
Inhibition of MurA Enzyme from Escherichia coli and Staphylococcus aureus by Diterpenes from Lepechinia meyenii and Their Synthetic Analogs

 ,
,  , ,
, ,  and
and
Abstract
:1. Introduction
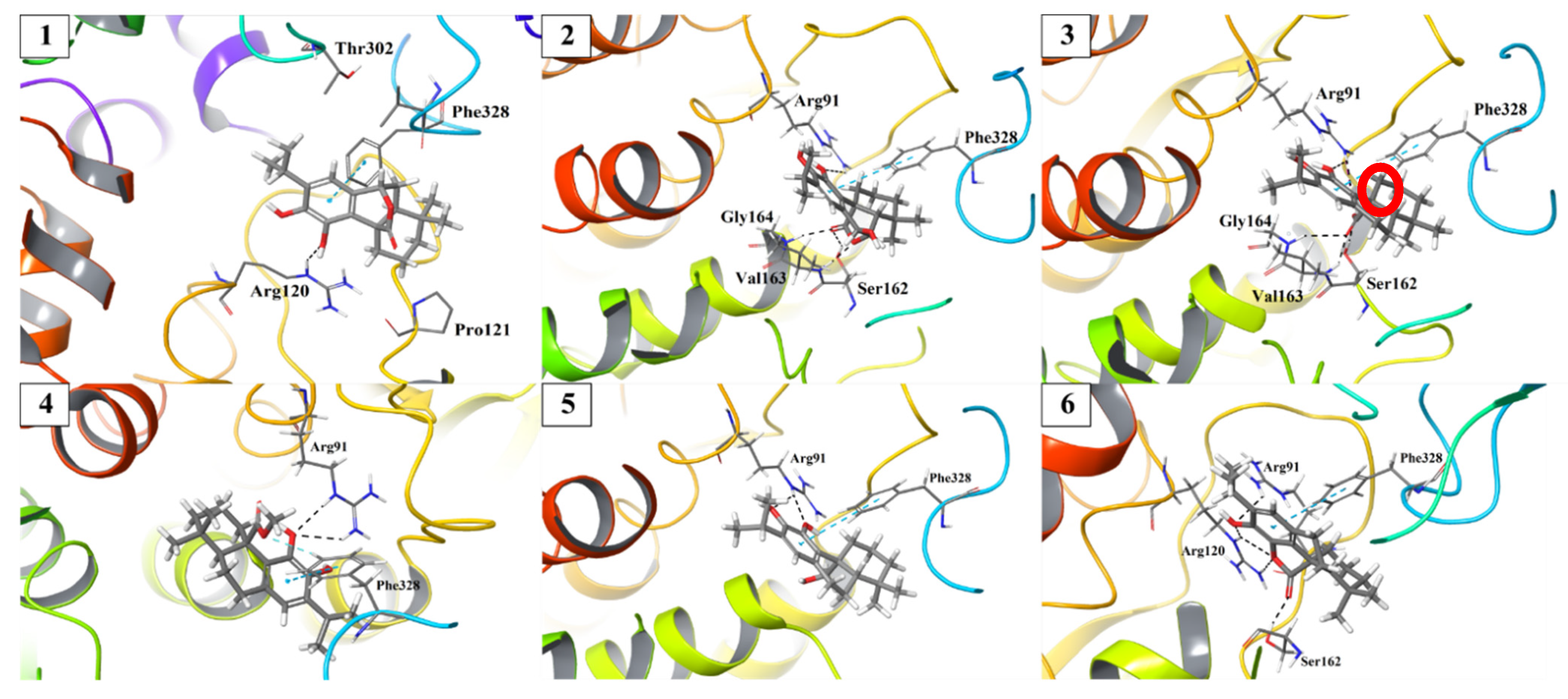
2. Results
3. Discussion
4. Materials and Methods
4.1. Reagents
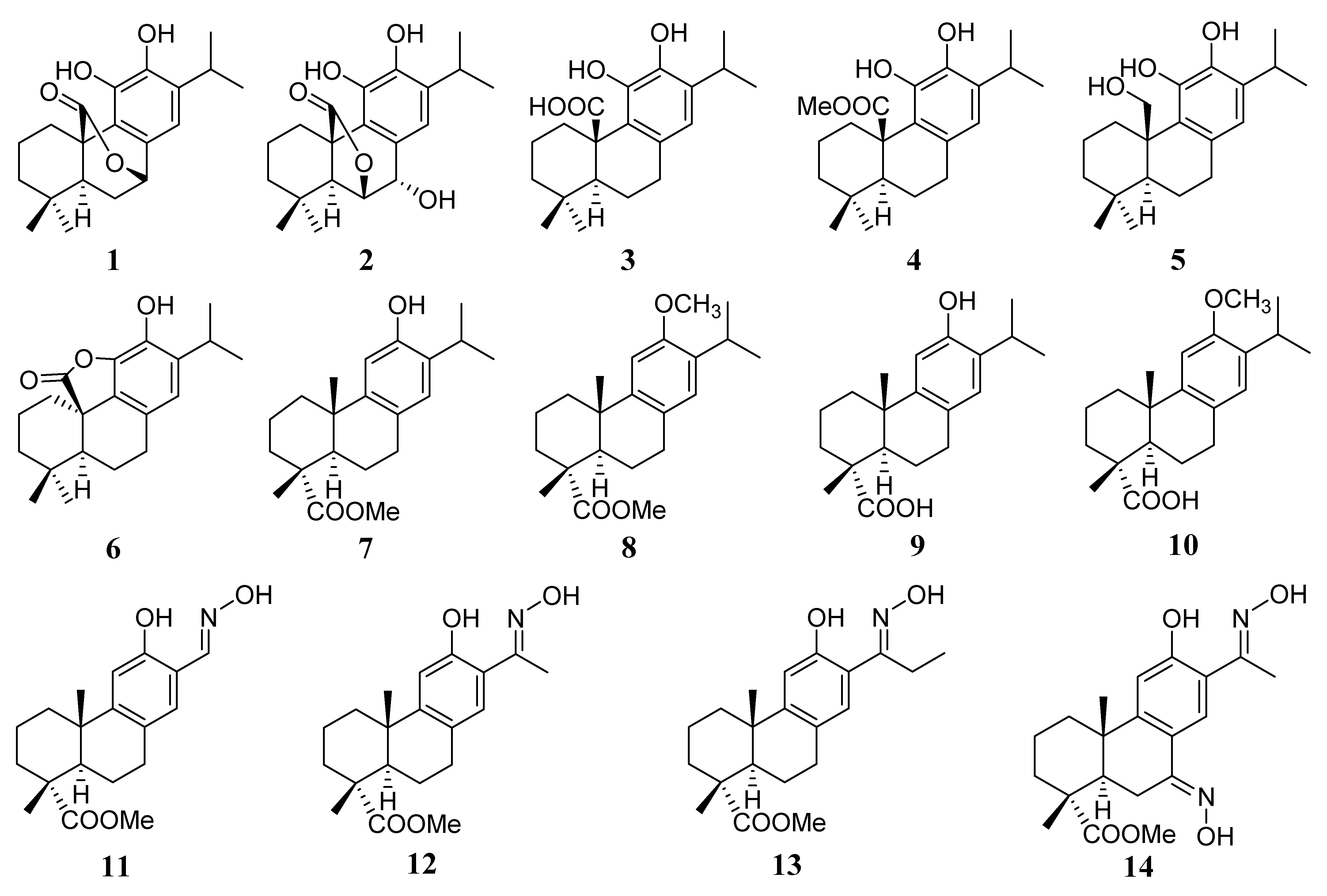
4.2. Synthesis of Carnosic and Dehydroabietic Acid Derivatives
4.3. Bacterial Isolates and Cultures
4.4. Antibacterial Susceptibility Testing
4.5. MurA and MurF Inhibition Assays
4.6. Preparation of Ligands
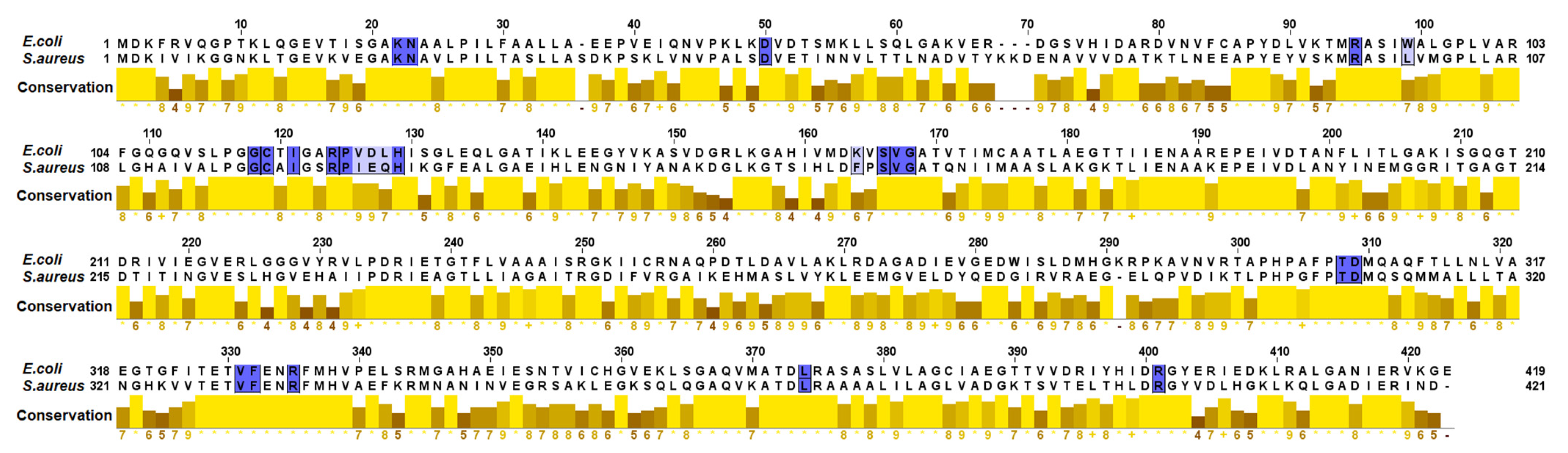
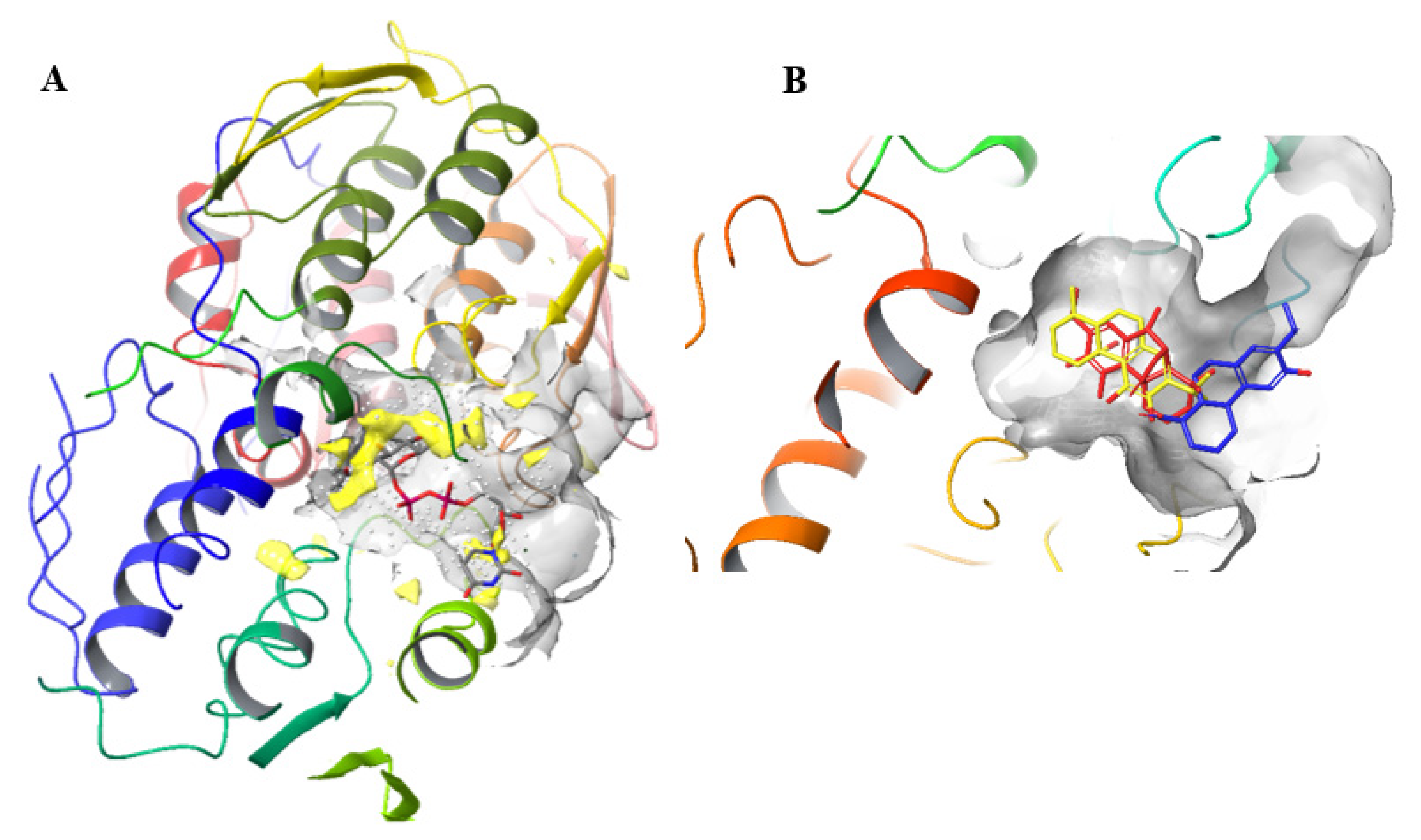
4.7. Comparison between the Active Sites of MurA
4.8. Protein Preparation
4.9. Molecular Modeling
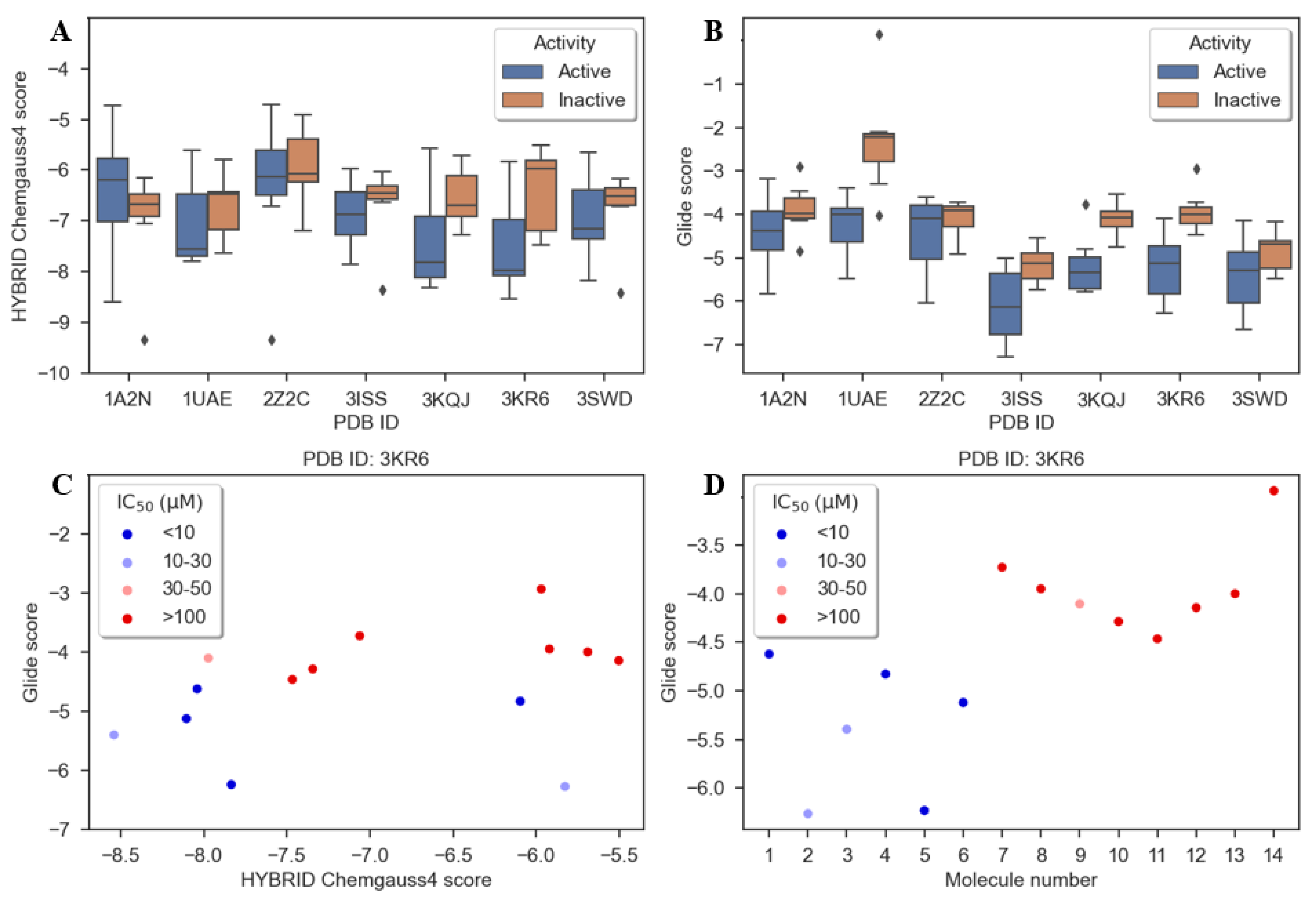
4.10. Docking with Hybrid
4.11. Docking with Glide
4.12. Generation of Figures
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hutchings, M.I.; Truman, A.W.; Wilkinson, B. Antibiotics: Past, present and future. Curr. Opin. Microbiol. 2019, 51, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Joray, M.B.; González, M.L.; Palacios, S.M.; Carpinella, M.C. Antibacterial activity of the plant-derived compounds 23-methyl-6-O-desmethylauricepyrone and (Z,Z)-5-(trideca-4,7-dienyl)resorcinol and their synergy with antibiotics against methicillin-susceptible and -resistant Staphylococcus aureus. J. Agric. Food Chem. 2011, 59, 11534–11542. [Google Scholar] [CrossRef]
- Funes Chabán, M.; Karagianni, C.; Joray, M.B.; Toumpa, D.; Sola, C.; Crespo, M.I.; Palacios, S.M.; Athanassopoulos, C.M.; Carpinella, M.C. Antibacterial effects of extracts obtained from plants of Argentina: Bioguided isolation of compounds from the anti-infectious medicinal plant Lepechinia meyenii. J. Ethnopharmacol. 2019, 239, 111930. [Google Scholar] [CrossRef]
- Liu, Y.; Breukink, E. The membrane steps of bacterial cell wall synthesis as antibiotic targets. Antibiotics 2016, 5, 28. [Google Scholar] [CrossRef]
- Laddomada, F.; Miyachiro, M.M.; Dessen, A. Structural insights into protein-protein interactions involved in bacterial cell wall biogenesis. Antibiotics 2016, 5, 14. [Google Scholar] [CrossRef] [Green Version]
- Egan, A.J.; Errington, J.; Vollmer, W. Regulation of peptidoglycan synthesis and remodelling. Nat. Rev. Microbiol. 2020, 18, 446–460. [Google Scholar] [CrossRef] [PubMed]
- Vollmer, W.; Blanot, D.; De Pedro, M.A. Peptidoglycan structure and architecture. FEMS Microbiol. Rev. 2008, 32, 149–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, P.; Yarlagadda, V.; Ghosh, C.; Haldar, J. A review on cell wall synthesis inhibitors with an emphasis on glycopeptide antibiotics. Med. Chem. Comm. 2017, 8, 516–533. [Google Scholar] [CrossRef]
- Mihalovits, L.M.; Ferenczy, G.G.; Keserű, G.M. Catalytic mechanism and covalent inhibition of UDP-N-acetylglucosamine enolpyruvyl transferase (MurA): Implications to the design of novel antibacterials. J. Chem. Inf. Model. 2019, 59, 5161–5173. [Google Scholar] [CrossRef]
- Rožman, K.; Lešnik, S.; Brus, B.; Hrast, M.; Sova, M.; Patin, D.; Barreteau, H.; Konc, J.; Janežič, D.; Gobec, S. Discovery of new MurA inhibitors using induced-fit simulation and docking. Bioorg. Med. Chem. Lett. 2017, 27, 944–949. [Google Scholar] [CrossRef] [PubMed]
- Jukič, M.; Gobec, S.; Sova, M. Reaching toward underexplored targets in antibacterial drug design. Drug Dev. Res. 2019, 80, 6–10. [Google Scholar] [CrossRef] [Green Version]
- Keeley, A.; Ábrányi-Balogh, P.; Hrast, M.; Imre, T.; Ilaš, J.; Gobec, S.; Keserű, G.M. Heterocyclic electrophiles as new MurA inhibitors. Arch. Pharm. 2018, 351, 1800184. [Google Scholar] [CrossRef] [PubMed]
- Falagas, M.E.; Athanasaki, F.; Voulgaris, G.L.; Triarides, N.A.; Vardakas, K.Z. Resistance to fosfomycin: Mechanisms, frequency and clinical consequences. Int. J. Antimicrob. Agents 2019, 53, 22–28. [Google Scholar] [CrossRef]
- Sorlozano-Puerto, A.; Lopez-Machado, I.; Albertuz-Crespo, M.; Martinez-Gonzalez, L.J.; Gutierrez-Fernandez, J. Characterization of fosfomycin and nitrofurantoin resistance mechanisms in Escherichia coli isolated in clinical urine samples. Antibiotics 2020, 9, 534. [Google Scholar] [CrossRef] [PubMed]
- Funes Chabán, M.; Antoniou, A.; Karagianni, C.; Toumpa, D.; Belén Joray, M.; Bocco, J.L.; Sola, C.; Athanassopoulos, C.; Carpinella, M.C. Synthesis and structure-activity relationships of novel abietane diterpenoids with activity against Staphylococcus aureus. Future Med. Chem. 2019, 11, 3109–3124. [Google Scholar] [CrossRef]
- Schrödinger Release 2021-3: Glide; Schrödinger, LLC: New York, NY, USA, 2021.
- McGann, M. FRED and HYBRID docking performance on standardized datasets. J. Comput. Aided Mol. Des. 2012, 26, 897–906. [Google Scholar] [CrossRef] [PubMed]
- Eschenburg, S.; Kabsch, W.; Healy, M.L.; Schönbrunn, E. A new view of the mechanisms of UDP-N-acetylglucosamine enolpyruvyl transferase (MurA) and 5-enolpyruvylshikimate-3-phosphate synthase (AroA) derived from X-ray structures of their tetrahedral reaction intermediate states. J. Biol. Chem. 2003, 278, 49215–49222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isa, M.A. Homology modeling and molecular dynamic simulation of UDP-N-acetylmuramoyl-l-alanine-d-glutamate ligase (MurD) from Mycobacterium tuberculosis H37Rv using in silico approach. Comput. Biol. Chem. 2019, 78, 116–126. [Google Scholar] [CrossRef]
- Joray, M.B.; del Rollán, M.R.; Ruiz, G.M.; Palacios, S.M.; Carpinella, M.C. Antibacterial activity of extracts from plants of central Argentina—Isolation of an active principle from Achyrocline satureioides. Planta Med. 2011, 77, 95–100. [Google Scholar] [CrossRef]
- Choi, U.; Lee, C.-R. Distinct Roles of Outer Membrane Porins in Antibiotic Resistance and Membrane Integrity in Escherichia coli. Front. Microbiol. 2019, 10, 953. [Google Scholar] [CrossRef]
- Gravel, J.; Paradis-Bleau, C.; Schmitzer, A.R. Adaptation of a bacterial membrane permeabilization assay for quantitative evaluation of benzalkonium chloride as a membrane-disrupting agent. Medchemcomm 2017, 8, 1408–1413. [Google Scholar] [CrossRef]
- Fu, L.; Wan, M.; Zhang, S.; Gao, L.; Fang, W. Polymyxin B loosens lipopolysaccharide bilayer but stiffens phospholipid bilayer. Biophys. J. 2020, 118, 138–150. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Martínez, F.J.; Barrajón-Catalán, E.; Herranz-López, M.; Micol, V. Antibacterial plant compounds, extracts and essential oils: An updated review on their effects and putative mechanisms of action. Phytomedicine 2021, 90, 153626. [Google Scholar] [CrossRef]
- Mendgen, T.; Scholz, T.; Klein, C.D. Structure–activity relationships of tulipalines, tuliposides, and related compounds as inhibitors of MurA. Bioorg. Med. Chem. Lett. 2010, 20, 5757–5762. [Google Scholar] [CrossRef] [PubMed]
- Gautam, A.; Rishi, P.; Tewari, R. UDP-N-acetylglucosamine enolpyruvyl transferase as a potential target for antibacterial chemotherapy: Recent developments. Appl. Microbiol. Biotechnol. 2011, 92, 211–225. [Google Scholar] [CrossRef] [PubMed]
- Moliner, C.; López, V.; Barros, L.; Dias, M.I.; Ferreira, I.C.; Langa, E.; Gómez-Rincón, C. Rosemary Flowers as Edible Plant Foods: Phenolic Composition and Antioxidant Properties in Caenorhabditis elegans. Antioxidants 2020, 9, 811. [Google Scholar] [CrossRef]
- Minnunni, M.; Wolleb, U.; Mueller, O.; Pfeifer, A.; Aeschbacher, H. Natural antioxidants as inhibitors of oxygen species induced mutagenicity. Mutat. Res./Fundam. Mol. Mech. Mutagenesis 1992, 269, 193–200. [Google Scholar] [CrossRef]
- Lin, C.; Zhang, X.; Xiao, J.; Zhong, Q.; Kuang, Y.; Cao, Y.; Chen, Y. Effects on longevity extension and mechanism of action of carnosic acid in Caenorhabditis elegans. Food Funct. 2019, 10, 1398–1410. [Google Scholar] [CrossRef]
- Holdgate, G.A.; Meek, T.D.; Grimley, R.L. Mechanistic enzymology in drug discovery: A fresh perspective. Nat. Rev. Drug Discov. 2018, 17, 115–132. [Google Scholar] [CrossRef] [PubMed]
- Bachelier, A.; Mayer, R.; Klein, C.D. Sesquiterpene lactones are potent and irreversible inhibitors of the antibacterial target enzyme MurA. Bioorg. Med. Chem. Lett. 2006, 16, 5605–5609. [Google Scholar] [CrossRef]
- Steinbach, A.; Scheidig, A.J.; Klein, C.D. The unusual binding mode of cnicin to the antibacterial target enzyme MurA revealed by X-ray crystallography. J. Med. Chem. 2008, 51, 5143–5147. [Google Scholar] [CrossRef]
- Ábrányi-Balogh, P.; Petri, L.; Imre, T.; Szijj, P.; Scarpino, A.; Hrast, M.; Mitrović, A.; Fonovič, U.P.; Németh, K.; Barreteau, H.; et al. A road map for prioritizing warheads for cysteine targeting covalent inhibitors. Eur. J. Med. Chem. 2018, 160, 94–107. [Google Scholar] [CrossRef]
- Raina, D.; Tiwari, H.; Sharma, S.; Chinthakindi, P.K.; Nargotra, A.; Sangwan, P.L.; Eniyan, K.; Bajpai, U.; Vishwakarma, R.A.; Khan, F.G. Screening of compound library identifies novel inhibitors against the MurA enzyme of Escherichia coli. Appl. Microbiol. Biotechnol. 2021, 105, 3611–3623. [Google Scholar] [CrossRef]
- Cao, Y.; Peng, Q.; Li, S.; Deng, Z.; Gao, J. The intriguing biology and chemistry of fosfomycin: The only marketed phosphonate antibiotic. RSC Adv. 2019, 9, 42204–42218. [Google Scholar] [CrossRef] [Green Version]
- Garallah, E.T.; Al-Jubori, S.S. Surveillance of murA and the plasmid-mediated fosfomycin resistance fosA gene in uropathogenic E. coli isolates from UTI patients. Gene Rep. 2020, 21, 100872. [Google Scholar] [CrossRef]
- Yoon, H.J.; Lee, S.J.; Mikami, B.; Park, H.J.; Yoo, J.; Suh, S.W. Crystal structure of UDP-N-acetylglucosamine enolpyruvyl transferase from Haemophilus influenzae in complex with UDP-N-acetylglucosamine and fosfomycin. Proteins Struct. Funct. Bioinform. 2008, 71, 1032–1037. [Google Scholar] [CrossRef] [PubMed]
- McCoy, A.J.; Maurelli, A.T. Building the invisible wall: Updating the chlamydial peptidoglycan anomaly. Trends Microbiol. 2006, 14, 70–77. [Google Scholar] [CrossRef]
- Mazlan, M.K.N.; Mohd Tazizi, M.H.D.; Ahmad, R.; Noh, M.A.A.; Bakhtiar, A.; Wahab, H.A.; Mohd Gazzali, A. Antituberculosis Targeted Drug Delivery as a Potential Future Treatment Approach. Antibiotics 2021, 10, 908. [Google Scholar] [CrossRef]
- Dementin, S.; Bouhss, A.; Auger, G.; Parquet, C.; Mengin-Lecreulx, D.; Dideberg, O.; van Heijenoort, J.; Blanot, D. Evidence of a functional requirement for a carbamoylated lysine residue in MurD, MurE and MurF synthetases as established by chemical rescue experiments. Eur. J. Biochem. 2001, 268, 5800–5807. [Google Scholar] [CrossRef]
- Patin, D.; Boniface, A.; Kovač, A.; Hervé, M.; Dementin, S.; Barreteau, H.; Mengin-Lecreulx, D.; Blanot, D. Purification and biochemical characterization of Mur ligases from Staphylococcus aureus. Biochimie 2010, 92, 1793–1800. [Google Scholar] [CrossRef]
- van Groesen, E.; Slingerland, C.J.; Innocenti, P.; Mihajlovic, M.; Masereeuw, R.; Martin, N.I. Vancomyxins: Vancomycin-polymyxin nonapeptide conjugates that retain anti-Gram-positive activity with enhanced potency against Gram-negative strains. ACS Infect. Dis. 2021, 7, 2746–2754. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Cebrián, R.; Montalbán-López, M.; Ren, H.; Wu, W.; Kuipers, O.P. Outer-membrane-acting peptides and lipid II-targeting antibiotics cooperatively kill Gram-negative pathogens. Commun. Biol. 2021, 4, 31. [Google Scholar] [CrossRef] [PubMed]
- Zhou, A.; Kang, T.M.; Yuan, J.; Beppler, C.; Nguyen, C.; Mao, Z.; Nguyen, M.Q.; Yeh, P.; Miller, J.H. Synergistic interactions of vancomycin with different antibiotics against Escherichia coli: Trimethoprim and nitrofurantoin display strong synergies with vancomycin against wild-type E. coli. Antimicrob. Agents Chemother. 2015, 59, 276–281. [Google Scholar] [CrossRef] [Green Version]
- Shinohara, D.R.; Menegucci, T.C.; Fedrigo, N.H.; Migliorini, L.B.; Carrara-Marroni, F.E.; Dos Anjos, M.M.; Cardoso, C.L.; Nishiyama, S.A.B.; Tognim, M.C.B. Synergistic activity of polymyxin B combined with vancomycin against carbapenem-resistant and polymyxin-resistant Acinetobacter baumannii: First in vitro study. J. Med. Microbiol. 2019, 68, 309–315. [Google Scholar] [CrossRef]
- Perdih, A.; Hrast, M.; Barreteau, H.; Gobec, S.; Wolber, G.; Solmajer, T. Benzene-1, 3-dicarboxylic acid 2, 5-dimethylpyrrole derivatives as multiple inhibitors of bacterial Mur ligases (MurC–MurF). Bioorg. Med. Chem. 2014, 22, 4124–4134. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Chemoinformatics 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Open Babel Package, Version 2.3.1. Available online: http://Openbabel.Org (accessed on 23 September 2021).
- Schrödinger Release 2021-3: Maestro; Schrödinger, LLC: New York, NY, USA, 2021.
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pK a prediction and protonation state generation for drug-like molecules. J. Comput. Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2-a multiple sequence alignment editor and analysis workbench. Bioinformatics 2021, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skarzynski, T.; Mistry, A.; Wonacott, A.; Hutchinson, S.E.; Kelly, V.A.; Duncan, K. Structure of UDP-N-acetylglucosamine enolpyruvyl transferase, an enzyme essential for the synthesis of bacterial peptidoglycan, complexed with substrate UDP-N-acetylglucosamine and the drug fosfomycin. Structure 1996, 4, 1465–1474. [Google Scholar] [CrossRef] [Green Version]
- Schonbrunn, E. MurA Binary Complex with UDP-N-Acetylglucosamine. 2009. Available online: https://www.rcsb.org/structure/3KQJ (accessed on 23 September 2021).
- Han, H.; Yang, Y.; Olesen, S.H.; Becker, A.; Betzi, S.; Schönbrunn, E. The fungal product terreic acid is a covalent inhibitor of the bacterial cell wall biosynthetic enzyme UDP-N-acetylglucosamine 1-carboxyvinyltransferase (MurA). Biochemistry 2010, 49, 4276–4282. [Google Scholar] [CrossRef] [Green Version]
- Jackson, S.G.; Zhang, F.; Chindemi, P.; Junop, M.S.; Berti, P.J. Evidence of kinetic control of ligand binding and staged product release in MurA (enolpyruvyl UDP-GlcNAc synthase)-catalyzed reactions. Biochemistry 2009, 48, 11715–11723. [Google Scholar] [CrossRef]
- Zhu, J.-Y.; Yang, Y.; Han, H.; Betzi, S.; Olesen, S.H.; Marsilio, F.; Schönbrunn, E. Functional consequence of covalent reaction of phosphoenolpyruvate with UDP-N-acetylglucosamine 1-carboxyvinyltransferase (MurA). J. Biol. Chem. 2012, 287, 12657–12667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skarzynski, T.; Kim, D.H.; Lees, W.J.; Walsh, C.T.; Duncan, K. Stereochemical course of enzymatic enolpyruvyl transfer and catalytic conformation of the active site revealed by the crystal structure of the fluorinated analogue of the reaction tetrahedral intermediate bound to the active site of the C115A mutant of MurA. Biochemistry 1998, 37, 2572–2577. [Google Scholar]
- Steinbach, A.; Skarzynski, T.; Scheidig, A.J.; Klein, C.D. MURA Inhibited by Unag-Cnicin Adduct. 2007. Available online: https://www.rcsb.org/structure/2Z2C (accessed on 23 September 2021).
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Schrödinger Release 2021-3: Protein Preparation Wizard: Epik; Schrödinger, LLC: New York, NY, USA, 2021.
- OEDOCKING 4.1.0.1; OpenEye Scientific Software, Inc.: Santa Fe, NM, USA.
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein—ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waskom, M.L. Seaborn: Statistical data visualization. J. Open Res. Softw. 2021, 6, 3021. [Google Scholar] [CrossRef]
- Hunter, J.D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Kluyver, T.; Ragan-Kelley, B.; Pérez, F.; Granger, B.E.; Bussonnier, M.; Frederic, J.; Kelley, K.; Hamrick, J.B.; Grout, J.; Corlay, S.; et al. Jupyter Notebooks-A Publishing Format for Reproducible Computational Workflows. In Positioning and Power in Academic Publishing: Players, Agents and Agendas; IOS Press: Amsterdam, The Netherlands, 2016; pp. 87–90. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | RA % (IC50 µM) Escherichia coli MurA a | RA % (IC50 µM) Escherichia coli MurA b | RA % (IC50 µM) Staphylococcus aureus MurA a | RA % (IC50 µM) Staphylococcus aureus MurA b |
|---|---|---|---|---|
| 1 | 38 ± 2 (66 ± 8) | 5 ± 1 (2.8 ± 0.7) | 32 ± 3 (61 ± 7) | 3 ± 1 (1.1 ± 0.8) |
| 2 | 72 ± 2 | 9 ± 2 (12.9 ± 3.4) | 70 ± 3 | 8 ± 1.5 (5.7 ± 2.1) |
| 3 | 74 ± 4 | 8 ± 2 (25.1 ± 6.5) | 72 ± 3 | 7 ± 1 (12.3 ± 2.5) |
| 4 | 30 ± 2 (48 ± 5) | 0 (2.8 ± 0.4) | 41 ± 2 (67 ± 7) | 0 (3.4 ± 0.3) |
| 5 | 96 ± 5 | 0 (6.1 ± 0.7) | 91 ± 4 | 3 ± 1 (7.4 ± 0.9) |
| 6 | 78 ± 4 | 5 ± 1 (4.8 ± 0.4) | 77 ± 3 | 8 ± 1 (7.9 ± 0.6) |
| 7 | 98 ± 4 | 100 ± 5 | 99 ± 5 | 96 ± 4 |
| 8 | 100 ± 5 | 96 ± 4 | 98 ± 5 | 98 ± 4 |
| 9 | 85 ± 4 | 23 ± 2 (49.4 ± 5.1) | 87 ± 4 | 28 ± 1 (55.2 ± 6.3) |
| 10 | 98 ± 4 | 98 ± 4 | 99 ± 5 | 100 ± 5 |
| 11 | 100 ± 5 | 100 ± 5 | 96 ± 4 | 93 ± 4 |
| 12 | 98 ± 4 | 100 ± 5 | 97 ± 4 | 90 ± 4 |
| 13 | 97 ± 4 | 98 ± 5 | 96 ± 4 | 72 ± 3 |
| 14 | 100 ± 4 | 94 ± 4 | 98 ± 5 | 89 ± 5 |
| fosfomycin | N.d. | 0 (0.21 ± 0.04) | N.d. | 0 (0.30 ± 0.05) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Funes Chabán, M.; Hrast, M.; Frlan, R.; Graikioti, D.G.; Athanassopoulos, C.M.; Carpinella, M.C. Inhibition of MurA Enzyme from Escherichia coli and Staphylococcus aureus by Diterpenes from Lepechinia meyenii and Their Synthetic Analogs. Antibiotics 2021, 10, 1535. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics10121535
Funes Chabán M, Hrast M, Frlan R, Graikioti DG, Athanassopoulos CM, Carpinella MC. Inhibition of MurA Enzyme from Escherichia coli and Staphylococcus aureus by Diterpenes from Lepechinia meyenii and Their Synthetic Analogs. Antibiotics. 2021; 10(12):1535. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics10121535
Chicago/Turabian StyleFunes Chabán, Macarena, Martina Hrast, Rok Frlan, Dafni G. Graikioti, Constantinos M. Athanassopoulos, and María Cecilia Carpinella. 2021. "Inhibition of MurA Enzyme from Escherichia coli and Staphylococcus aureus by Diterpenes from Lepechinia meyenii and Their Synthetic Analogs" Antibiotics 10, no. 12: 1535. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics10121535