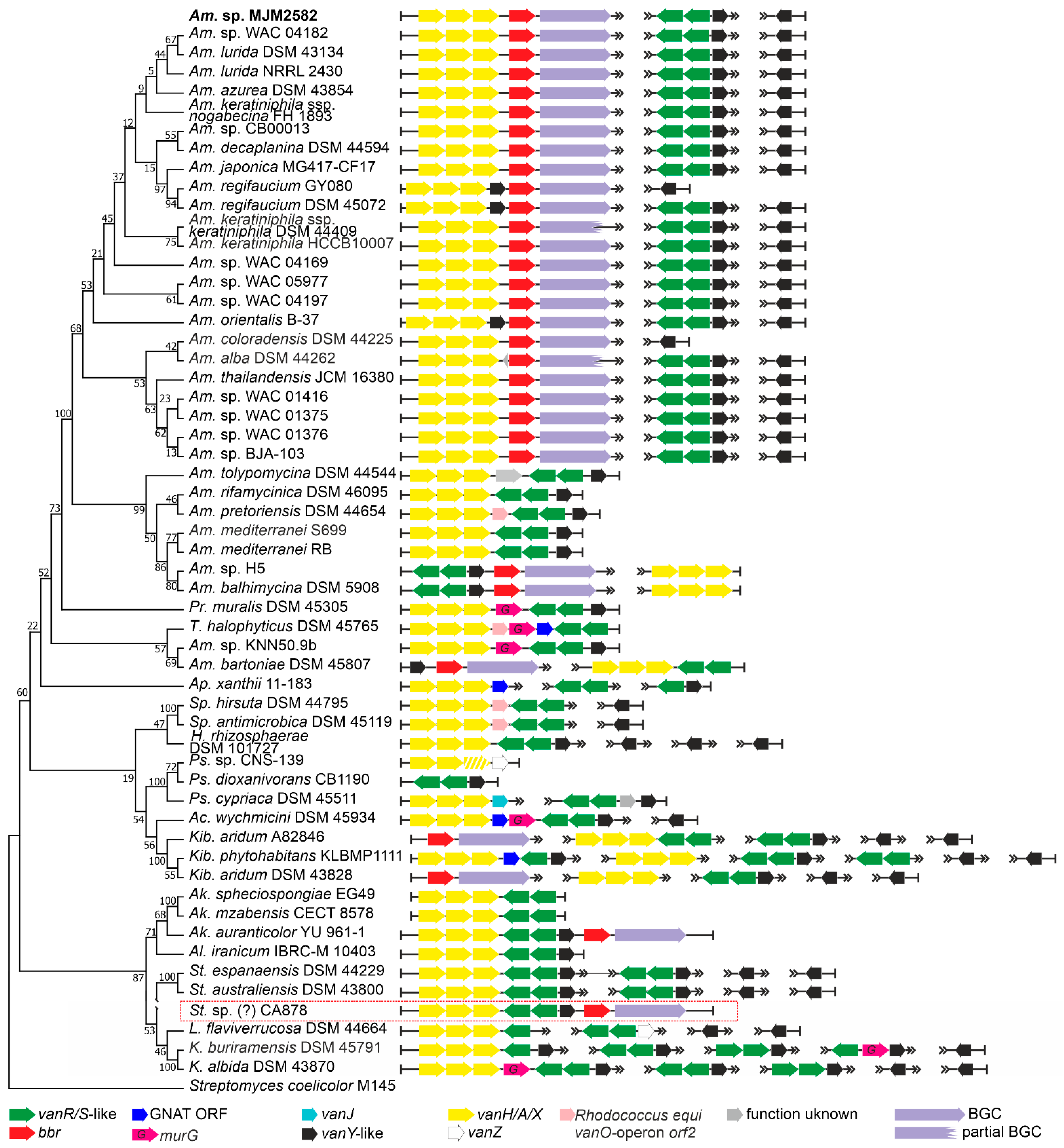
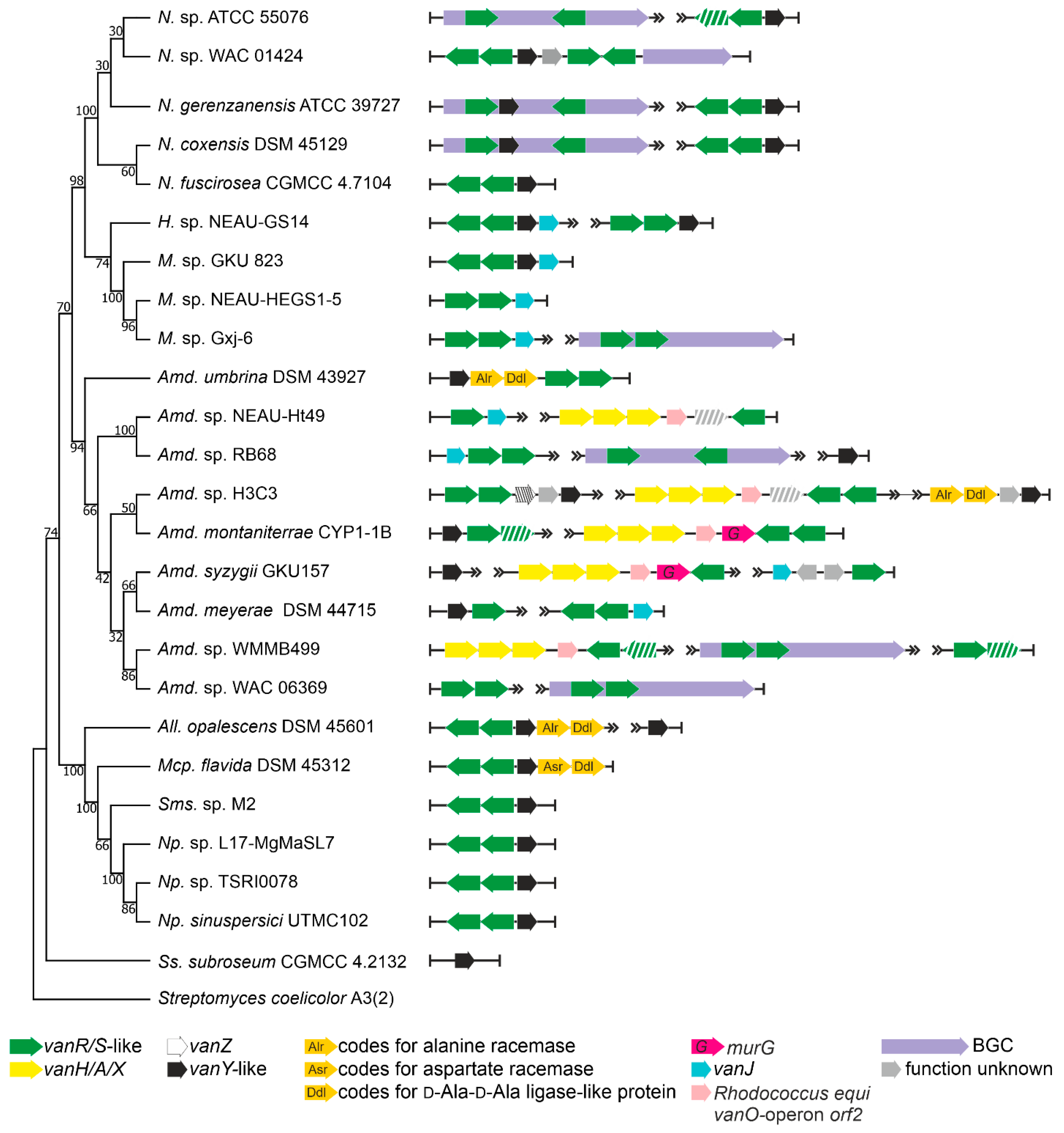
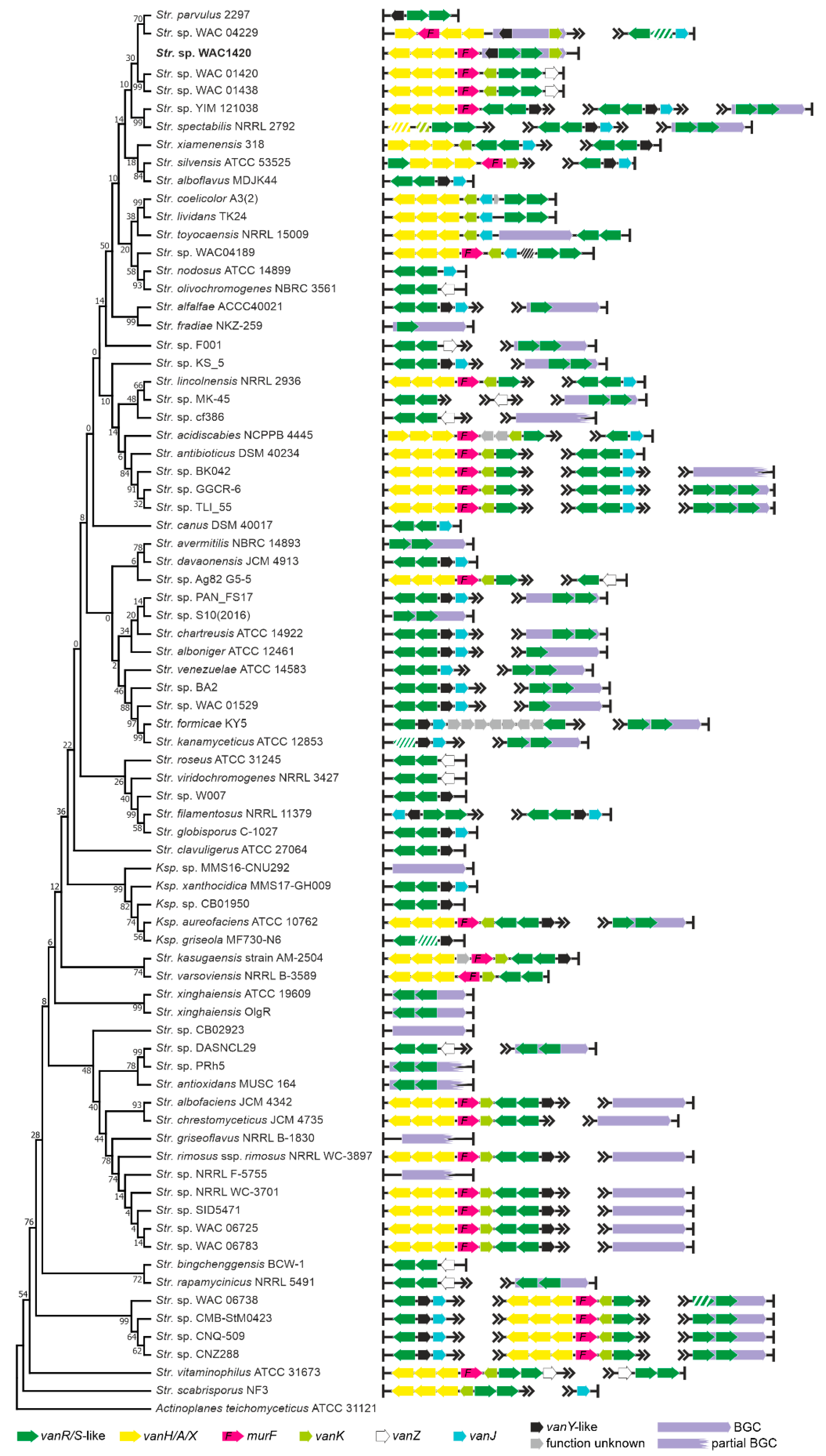
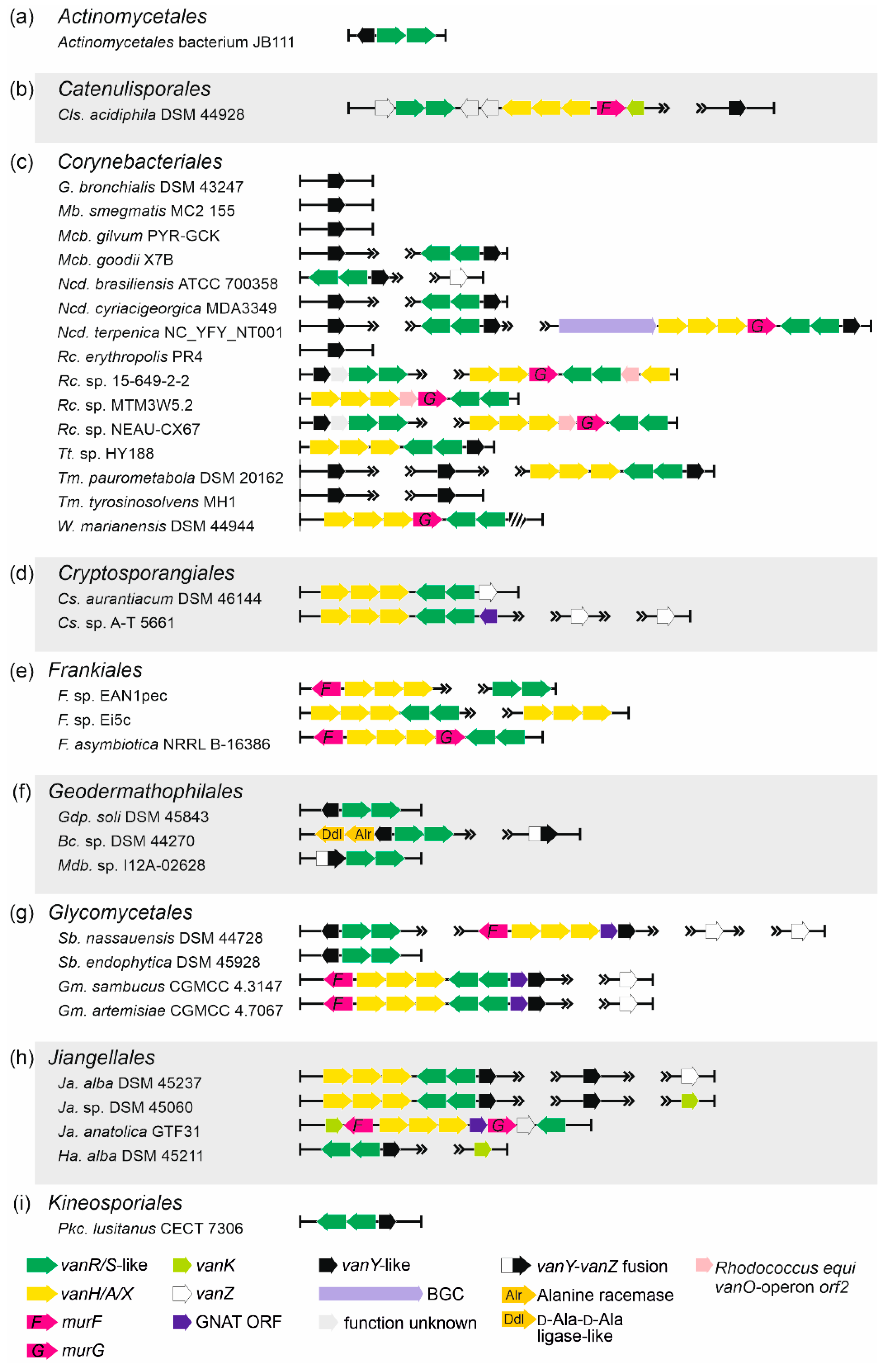
2.1. Organization of vlgs in GPA Producers and Beyond
2.1.1. Order Pseudonocardiales
2.1.2. Order Streptosporangiales
2.1.3. Order Micromonosporales
2.1.4. Order Streptomycetales
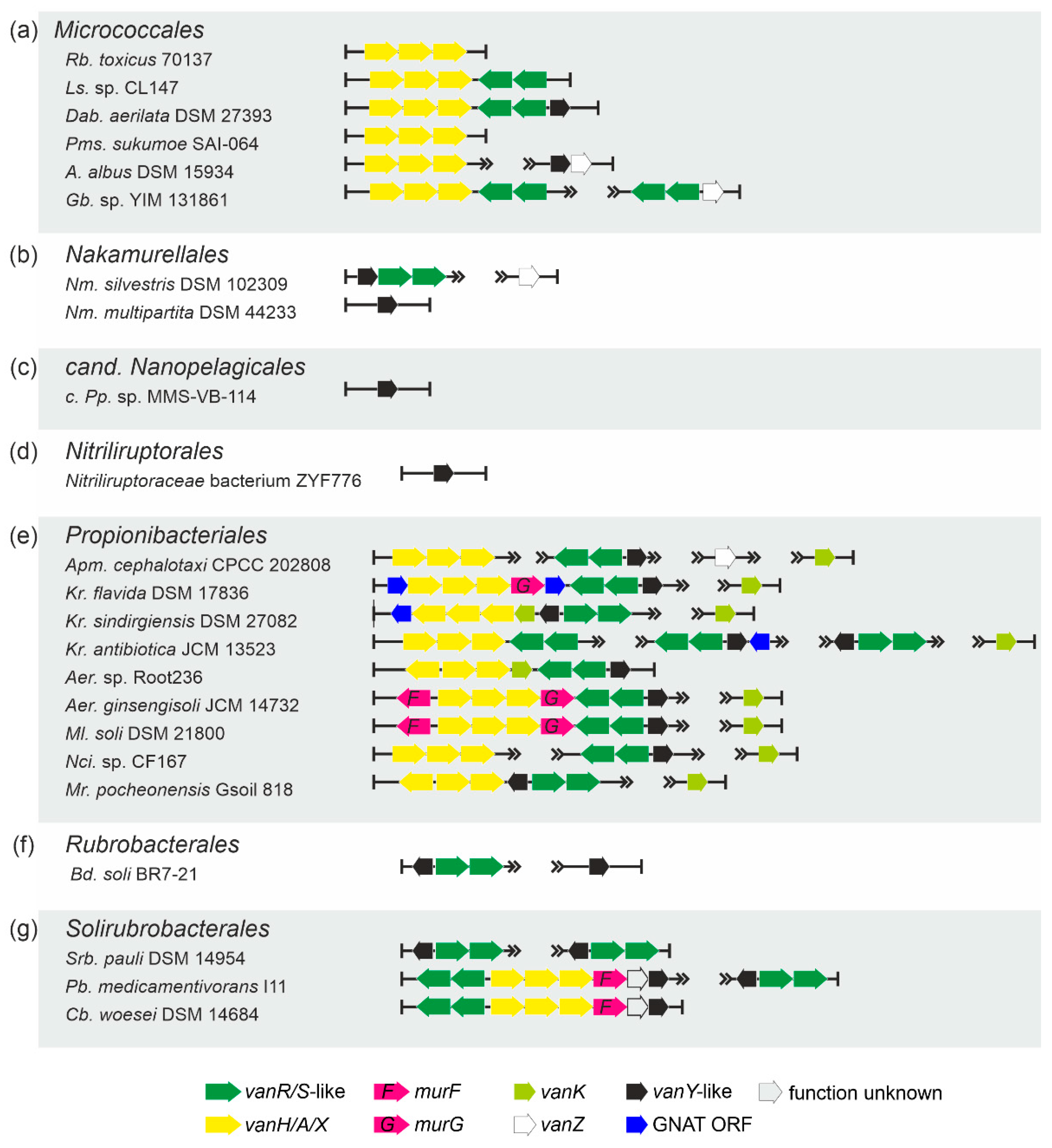
2.1.5. Occurrence of vlgs in GPA Non-Producing Groups
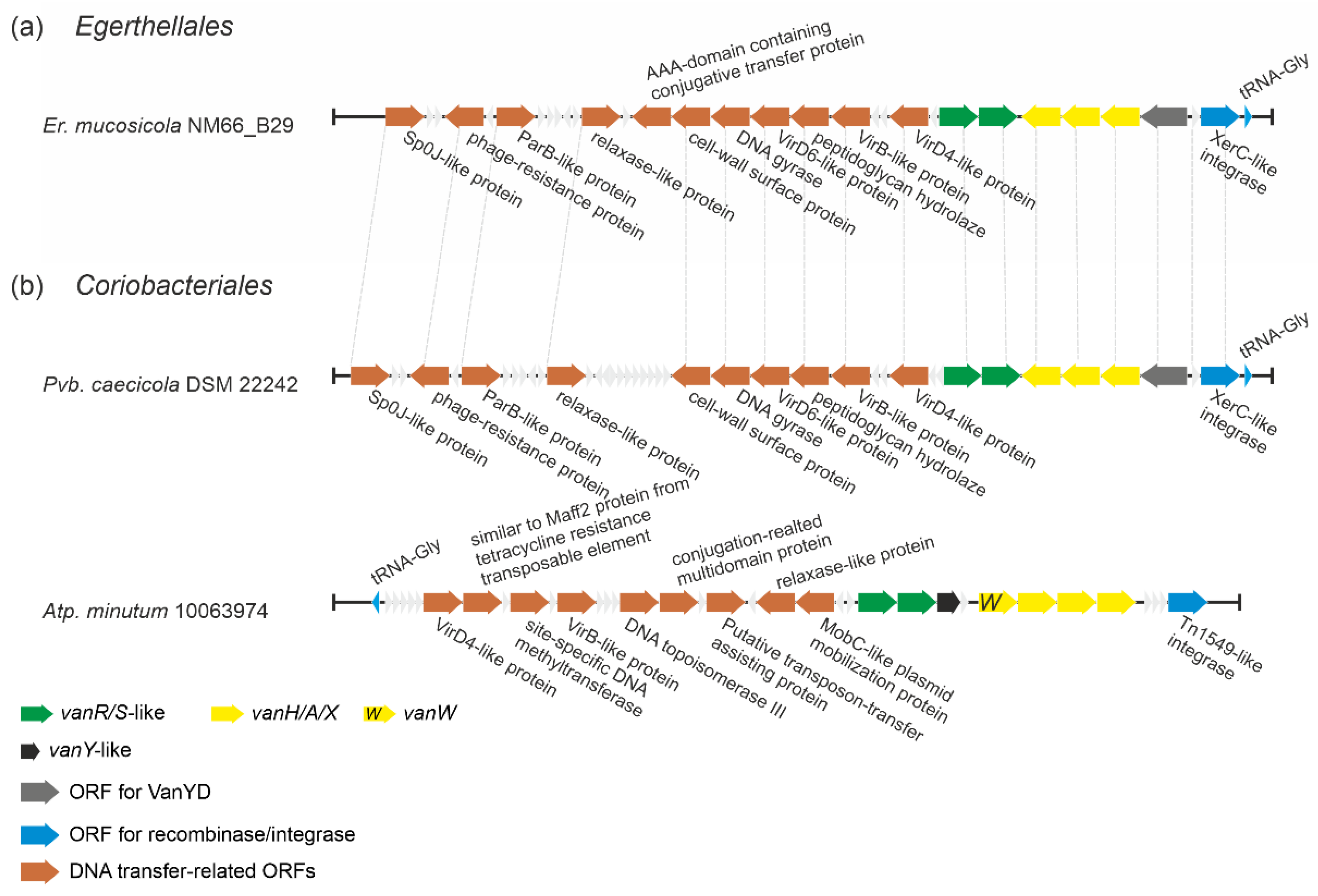
2.1.6. Putatively Novel Transposable Elements Carrying vlgs in Eggerthellales and Coriobacteriales spp.
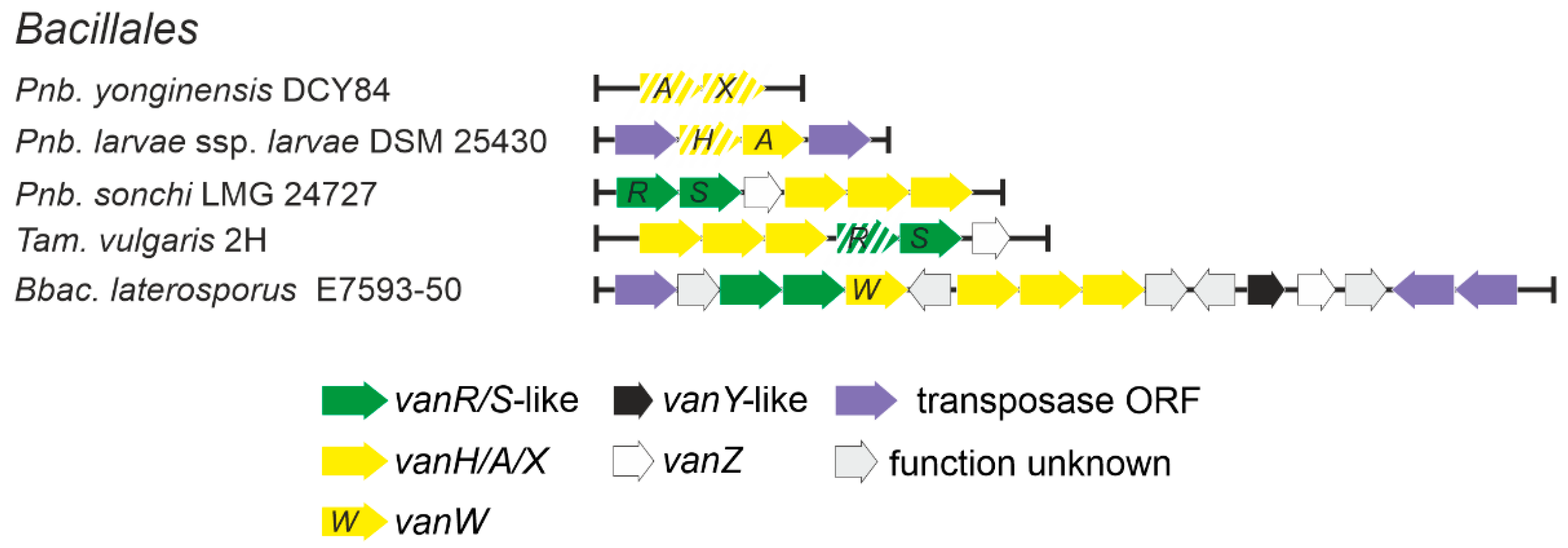
2.1.7. Occurrence of vlgs in Bacillales spp. (Firmicutes phylum)
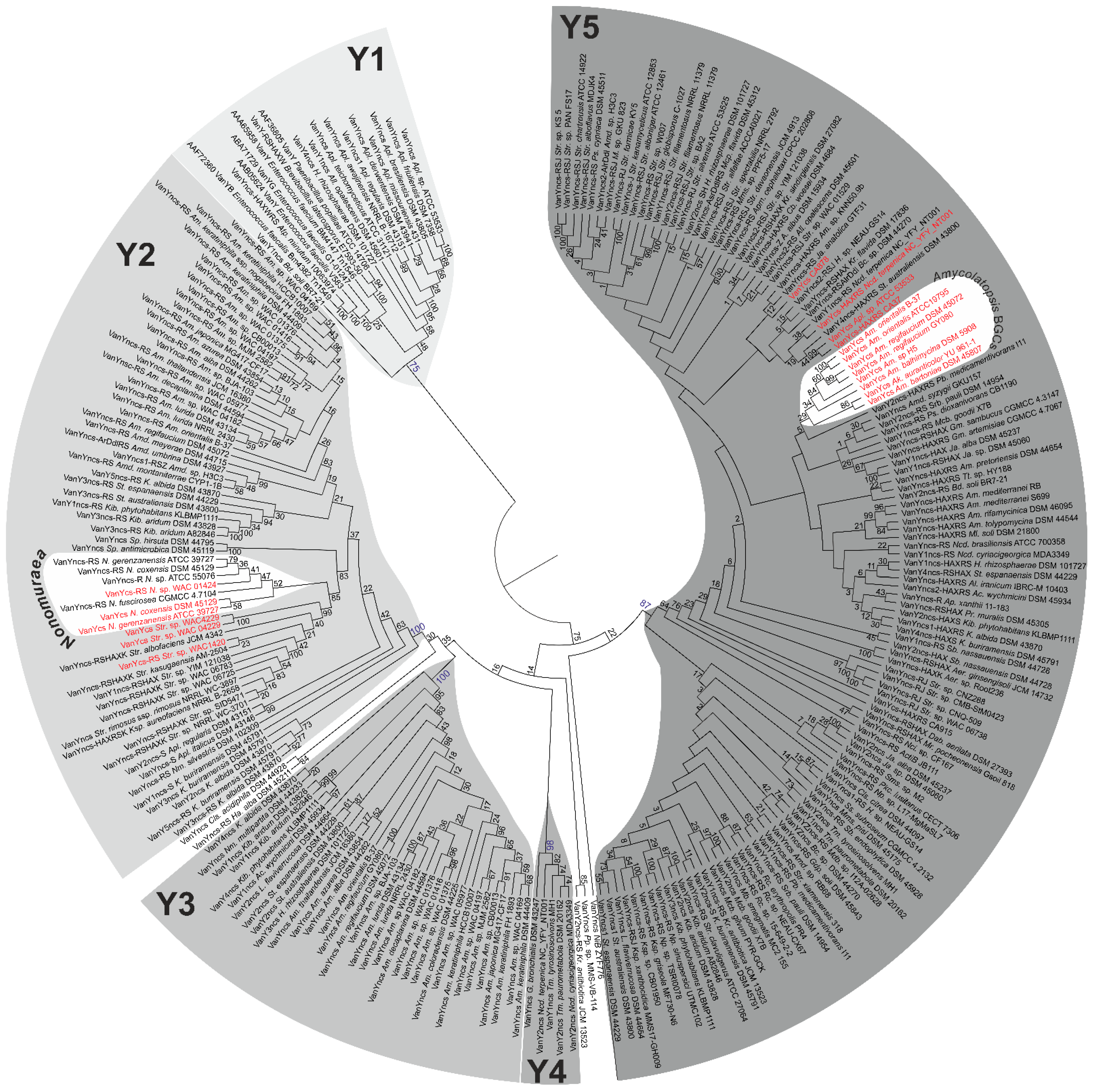
2.2. Phylogeny of VanY-like Carboxypeptidases
2.3. Phylogeny of VanHAX
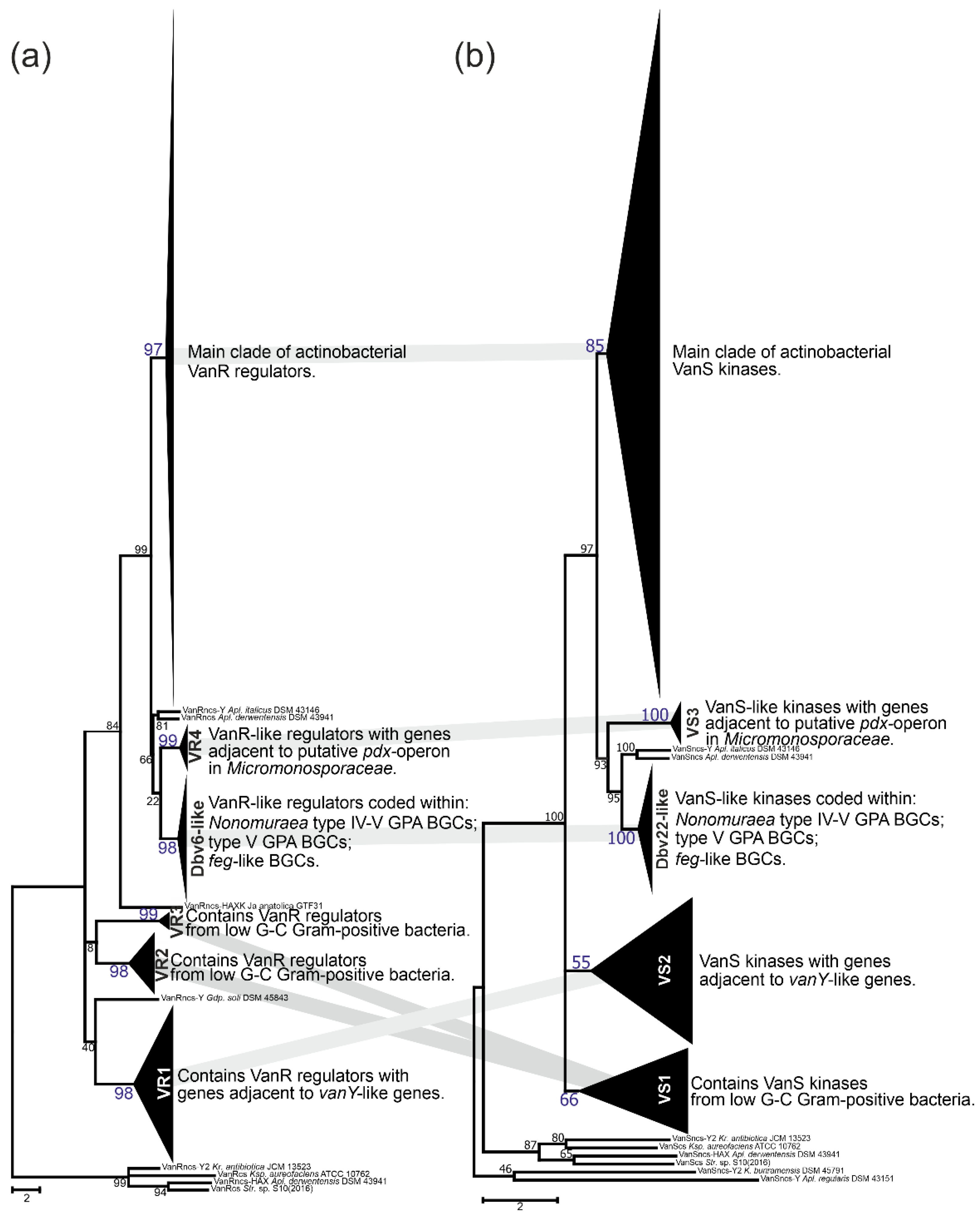
2.4. Phylogeny of VanRS-like Two-Component Regulatory Pairs
3. Discussion
4. Methods
4.1. Routine Analysis of Nucleic and Amino Acid Sequences
4.2. vlgs Search Pipeline
4.3. Search for Putative GPA-like BGCs
4.4. Phylogenetic Reconstruction
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fleming, A. On the antibacterial action of cultures of a penicillium, with special reference to their use in the isolation of B. influenzae. 1929. Bull. World Health Organ. 2001, 79, 780–790. [Google Scholar] [PubMed]
- O’ Neil, J. Review on Antibiotic resistance. Antimicrobial resistance: Tackling a crisis for the health and wealth of nations. Health Wealth Nations 2014, 1–16. Available online: https://wellcomecollection.org/works/rdpck35v (accessed on 21 October 2021).
- Ghosh, S.; Bornman, C.; Zafer, M.M. Antimicrobial resistance threats in the emerging COVID-19 pandemic: Where do we stand? J. Infect. Public Health 2021, 14, 555–560. [Google Scholar] [CrossRef]
- Marcone, G.L.; Binda, E.; Berini, F.; Marinelli, F. Old and new glycopeptide antibiotics: From product to gene and back in the post-genomic era. Biotechnol. Adv. 2018, 36, 534–554. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, K.C.; Boddy, C.N.C.; Bräse, S.; Winssinger, N. Chemistry, biology, and medicine of the glycopeptide antibiotics. Angew. Chem. Int. Ed. 1999, 38, 2096–2152. [Google Scholar] [CrossRef]
- Sarkar, P.; Yarlagadda, V.; Ghosh, C.; Haldar, J. A review on cell wall synthesis inhibitors with an emphasis on glycopeptide antibiotics. Medchemcomm 2017, 8, 516–533. [Google Scholar] [CrossRef] [PubMed]
- Vollmer, W.; Blanot, D.; De Pedro, M.A. Peptidoglycan structure and architecture. FEMS Microbiol. Rev. 2008, 32, 149–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perkins, H.R. Specificity of combination between mucopeptide precursors and vancomycin or ristocetin. Biochem. J. 1969, 111, 195–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nitanai, Y.; Kikuchi, T.; Kakoi, K.; Hanamaki, S.; Fujisawa, I.; Aoki, K. Crystal structures of the complexes between vancomycin and cell-wall precursor analogs. J. Mol. Biol. 2009, 385, 1422–1432. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.H. The glycopeptide story-how to kill the deadly “Superbugs”. Nat. Prod. Rep. 1996, 13, 469–477. [Google Scholar] [CrossRef]
- Marschall, E.; Cryle, M.J.; Tailhades, J. Biological, chemical, and biochemical strategies for modifying glycopeptide antibiotics. J. Biol. Chem. 2019, 294, 18769–18783. [Google Scholar] [CrossRef] [Green Version]
- Parenti, F.; Cavalleri, B. Proposal to name the vancomycin-ristocetin like glycopeptides as dalbaheptides. J. Antibiot. 1989, 42, 1882–1883. [Google Scholar] [CrossRef] [PubMed]
- Yushchuk, O.; Binda, E.; Marinelli, F. Glycopeptide antibiotic resistance genes: Distribution and function in the producer actinomycetes. Front. Microbiol. 2020, 11, 1173. [Google Scholar] [CrossRef] [PubMed]
- Binda, E.; Marinelli, F.; Marcone, G.L. Old and new glycopeptide antibiotics: Action and resistance. Antibiotics 2014, 3, 572–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arthur, M.; Quintiliani, R.J. Regulation of VanA-and VanB-type glycopeptide resistance in enterococci. Antimicrob. Agents Chemother. 2001, 45, 375–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arthur, M.; Molinas, C.; Bugg, T.D.H.; Wright, G.D.; Walsh, C.T.; Courvalin, P. Evidence for in vivo incorporation of d-lactate into peptidoglycan precursors of vancomycin-resistant enterococci. Antimicrob. Agents Chemother. 1992, 36, 867–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stegmann, E.; Frasch, H.J.; Kilian, R.; Pozzi, R. Self-resistance mechanisms of actinomycetes producing lipid II-targeting antibiotics. Int. J. Med. Microbiol. 2015, 305, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Waglechner, N.; McArthur, A.G.; Wright, G.D. Phylogenetic reconciliation reveals the natural history of glycopeptide antibiotic biosynthesis and resistance. Nat. Microbiol. 2019, 4, 1862–1871. [Google Scholar] [CrossRef] [PubMed]
- Culp, E.J.; Waglechner, N.; Wang, W.; Fiebig-Comyn, A.A.; Hsu, Y.P.; Koteva, K.; Sychantha, D.; Coombes, B.K.; Van Nieuwenhze, M.S.; Brun, Y.V.; et al. Evolution-guided discovery of antibiotics that inhibit peptidoglycan remodelling. Nature 2020, 578, 582–587. [Google Scholar] [CrossRef]
- Xu, M.; Wang, W.; Waglechner, N.; Culp, E.J.; Guitor, A.K.; Wright, G.D. GPAHex-A synthetic biology platform for Type IV–V glycopeptide antibiotic production and discovery. Nat. Commun. 2020, 11, 5232. [Google Scholar] [CrossRef]
- Mitchell, S.J.; Verma, D.; Griswold, K.E.; Bailey-Kellogg, C. Building blocks and blueprints for bacterial autolysins. PLoS Comput. Biol. 2021, 17, e1008889. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.V.; Wuest, W.M. Phylogeny-guided approach yields glycopeptides with unique action. Trends Pharmacol. Sci. 2020, 41, 297–299. [Google Scholar] [CrossRef] [PubMed]
- Gonsior, M.; Mühlenweg, A.; Tietzmann, M.; Rausch, S.; Poch, A.; Süssmuth, R.D. Biosynthesis of the peptide antibiotic feglymycin by a linear nonribosomal peptide synthetase mechanism. Chem. Biol. Chem. 2015, 16, 2610–2614. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.J.; Paget, M.S.B.; Buttner, M.J. A signal transduction system in Streptomyces coelicolor that activates expression of a putative cell wall glycan operon in response to vancomycin and other cell wall-specific antibiotics. Mol. Microbiol. 2002, 44, 1199–1211. [Google Scholar] [CrossRef] [Green Version]
- Patel, R.; Piper, K.; Cockerill, F.R.; Steckelberg, J.M.; Yousten, A.A. The biopesticide Paenibacillus popilliae has a vancomycin resistance gene cluster homologous to the enterococcal VanA vancomycin resistance gene cluster. Antimicrob. Agents Chemother. 2000, 44, 705–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontana, R.; Ligozzi, M.; Pedrotti, C.; Padovani, E.M.; Cornaglia, G. Vancomycin-resistant Bacillus circulans carrying the vanA gene responsible for vancomycin resistance in enterococci. Eur. J. Clin. Microbiol. Infect. Dis. 1997, 16, 473–474. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C.G.; Broadhead, G.; Leskiw, B.K.; Wright, G.D. d-Ala-d-Ala ligases from glycopeptide antibiotic-producing organisms are highly homologous to the enterococcal vancomycin-resistance ligases VanA and VanB. Proc. Natl. Acad. Sci. USA 1997, 94, 6480–6483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aminov, R.I.; Mackie, R.I. Evolution and ecology of antibiotic resistance genes. FEMS Microbiol. Lett. 2007, 271, 147–161. [Google Scholar] [CrossRef] [PubMed]
- One Health. Available online: https://www.who.int/news-room/q-a-detail/one-health (accessed on 21 October 2021).
- Hutchings, M.I.; Hong, H.J.; Buttner, M.J. The vancomycin resistance VanRS two-component signal transduction system of Streptomyces coelicolor. Mol. Microbiol. 2006, 59, 923–935. [Google Scholar] [CrossRef]
- Hong, H.J.; Hutchings, M.I.; Neu, J.M.; Wright, G.D.; Paget, M.S.B.; Buttner, M.J. Characterization of an inducible vancomycin resistance system in Streptomyces coelicolor reveals a novel gene (vanK) required for drug resistance. Mol. Microbiol. 2004, 52, 1107–1121. [Google Scholar] [CrossRef] [PubMed]
- Adamek, M.; Alanjary, M.; Sales-Ortells, H.; Goodfellow, M.; Bull, A.T.; Winkler, A.; Wibberg, D.; Kalinowski, J.; Ziemert, N. Comparative genomics reveals phylogenetic distribution patterns of secondary metabolites in Amycolatopsis species. BMC Genom. 2018, 19, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Shawky, R.M.; Puk, O.; Wietzorrek, A.; Pelzer, S.; Takano, E.; Wohlleben, W.; Stegmann, E. The border sequence of the balhimycin biosynthesis gene cluster from Amycolatopsis balhimycina contains bbr, encoding a StrR-like pathway-specific regulator. J. Mol. Microbiol. Biotechnol. 2007, 13, 76–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gudeta, D.D.; Moodley, A.; Bortolaia, V.; Guardabassi, L. VanO, a new glycopeptide resistance operon in environmental Rhodococcus equi isolates. Antimicrob. Agents Chemother. 2014, 58, 1768–1770. [Google Scholar] [CrossRef] [Green Version]
- Mengin-Lecreulx, D.; Texier, L.; Rousseau, M.; Van Heijenoort, J. The murG gene of Escherichia coli codes for the UDP-N-acetylglucosamine: N-acetylmuramyl-(pentapeptide) pyrophosphoryl-undecaprenol N-acetylglucosamine transferase involved in the membrane steps of peptidoglycan synthesis. J. Bacteriol. 1991, 173, 4625–4636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banik, J.J.; Craig, J.W.; Calle, P.Y.; Brady, S.F. Tailoring enzyme-rich environmental DNA clones: A source of enzymes for generating libraries of unnatural natural products. J. Am. Chem. Soc. 2010, 132, 15661–15670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstein, B.P.; Selva, E.; Gastaldo, L.; Berti, M.; Pallanza, R.; Ripamonti, F.; Ferrari, P.; Denaro, M.; Arioli, V.; Cassani, G. A40926, a new glycopeptide antibiotic with anti-Neisseria activity. Antimicrob. Agents Chemother. 1987, 31, 1961–1966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binda, E.; Marcone, G.L.; Pollegioni, L.; Marinelli, F. Characterization of VanYn, a novel d, d-peptidase/d, d-carboxypeptidase involved in glycopeptide antibiotic resistance in Nonomuraea sp. ATCC 39727. FEBS J. 2012, 279, 3203–3213. [Google Scholar] [CrossRef]
- Binda, E.; Marcone, G.L.; Berini, F.; Pollegioni, L.; Marinelli, F. Streptomyces spp. as efficient expression system for a d,d-peptidase/d,d-carboxypeptidase involved in glycopeptide antibiotic resistance. BMC Biotechnol. 2013, 13, 24. [Google Scholar] [CrossRef]
- Yushchuk, O.; Vior, N.M.; Andreo-Vidal, A.; Berini, F.; Rückert, C.; Busche, T.; Binda, E.; Kalinowski, J.; Truman, A.W.; Marinelli, F. Genomic-led discovery of a novel glycopeptide antibiotic by Nonomuraea coxensis DSM 45129. ACS Chem. Biol. 2021, 16, 915–928. [Google Scholar] [CrossRef]
- Nazari, B.; Forneris, C.C.; Gibson, M.I.; Moon, K.; Schramma, K.R.; Seyedsayamdost, M.R. Nonomuraea sp. ATCC 55076 harbours the largest actinomycete chromosome to date and the kistamicin biosynthetic gene cluster. Medchemcomm 2017, 8, 780–788. [Google Scholar] [CrossRef] [Green Version]
- Yushchuk, O.; Ostash, B.; Truman, A.W.; Marinelli, F.; Fedorenko, V. Teicoplanin biosynthesis: Unraveling the interplay of structural, regulatory, and resistance genes. Appl. Microbiol. Biotechnol. 2020, 104, 3279–3291. [Google Scholar] [CrossRef]
- Bardone, M.R.; Paternoster, M.; Coronelli, C. Teichomycins, new antibiotics from Actinoplanes teichomyceticus nov. sp. II. Extraction and chemical characterization. J. Antibiot. 1978, 31, 170–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yim, G.; Kalan, L.; Koteva, K.; Thaker, M.N.; Waglechner, N.; Tang, I.; Wright, G.D. Harnessing the synthetic capabilities of glycopeptide antibiotic tailoring enzymes: Characterization of the UK-68, 597 biosynthetic cluster. Chem. Biol. Chem. 2014, 15, 2613–2623. [Google Scholar] [CrossRef]
- Debono, M.; Merkel, K.E.; Molloy, R.M.; Barnhart, M.; Prestí, E.; Hunt, A.H.; Hamill, R.L. Actaplanin, new glycopeptide antibiotics produced by Actinoplanes missouriensis the isolation and preliminary chemical characterization of actaplanin. J. Antibiot. 1984, 37, 85–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yushchuk, O.; Homoniuk, V.; Ostash, B.; Marinelli, F.; Fedorenko, V. Genetic insights into the mechanism of teicoplanin self-resistance in Actinoplanes teichomyceticus. J. Antibiot. 2020, 73, 255–259. [Google Scholar] [CrossRef]
- Owen, J.G.; Reddy, B.V.B.; Ternei, M.A.; Charlop-Powers, Z.; Calle, P.Y.; Kim, J.H.; Brady, S.F. Mapping gene clusters within arrayed metagenomic libraries to expand the structural diversity of biomedically relevant natural products. Proc. Natl. Acad. Sci. USA 2013, 110, 11797–11802. [Google Scholar] [CrossRef] [Green Version]
- Pootoolal, J.; Thomas, M.G.; Marshall, C.G.; Neu, J.M.; Hubbard, B.K.; Walsh, C.T.; Wright, G.D. Assembling the glycopeptide antibiotic scaffold: The biosynthesis of A47934 from Streptomyces toyocaensis NRRL15009. Proc. Natl. Acad. Sci. USA 2002, 99, 8962–8967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thaker, M.N.; Wang, W.; Spanogiannopoulos, P.; Waglechner, N.; King, A.M.; Medina, R.; Wright, G.D. Identifying producers of antibacterial compounds by screening for antibiotic resistance. Nat. Biotechnol. 2013, 31, 922–927. [Google Scholar] [CrossRef]
- Hong, H.J.; Hutchings, M.I.; Hill, L.M.; Buttner, M.J. The role of the novel fem protein VanK in vancomycin resistance in Streptomyces coelicolor. J. Biol. Chem. 2005, 280, 13055–13061. [Google Scholar] [CrossRef] [Green Version]
- Pathom-Aree, W.; Nogi, Y.; Sutcliffe, I.C.; Ward, A.C.; Horikoshi, K.; Bull, A.T.; Goodfellow, M. Williamsia marianensis sp. nov., a novel actinomycete isolated from the Mariana Trench. Int. J. Syst. Evol. Microbiol. 2006, 56, 1123–1126. [Google Scholar] [CrossRef] [Green Version]
- Hegstad, K.; Mikalsen, T.; Coque, T.M.; Werner, G.; Sundsfjord, A. Mobile genetic elements and their contribution to the emergence of antimicrobial resistant Enterococcus faecalis and Enterococcus faecium. Clin. Microbiol. Infect. 2010, 16, 541–554. [Google Scholar] [CrossRef] [PubMed]
- Garnier, F.; Taourit, S.; Glaser, P.; Courvalin, P.; Galimand, M. Characterization of transposon Tn1549, conferring VanB-type resistance in Enterococcus spp. Microbiol. 2000, 146, 1481–1489. [Google Scholar] [CrossRef] [Green Version]
- Handwerger, S.; Skoble, J. Identification of chromosomal mobile element conferring high-level vancomycin resistance in Enterococcus faecium. Antimicrob. Agents Chemother. 1995, 39, 2446–2453. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, P.E.; Ambur, O.H.; Casadewall, B.; Courvalin, P. The VanYD dd-carboxypeptidase of Enterococcus faecium BM4339 is a penicillin-binding protein. Microbiol. 2001, 147, 2571–2578. [Google Scholar] [CrossRef] [Green Version]
- Hopwood, D.A.; Wright, H.M. Transformation in Thermoactinomyces vulgaris. J. Gen. Microbiol. 1972, 71, 383–398. [Google Scholar] [CrossRef] [Green Version]
- Meziane-Cherif, D.; Stogios, P.J.; Evdokimova, E.; Savchenko, A.; Courvalin, P. Structural basis for the evolution of vancomycin resistance d,d-peptidases. Proc. Natl. Acad. Sci. USA 2014, 111, 5872–5877. [Google Scholar] [CrossRef] [Green Version]
- Rawlings, N.D.; Waller, M.; Barrett, A.J.; Bateman, A. MEROPS: The database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2014, 42, D503–D509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charlier, P.; Wery, J.-P.; Dideberg, O.; Frère, J.-M. Streptomyces albus G d-Ala-d-Ala carboxypeptidase. Handb. Met. 2006. [Google Scholar] [CrossRef]
- Marshall, C.G.; Lessard, I.A.D.; Park, I.S.; Wright, G.D. Glycopeptide antibiotic resistance genes in glycopeptide-producing organisms. Antimicrob. Agents Chemother. 1998, 42, 2215–2220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bussiere, D.E.; Pratt, S.D.; Katz, L.; Severin, J.M.; Holzman, T.; Park, C.H. The structure of VanX reveals a novel amino-dipeptidase involved in mediating transposon-based vancomycin resistance. Mol. Cell 1998, 2, 75–84. [Google Scholar] [CrossRef]
- Marcone, G.L.; Beltrametti, F.; Binda, E.; Carrano, L.; Foulston, L.; Hesketh, A.; Bibb, M.; Marinelli, F. Novel mechanism of glycopeptide resistance in the A40926 producer Nonomuraea sp. ATCC 39727. Antimicrob. Agents Chemother. 2010, 54, 2465–2472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilian, R.; Frasch, H.J.; Kulik, A.; Wohlleben, W.; Stegmann, E. The VanRS homologous two-component system VnlRSAb of the glycopeptide producer Amycolatopsis balhimycina activates transcription of the vanHAXSc genes in Streptomyces coelicolor, but not in A. balhimycina. Microb. Drug Resist. 2016, 22, 499–509. [Google Scholar] [CrossRef] [Green Version]
- Mainardi, J.-L.; Villet, R.; Bugg, T.D.; Mayer, C.; Arthur, M. Evolution of peptidoglycan biosynthesis under the selective pressure of antibiotics in Gram-positive bacteria. FEMS Microbiol. Rev. 2008, 32, 386–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Podmore, A.H.B.; Reynolds, P.E. Purification and characterization of VanXYc, a d,d-dipeptidase/d,d-carboxypeptidase in vancomycin-resistant Enterococcus gallinarum BM4174. Eur. J. Biochem. 2002, 269, 2740–2746. [Google Scholar] [CrossRef] [PubMed]
- Alduina, R.; Tocchetti, A.; Costa, S.; Ferraro, C.; Cancemi, P.; Sosio, M.; Donadio, S. A two-component regulatory system with opposite effects on glycopeptide antibiotic biosynthesis and resistance. Sci. Rep. 2020, 10, 6200. [Google Scholar] [CrossRef]
- D’Argenio, V.; Petrillo, M.; Pasanisi, D.; Pagliarulo, C.; Colicchio, R.; Talà, A.; De Biase, M.S.; Zanfardino, M.; Scolamiero, E.; Pagliuca, C.; et al. The complete 12 Mb genome and transcriptome of Nonomuraea gerenzanensis with new insights into its duplicated “magic” RNA polymerase. Sci. Rep. 2016, 6, 18. [Google Scholar] [CrossRef] [PubMed]
- Yushchuk, O.; Andreo-Vidal, A.; Marcone, G.L.; Bibb, M.; Marinelli, F.; Binda, E. New molecular tools for regulation and improvement of A40926 glycopeptide antibiotic production in Nonomuraea gerenzanensis ATCC 39727. Front. Microbiol. 2020, 11, 8. [Google Scholar] [CrossRef] [Green Version]
- Rioseras, B.; Yaguë, P.; López-Garciá, M.T.; Gonzalez-Quinõnez, N.; Binda, E.; Marinelli, F.; Manteca, A. Characterization of SCO4439, a d-alanyl-d-alanine carboxypeptidase involved in spore cell wall maturation, resistance, and germination in Streptomyces Coelicolor. Sci. Rep. 2016, 6, 21659. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Higgins, D.G. Clustal Omega. Curr. Protoc. Bioinform. 2014, 2014, 3.13.1–3.13.16. [Google Scholar] [CrossRef]
- Medema, M.H.; Takano, E.; Breitling, R. Detecting sequence homology at the gene cluster level with MultiGeneBlast. Mol. Biol. Evol. 2013, 30, 1218–1223. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Li, T.L.; Huang, F.; Haydock, S.F.; Mironenko, T.; Leadlay, P.F.; Spencer, J.B. Biosynthetic gene cluster of the glycopeptide antibiotic teicoplanin: Characterization of two glycosyltransferases and the key acyltransferase. Chem. Biol. 2004, 11, 107–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S.Y.; Medema, M.H.; Weber, T. AntiSMASH 5.0: Updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019, 47, W81–W87. [Google Scholar] [CrossRef] [Green Version]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Order | Number of Genome Assemblies Analyzed | Number of Genome Assembling Containing at Least One Vlg | Occurrence (%) | vanY-like | vanR-like | vanS-like | vanH | vanA | vanX |
|---|---|---|---|---|---|---|---|---|---|
| Acidimicrobiales | 216 | 0 | 0 | - | - | - | - | - | - |
| Actinomycetales | 223 | 1 | 0.45 | 1 | 1 | 1 | - | - | - |
| Actinopolysporales | 10 | 0 | 0 | - | - | - | - | - | - |
| Bifidobacteriales | 1028 | 0 | 0 | - | - | - | - | - | - |
| candidatus Actinomarinales | 214 | 0 | 0 | - | - | - | - | - | - |
| candidatus Nanopelagicales | 26 | 1 | 3.85 | 1 | - | - | - | - | - |
| Catenulisporales | 3 | 1 | 34 | 1 | 1 | 1 | 1 | 1 | 1 |
| Coriobacteriales | 217 | 2 | 0.9 | 1 | 2 | 2 | 2 | 2 | 2 |
| Corynebacteriales | 707 | 110 | 15.6 | 122 | 23 | 25 | 14 | 14 | 14 |
| Cryptosporangiales | 3 | 2 | 67 | - | 1 | 1 | 1 | 1 | 1 |
| Eggerthellales | 106 | 1 | 0.9 | - | 1 | 1 | 1 | 1 | 1 |
| Egibacterales | 3 | 0 | 0 | - | - | - | - | - | - |
| Frankiales | 46 | 7 | 15 | - | 6 | 7 | 8 | 8 | 8 |
| Gaiellales | 3 | 0 | 0 | - | - | - | - | - | - |
| Geodermatophilales | 60 | 11 | 18 | 12 | 10 | 9 | - | - | - |
| Glycomycetales | 12 | 11 | 92 | 10 | 10 | 10 | 8 | 8 | 8 |
| Jiangellales | 11 | 10 | 91 | 12 | 11 | 10 | 7 | 7 | 7 |
| Kineosporiales | 12 | 1 | 8.4 | 1 | 1 | 1 | - | - | - |
| Micrococcales | 1741 | 15 | 0.86 | 2 | 13 | 13 | 15 | 15 | 14 |
| Micromonosporales | 200 | 83 | 42 | 14 | 86 | 88 | 5 | 57 | 56 |
| Nakamurellales | 6 | 6 | 100 | 6 | 1 | 1 | - | - | - |
| Nitriliruptorales | 6 | 1 | 17 | 1 | - | - | - | - | - |
| Propionibacteriales | 593 | 16 | 2.7 | 13 | 16 | 16 | 16 | 16 | 16 |
| Pseudonocardiales | 243 | 135 | 56 | 141 | 129 | 139 | 105 | 105 | 105 |
| Rubrobacterales | 8 | 1 | 12.5 | 2 | 1 | 1 | - | - | - |
| Solirubrobacterales | 45 | 4 | 8.9 | 6 | 6 | 6 | 2 | 2 | 2 |
| Streptomycetales | 1138 | 418 | 37 | 126 | 414 | 429 | 93 | 92 | 94 |
| Streptosporangiales | 228 | 63 | 28 | 52 | 67 | 63 | 7 | 7 | 6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andreo-Vidal, A.; Binda, E.; Fedorenko, V.; Marinelli, F.; Yushchuk, O. Genomic Insights into the Distribution and Phylogeny of Glycopeptide Resistance Determinants within the Actinobacteria Phylum. Antibiotics 2021, 10, 1533. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics10121533
Andreo-Vidal A, Binda E, Fedorenko V, Marinelli F, Yushchuk O. Genomic Insights into the Distribution and Phylogeny of Glycopeptide Resistance Determinants within the Actinobacteria Phylum. Antibiotics. 2021; 10(12):1533. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics10121533
Chicago/Turabian StyleAndreo-Vidal, Andrés, Elisa Binda, Victor Fedorenko, Flavia Marinelli, and Oleksandr Yushchuk. 2021. "Genomic Insights into the Distribution and Phylogeny of Glycopeptide Resistance Determinants within the Actinobacteria Phylum" Antibiotics 10, no. 12: 1533. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics10121533