
Sorting out the Superbugs: Potential of Sortase A Inhibitors among Other Antimicrobial Strategies to Tackle the Problem of Antibiotic Resistance



{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Emergence of Antibiotic-Resistant Gram-Positive Pathogens of Healthcare Importance
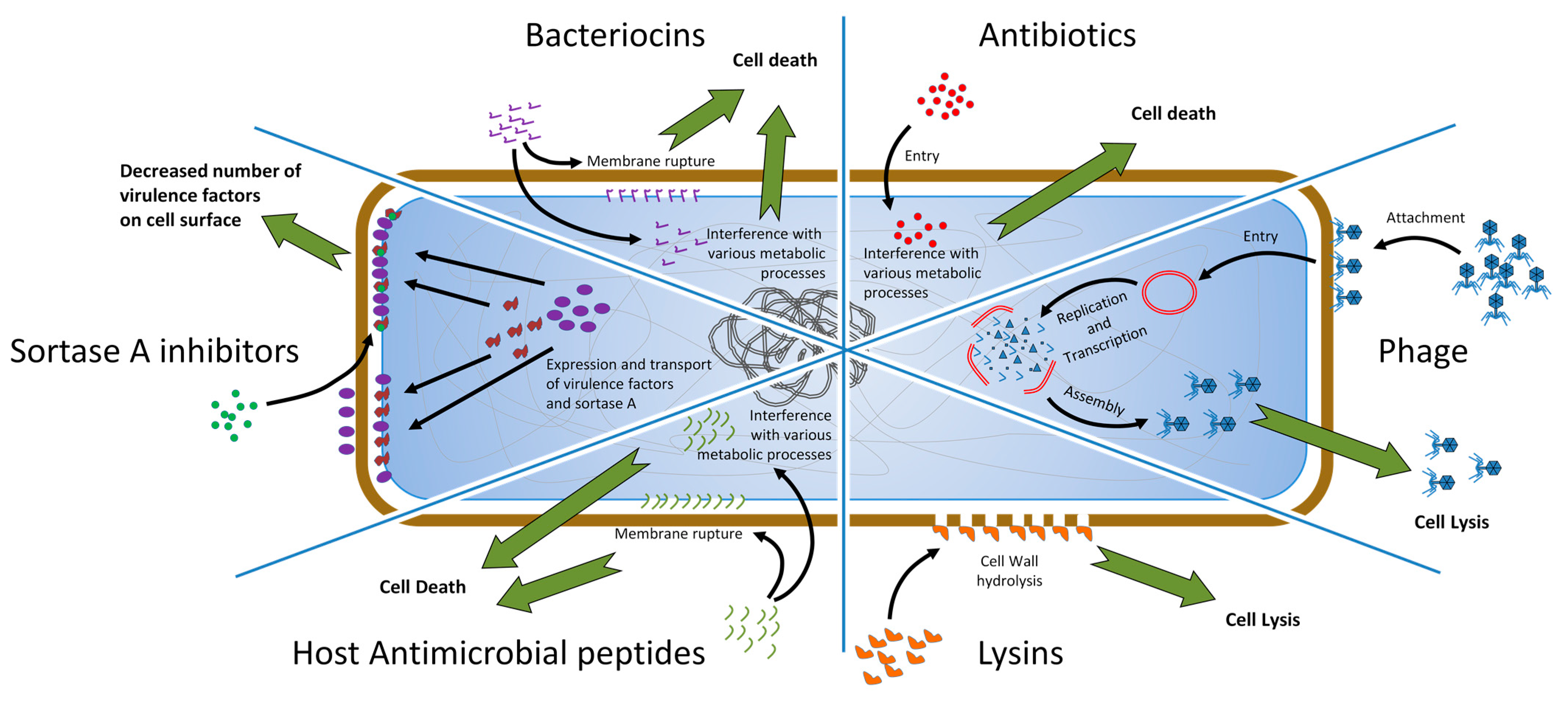
3. Alternative Options
3.1. Phage Therapy
3.2. Lysins
3.3. Antimicrobial Peptides
3.4. Bacteriocins
3.5. Antivirulence Strategies
4. Sortase A (SrtA)
5. SrtA Inhibitors
6. Materials and Methods
6.1. SrtA Expression and Purification
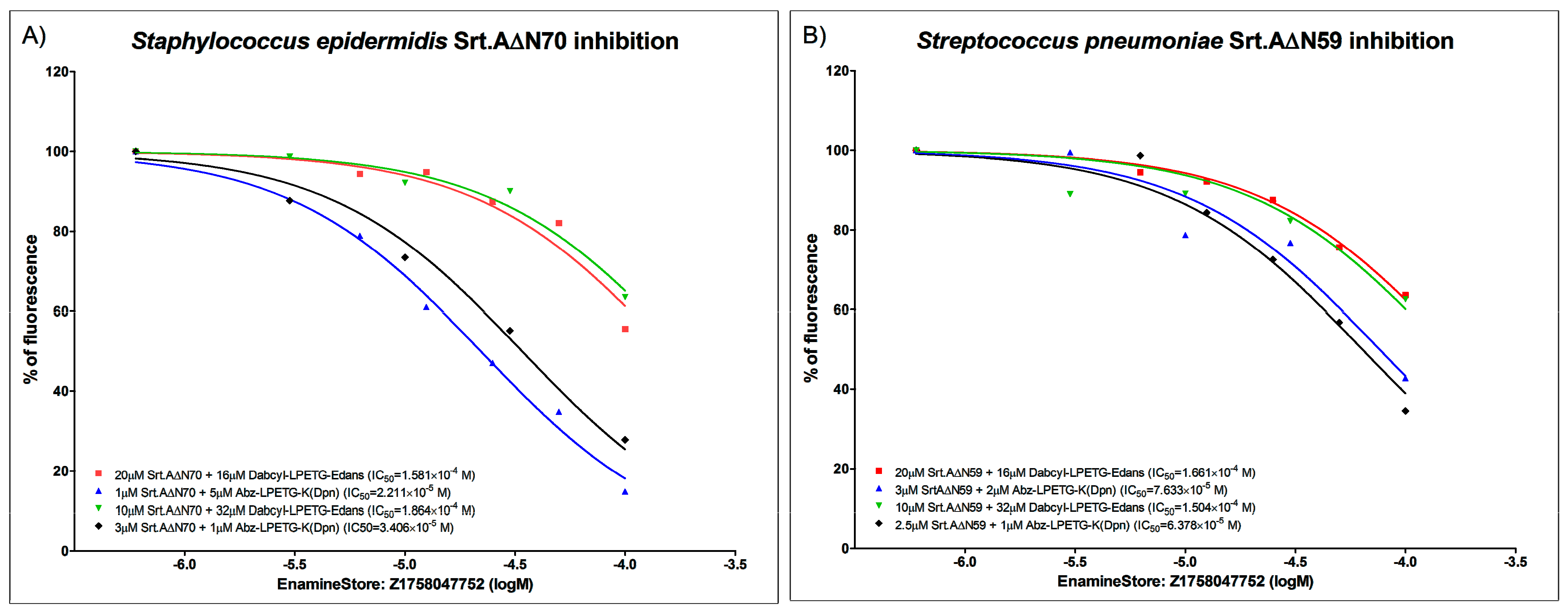
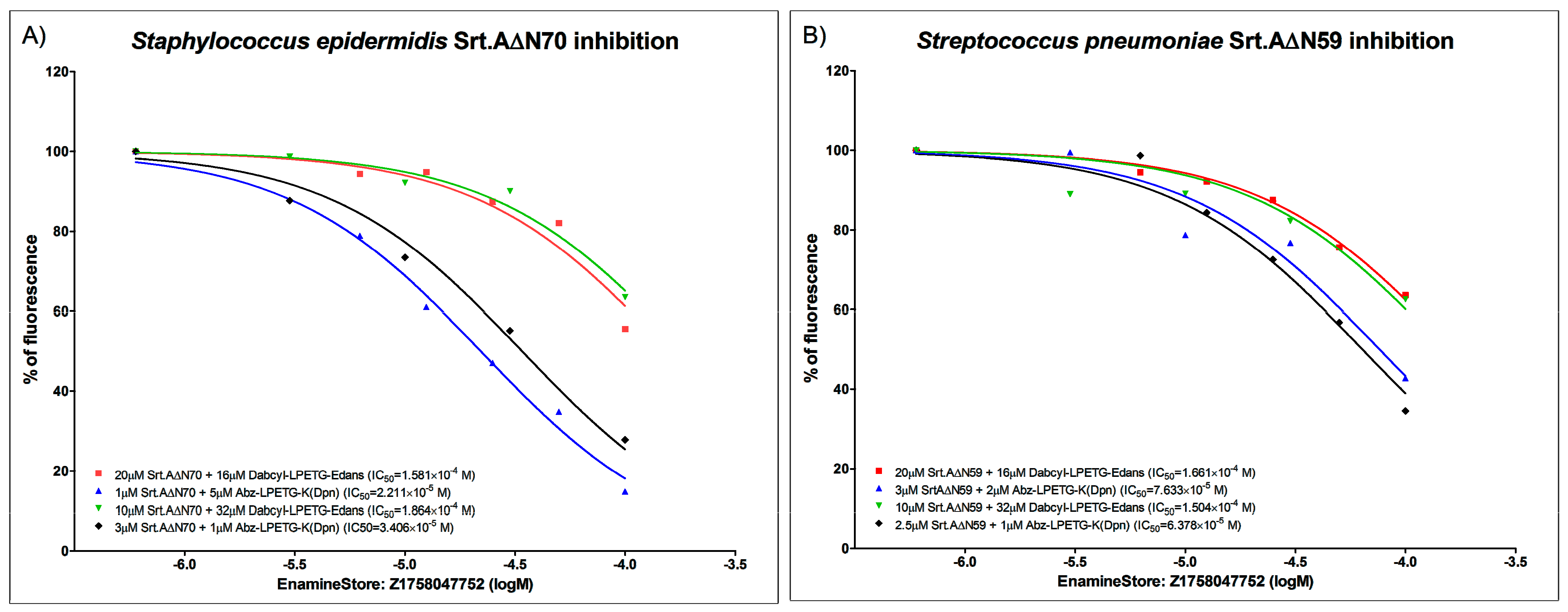
6.2. FRET Enzymatic Assay and IC50 Determination
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Czaplewski, L.; Bax, R.; Clokie, M.; Dawson, M.; Fairhead, H.; Fischetti, V.A.; Foster, S.; Gilmore, B.F.; Hancock, R.E.W.; Harper, D.; et al. Alternatives to antibiotics-a pipeline portfolio review. Lancet Infect. Dis. 2016, 16, 239–251. [Google Scholar] [CrossRef] [Green Version]
- David, M.Z.; Dryden, M.; Gottlieb, T.; Tattevin, P.; Gould, I.M. Recently approved antibacterials for methicillin-resistant Staphylococcus aureus (MRSA) and other Gram-positive pathogens: The shock of the new. Int. J. Antimicrob. Agents 2017, 50, 303–307. [Google Scholar] [CrossRef]
- Waldetoft, K.W.; Brown, S.P. Alternative therapeutics for self-limiting infections—An indirect approach to the antibiotic resistance challenge. PLoS Biol. 2017, 15, 1–10. [Google Scholar] [CrossRef] [Green Version]
- De Oliveira, D.M.P.; Forde, B.M.; Kidd, T.J.; Harris, P.N.A.; Schembri, M.A.; Beatson, S.A.; Paterson, D.L.; Walker, M.J. Antimicrobial resistance in ESKAPE pathogens. Clin. Microbiol. Rev. 2020, 33, e00181-19. [Google Scholar] [CrossRef] [PubMed]
- Vouga, M.; Greub, G. Emerging bacterial pathogens: The past and beyond. Clin. Microbiol. Infect. 2016, 22, 12–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, S.Y.C.; Davis, J.S.; Eichenberger, E.; Holland, T.L.; Fowler, V.G. Staphylococcus aureus infections: Epidemiology, pathophysiology, clinical manifestations, and management. Clin. Microbiol. Rev. 2015, 28, 603–661. [Google Scholar] [CrossRef] [Green Version]
- Namvar, A.E.; Bastarahang, S.; Abbasi, N.; Ghehi, G.S.; Farhadbakhtiarian, S.; Arezi, P.; Hosseini, M.; Baravati, S.Z.; Jokar, Z.; Chermahin, S.G. Clinical characteristics of Staphylococcus epidermidis: A systematic review. GMS Hyg. Infect. Control 2014, 9. [Google Scholar] [CrossRef]
- Peng, Z.; Jin, D.; Kim, H.B.; Stratton, C.W.; Wu, B.; Tang, Y.W.; Suna, X. Update on antimicrobial resistance in Clostridium difficile: Resistance mechanisms and antimicrobial susceptibility testing. J. Clin. Microbiol. 2017, 55, 1998–2008. [Google Scholar] [CrossRef] [Green Version]
- Spigaglia, P. Recent advances in the understanding of antibiotic resistance in Clostridium difficile infection. Ther. Adv. Infect. Dis. 2016, 3, 23–42. [Google Scholar] [CrossRef] [Green Version]
- Agudelo Higuita, N.I.; Huycke, M.M. Enterococcal disease, epidemiology, and implications for treatment. In Enterococci—From Commensals to Leading Causes to Drug Resistant Infection; Massachusetts Eye and Ear Infirmary: Boston, MA, USA, 2014; pp. 1–35. [Google Scholar]
- Karikalan, S.; Mohankumar, A. Antibiogram of streptococcus mutans isolated from dental caries patients. Int. J. Med. Health Res. 2016, 2, 79–83. [Google Scholar]
- Salehi, B.; Kregiel, D.; Mahady, G.; Sharifi-Rad, J.; Martins, N.; Rodrigues, C.F. Management of Streptococcus mutans-Candida spp. oral biofilms’ infections: Paving the way for effective clinical interventions. J. Clin. Med. 2020, 9, 517. [Google Scholar] [CrossRef] [Green Version]
- Cherazard, R.; Epstein, M.; Doan, T.L.; Salim, T.; Bharti, S.; Smith, M.A. Antimicrobial resistant Streptococcus pneumoniae: Prevalence, mechanisms, and clinical implications. Am. J. Ther. 2017, 24, e361–e369. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, R.L.; Gorris, L.G.M.; Hayman, M.M.; Jackson, T.C.; Whiting, R.C. A review of Listeria monocytogenes: An update on outbreaks, virulence, dose-response, ecology, and risk assessments. Food Control 2017, 75, 1–13. [Google Scholar] [CrossRef]
- Olaimat, A.N.; Al-Holy, M.A.; Shahbaz, H.M.; Al-Nabulsi, A.A.; Abu Ghoush, M.H.; Osaili, T.M.; Ayyash, M.M.; Holley, R.A. Emergence of antibiotic resistance in Listeria monocytogenes isolated from food products: A comprehensive review. Compr. Rev. Food Sci. Food Saf. 2018, 17, 1277–1292. [Google Scholar] [CrossRef] [Green Version]
- Athamna, A.; Athamna, M.; Abu-Rashed, N.; Medlej, B.; Bast, D.J.; Rubinstein, E. Selection of Bacillus anthracis isolates resistant to antibiotics. J. Antimicrob. Chemother. 2004, 54, 424–428. [Google Scholar] [CrossRef] [Green Version]
- Cavallo, J.D.; Ramisse, F.; Girardet, M.; Vaissaire, J.; Mock, M.; Hernandez, E. Antibiotic susceptibilities of 96 isolates of Bacillus anthracis isolated in France between 1994 and 2000. Antimicrob. Agents Chemother. 2002, 46, 2307–2309. [Google Scholar] [CrossRef] [Green Version]
- Munita, J.M.; Bayer, A.S.; Arias, C.A. Evolving resistance among gram-positive pathogens. Clin. Infect. Dis. 2015, 61, S48–S57. [Google Scholar] [CrossRef] [Green Version]
- Koulenti, D.; Xu, E.; Mok, Y.S.I.; Song, A.; Karageorgopoulos, D.E.; Armaganidis, A.; Lipman, J.; Tsiodras, S. Novel antibiotics for multidrug-resistant gram-positive microorganisms. Microorganisms 2019, 7, 270. [Google Scholar] [CrossRef] [Green Version]
- Tan, S.Y.; Tatsumura, Y. Alexander Fleming (1881–1955): Discoverer of penicillin. Singap. Med. J. 2015, 56, 366–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rios, A.C.; Moutinho, C.G.; Pinto, F.C.; del Fiol, F.S.; Jozala, A.; Chaud, M.V.; Vila, M.M.D.C.; Teixeira, J.A.; Balcão, V.M. Alternatives to overcoming bacterial resistances: State-of-the-art. Microbiol. Res. 2016, 191, 51–80. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, C.; Sarkar, P.; Issa, R.; Haldar, J. Alternatives to conventional antibiotics in the era of antimicrobial resistance. Trends Microbiol. 2019, 27, 323–338. [Google Scholar] [CrossRef] [PubMed]
- Pollock, J.; Low, A.S.; McHugh, R.E.; Muwonge, A.; Stevens, M.P.; Corbishley, A.; Gally, D.L. Alternatives to antibiotics in a one health context and the role genomics can play in reducing antimicrobial use. Clin. Microbiol. Infect. 2020. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.J.; Payne, D.J.; Rappuoli, R.; de Gregorio, E. Technologies to address antimicrobial resistance. Proc. Natl. Acad. Sci. USA 2018, 115, 12887–12895. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.-H.; Hsieh, Y.-H.; Powers, Z.M.; Kao, C.-Y. Defeating antibiotic-resistant bacteria: Exploring alternative therapies for a post-antibiotic era. Int. J. Mol. Sci. 2020, 21, 1061. [Google Scholar] [CrossRef] [Green Version]
- Hauser, A.R.; Mecsas, J.; Moir, D.T. Beyond antibiotics: New therapeutic approaches for bacterial infections. Clin. Infect. Dis. 2016, 63, 89–95. [Google Scholar] [CrossRef] [Green Version]
- Twort, F.W. An investigation on the nature of ultra-microscopic viruses. Lancet 1915, 186, 1241–1243. [Google Scholar] [CrossRef] [Green Version]
- D’Herelle, F. Sur un microbe invisible antagoniste des bacilles dysenteriques [An invisible microbe that is antagonistic to the dysentery bacillus]. Comptes Rendus 1917, 165, 373–375. [Google Scholar]
- Chanishvili, N. Phage Therapy-History from Twort and d’Herelle Through Soviet Experience to Current Approaches, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2012; Volume 83, ISBN 9780123944382. [Google Scholar]
- Abedon, S.T.; Kuhl, S.J.; Blasdel, B.G.; Kutter, E.M. Phage treatment of human infections. Bacteriophage 2011, 1, 66–85. [Google Scholar] [CrossRef] [Green Version]
- Loc-Carrillo, C.; Abedon, S.T. Pros and cons of phage therapy. Bacteriophage 2011, 1, 111–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dedrick, R.M.; Guerrero-Bustamante, C.A.; Garlena, R.A.; Russell, D.A.; Ford, K.; Harris, K.; Gilmour, K.C.; Soothill, J.; Jacobs-Sera, D.; Schooley, R.T.; et al. Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat. Med. 2019, 25, 730–733. [Google Scholar] [CrossRef]
- Fischetti, V.A.; Nelson, D.; Schuch, R. Reinventing phage therapy: Are the parts greater than the sum? Nat. Biotechnol. 2006, 24, 1508–1511. [Google Scholar] [CrossRef]
- Fenton, M.; Ross, P.; Mcauliffe, O.; O’Mahony, J.; Coffey, A. Recombinant bacteriophage lysins as antibacterials. Bioeng. Bugs 2010, 1, 9–16. [Google Scholar] [CrossRef]
- Nelson, D.; Loomis, L.; Fischetti, V.A. Prevention and elimination of upper respiratory colonization of mice by group A streptococci by using a bacteriophage lytic enzyme. Proc. Natl. Acad. Sci. USA 2001, 98, 4107–4112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Ruiz, I.; Coutinho, F.H.; Rodriguez-Valera, F. Thousands of novel endolysins discovered in uncultured phage genomes. Front. Microbiol. 2018, 9, 1–8. [Google Scholar] [CrossRef] [Green Version]
- De Maesschalck, V.; Gutiérrez, D.; Paeshuyse, J.; Lavigne, R.; Briers, Y. Advanced engineering of third-generation lysins and formulation strategies for clinical applications. Crit. Rev. Microbiol. 2020, 46, 548–564. [Google Scholar] [CrossRef] [PubMed]
- Gerstmans, H.; Rodríguez-Rubio, L.; Lavigne, R.; Briers, Y. From endolysins to Artilysin®s: Novel enzyme-based approaches to kill drug-resistant bacteria. Biochem. Soc. Trans. 2016, 44, 123–128. [Google Scholar] [CrossRef] [Green Version]
- Hancock, R.E.W.; Sahl, H.G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006, 24, 1551–1557. [Google Scholar] [CrossRef]
- Mahlapuu, M.; Håkansson, J.; Ringstad, L.; Björn, C. Antimicrobial peptides: An emerging category of therapeutic agents. Front. Cell. Infect. Microbiol. 2016, 6, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.J.; Gallo, R.L. Antimicrobial peptides. Curr. Biol. 2016, 26, R14–R19. [Google Scholar] [CrossRef]
- Fleming, A. On a remarkable bacteriolytic element found in tissues and secretions. Proc. R. Soc. Lond. Ser. B Contain. Pap. Biol. Character 1922, 93, 306–317. [Google Scholar] [CrossRef] [Green Version]
- Wang, G. Improved methods for classification, prediction, and design of antimicrobial peptides. Methods Mol. Biol. 2015, 1268, 43–66. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Li, X.; Wang, Z. APD3: The antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016, 44, D1087–D1093. [Google Scholar] [CrossRef] [Green Version]
- Martínez, B.; Rodríguez, A.; Suárez, E. Antimicrobial peptides produced by bacteria: The bacteriocins. In New Weapons to Control Bacterial Growth; Villa, T.G., Vinas, M., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 15–38. ISBN 978-3-319-28368-5. [Google Scholar]
- Gratia, A. Sur un remarquable exemple d’antagonisme entre deux souches de coilbacille. C. R. Seances Soc. Biol. Fil. 1925, 93, 1040–1041. [Google Scholar]
- Cotter, P.D.; Ross, R.P.; Hill, C. Bacteriocins-a viable alternative to antibiotics? Nat. Rev. Microbiol. 2013, 11, 95–105. [Google Scholar] [CrossRef]
- Dicks, L.M.T.; Dreyer, L.; Smith, C.; van Staden, A.D. A review: The fate of bacteriocins in the human gastro-intestinal tract: Do they cross the gut–blood barrier? Front. Microbiol. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Fahim, H.A.; Khairalla, A.S.; El-Gendy, A.O. Nanotechnology: A valuable strategy to improve bacteriocin formulations. Front. Microbiol. 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickey, S.W.; Cheung, G.Y.C.; Otto, M. Different drugs for bad bugs: Antivirulence strategies in the age of antibiotic resistance. Nat. Rev. Drug Discov. 2017, 16, 457–471. [Google Scholar] [CrossRef]
- Martínez, O.F.; Cardoso, M.H.; Ribeiro, S.M.; Franco, O.L. Recent advances in anti-virulence therapeutic strategies with a focus on dismantling bacterial membrane microdomains, toxin neutralization, quorum-sensing interference and biofilm inhibition. Front. Cell. Infect. Microbiol. 2019, 9, 1–24. [Google Scholar] [CrossRef]
- Nealson, K.H.; Platt, T.; Hastings, J.W. Cellular control of the synthesis and activity of the bacterial luminescent system. J. Bacteriol. 1970, 104, 313–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuqua, W.C.; Winans, S.C.; Greenberg, E.P. Quorum sensing in bacteria: The LuxR-LuxI family of cell density- responsive transcriptional regulators. J. Bacteriol. 1994, 176, 269–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassler, B.L.; Greenberg, E.P.; Stevens, A.M. Cross-species induction of luminescence in the quorum-sensing bacterium Vibrio harveyi. J. Bacteriol. 1997, 179, 4043–4045. [Google Scholar] [CrossRef] [Green Version]
- Deep, A.; Chaudhary, U.; Gupta, V. Quorum sensing and bacterial pathogenicity: From molecules to disease. J. Lab. Physicians 2011, 3, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Rémy, B.; Mion, S.; Plener, L.; Elias, M.; Chabrière, E.; Daudé, D. Interference in bacterial quorum sensing: A biopharmaceutical perspective. Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef]
- Kong, C.; Neoh, H.M.; Nathan, S. Targeting Staphylococcus aureus toxins: A potential form of anti-virulence therapy. Toxins 2016, 8, 72. [Google Scholar] [CrossRef] [Green Version]
- Fang, R.H.; Luk, B.T.; Hu, C.M.J.; Zhang, L. Engineered nanoparticles mimicking cell membranes for toxin neutralization. Adv. Drug Deliv. Rev. 2015, 90, 69–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López, D.; Kolter, R. Functional microdomains in bacterial membranes. Genes Dev. 2010, 24, 1893–1902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Fernández, E.; Koch, G.; Wagner, R.M.; Fekete, A.; Stengel, S.T.; Schneider, J.; Mielich-Süss, B.; Geibel, S.; Markert, S.M.; Stigloher, C.; et al. Membrane microdomain disassembly inhibits MRSA antibiotic resistance. Cell 2017, 171, 1354–1367. [Google Scholar] [CrossRef]
- Koch, G.; Wermser, C.; Acosta, I.C.; Kricks, L.; Stengel, S.T.; Yepes, A.; Lopez, D. Attenuating Staphylococcus aureus virulence by targeting flotillin protein scaffold activity. Cell Chem. Biol. 2017, 24, 845–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, D.; Misba, L.; Khan, A.U. Antibiotics versus biofilm: An emerging battleground in microbial communities. Antimicrob. Resist. Infect. Control 2019, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Bjarnsholt, T. The role of bacterial biofilms in chronic infections. APMIS Suppl. 2013, 1–51. [Google Scholar] [CrossRef]
- Ha, M.W.; Yi, S.W.; Paek, S.M. Design and synthesis of small molecules as potent staphylococcus aureus sortase a inhibitors. Antibiotics 2020, 9, 706. [Google Scholar] [CrossRef]
- Hendrickx, A.P.A.; Budzik, J.M.; Oh, S.Y.; Schneewind, O. Architects at the bacterial surface-sortases and the assembly of pili with isopeptide bonds. Nat. Rev. Microbiol. 2011, 9, 166–176. [Google Scholar] [CrossRef]
- Malik, A.; Kim, S.B. A comprehensive in silico analysis of sortase superfamily. J. Microbiol. 2019, 57, 431–443. [Google Scholar] [CrossRef]
- Zong, Y.; Bice, T.W.; Ton-That, H.; Schneewind, O.; Narayana, S.V.L. Crystal structures of Staphylococcus aureus Sortase A and its substrate complex. J. Biol. Chem. 2004, 279, 31383–31389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ton-That, H.; Liu, G.; Mazmanian, S.K.; Faull, K.F.; Schneewind, O. Purification and characterization of sortase, the transpeptidase that cleaves surface proteins of Staphylococcus aureus at the LPXTG motif. Proc. Natl. Acad. Sci. USA 1999, 96, 12424–12429. [Google Scholar] [CrossRef] [Green Version]
- Suree, N.; Liew, C.K.; Villareal, V.A.; Thieu, W.; Fadeev, E.A.; Clemens, J.J.; Jung, M.E.; Clubb, R.T. The structure of the Staphylococcus aureus sortase-substrate complex reveals how the universally conserved LPXTG sorting signal is recognized. J. Biol. Chem. 2009, 284, 24465–24477. [Google Scholar] [CrossRef] [Green Version]
- Mazmanian, S.K.; Liu, G.; Ton-That, H.; Schneewind, O. Staphylococcus aureus sortase, an enzyme that anchors surface proteins to the cell wall. Science 1999, 285, 760–763. [Google Scholar] [CrossRef]
- Mazmanian, S.K.; Ton-That, H.; Schneewind, O. Sortase-catalysed anchoring of surface proteins to the cell wall of Staphylococcus aureus. Mol. Microbiol. 2001, 40, 1049–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paterson, G.K.; Mitchell, T.J. The biology of gram-positive sortase enzymes. Trends Microbiol. 2004, 12, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Navarre, W.W.; Schneewind, O. Surface proteins of gram-positive bacteria and mechanisms of their targeting to the cell wall envelope. Microbiol. Mol. Biol. Rev. 1999, 63, 174–229. [Google Scholar] [CrossRef] [Green Version]
- Foster, T.J.; McDevitt, D. Surface-associated proteins of Staphylococcus aureus: Their possible roles in virulence. FEMS Microbiol. Lett. 1994, 118, 199–205. [Google Scholar] [CrossRef]
- Foster, T.J.; Höök, M. Surface protein adhesins of Staphylococcus aureus. Trends Microbiol. 1998, 6, 484–488. [Google Scholar] [CrossRef]
- Patel, A.H.; Nowlan, P.; Weavers, E.D.; Foster, T. Virulence of protein A-deficient and alpha-toxin-deficient mutants of Staphylococcus aureus isolated by allele replacement. Infect. Immun. 1987, 55, 3103–3110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazmanian, S.K.; Liu, G.; Jensen, E.R.; Lenoy, E.; Schneewind, O. Staphylococcus aureus sortase mutants defective in the display of surface proteins and in the pathogenesis of animal infections. Proc. Natl. Acad. Sci. USA 2000, 97, 5510–5515. [Google Scholar] [CrossRef] [Green Version]
- Bierne, H.; Mazmanian, S.K.; Trost, M.; Pucciarelli, M.G.; Liu, G.; Dehoux, P.; Jänsch, L.; Garcia-del Portillo, F.; Schneewind, O.; Cossart, P. Inactivation of the SrtA gene in Listeria monocytogenes inhibits anchoring of surface proteins and affects virulence. Mol. Microbiol. 2002, 43, 869–881. [Google Scholar] [CrossRef] [PubMed]
- Kharat, A.S.; Tomasz, A. Inactivation of the SrtA gene affects localization of surface proteins and decreases adhesion of Streptococcus pneumoniae to human pharyngeal cells in vitro. Infect. Immun. 2003, 71, 2758–2765. [Google Scholar] [CrossRef] [Green Version]
- Vanier, G.; Sekizaki, T.; Domínguez-Punaro, M.C.; Esgleas, M.; Osaki, M.; Takamatsu, D.; Segura, M.; Gottschalk, M. Disruption of SrtA gene in Streptococcus suis results in decreased interactions with endothelial cells and extracellular matrix proteins. Vet. Microbiol. 2008, 127, 417–424. [Google Scholar] [CrossRef]
- Hu, P.; Huang, P.; Chen, W.M. Curcumin inhibits the sortase a activity of the streptococcus mutans UA159. Appl. Biochem. Biotechnol. 2013, 171, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.C.; Popat, R.; Diggle, S.P.; Brown, S.P. Targeting virulence: Can we make evolution-proof drugs? Nat. Rev. Microbiol. 2014, 12, 300–308. [Google Scholar] [CrossRef]
- Nitulescu, G.; Nicorescu, I.M.; Olaru, O.T.; Ungurianu, A.; Mihai, D.P.; Zanfirescu, A.; Nitulescu, G.M.; Margina, D. Molecular docking and screening studies of new natural sortase A inhibitors. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [Green Version]
- Suree, N.; Yi, S.W.; Thieu, W.; Marohn, M.; Damoiseaux, R.; Chan, A.; Jung, M.E.; Clubb, R.T. Discovery and structure-activity relationship analysis of Staphylococcus aureus sortase A inhibitors. Bioorg. Med. Chem. 2009, 17, 7174–7185. [Google Scholar] [CrossRef] [Green Version]
- Selvaraj, C.; Sivakamavalli, J.; Baskaralingam, V.; Singh, S.K. Virtual screening of LPXTG competitive SrtA inhibitors targeting signal transduction mechanism in Bacillus anthracis: A combined experimental and theoretical study. J. Recept. Signal Transduct. 2014, 34, 221–232. [Google Scholar] [CrossRef]
- Davis, B.J.; Erlanson, D.A. Learning from our mistakes: The “unknown knowns” in fragment screening. Bioorg. Med. Chem. Lett. 2013, 23, 2844–2852. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Shin, D.S.; Oh, M.N.; Chung, S.C.; Lee, J.S.; Chang, I.M.; Oh, K.B. Inhibition of sortase, a bacterial surface protein anchoring transpeptidase, by β-sitosterol-3-O-glucopyranoside from Fritillaria verticillata. Biosci. Biotechnol. Biochem. 2003, 67, 2477–2479. [Google Scholar] [CrossRef]
- Kim, S.H.; Shin, D.S.; Oh, M.N.; Chung, S.C.; Lee, J.S.; Oh, K.B. Inhibition of the bacterial surface protein anchoring transpeptidase sortase by isoquinoline alkaloids. Biosci. Biotechnol. Biochem. 2004, 68, 421–424. [Google Scholar] [CrossRef]
- Oh, K.B.; Mar, W.; Kim, S.; Kim, J.Y.; Oh, M.N.; Kim, J.G.; Shin, D.; Sim, C.J.; Shin, J. Bis(indole) alkaloids as sortase A inhibitors from the sponge Spongosorites sp. Bioorg. Med. Chem. Lett. 2005, 15, 4927–4931. [Google Scholar] [CrossRef] [PubMed]
- Jang, K.H.; Chung, S.C.; Shin, J.; Lee, S.H.; Kim, T.I.; Lee, H.S.; Oh, K.B. Aaptamines as sortase A inhibitors from the tropical sponge Aaptos aaptos. Bioorg. Med. Chem. Lett. 2007, 17, 5366–5369. [Google Scholar] [CrossRef] [PubMed]
- Oh, I.; Yang, W.Y.; Chung, S.C.; Kim, T.Y.; Oh, K.B.; Shin, J. In vitro sortase A inhibitory and antimicrobial activity of flavonoids isolated from the roots of Sophora flavescens. Arch. Pharm. Res. 2011, 34, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Park, B.S.; Kim, J.G.; Kim, M.R.; Lee, S.E.; Takeoka, G.R.; Oh, K.B.; Kim, J.H. Curcuma longa L. constituents inhibit sortase A and Staphylococcus aureus cell adhesion to fibronectin. J. Agric. Food Chem. 2005, 53, 9005–9009. [Google Scholar] [CrossRef]
- Yang, W.Y.; Won, T.H.; Ahn, C.H.; Lee, S.H.; Yang, H.C.; Shin, J.; Oh, K.B. Streptococcus mutans sortase A inhibitory metabolites from the flowers of Sophora japonica. Bioorg. Med. Chem. Lett. 2015, 25, 1394–1397. [Google Scholar] [CrossRef]
- Lee, S.; Song, I.H.; Lee, J.H.; Yang, W.Y.; Oh, K.B.; Shin, J. Sortase A inhibitory metabolites from the roots of Pulsatilla koreana. Bioorg. Med. Chem. Lett. 2014, 24, 44–48. [Google Scholar] [CrossRef]
- Kang, S.S.; Kim, J.G.; Lee, T.H.; Oh, K.B. Flavonols inhibit sortases and sortase-mediated Staphylococcus aureus clumping to fibrinogen. Biol. Pharm. Bull. 2006, 29, 1751–1755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeon, J.E.; Na, Z.; Jung, M.; Lee, H.S.; Sim, C.J.; Nahm, K.; Oh, K.B.; Shin, J. Discorhabdins from the Korean marine sponge Sceptrella sp. J. Nat. Prod. 2010, 73, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.; Jeon, J.E.; Lee, Y.J.; Lee, H.S.; Sim, C.J.; Oh, K.B.; Shin, J. Sesterterpenes from the tropical sponge Coscinoderma sp. J. Nat. Prod. 2011, 74, 1805–1811. [Google Scholar] [CrossRef]
- Won, T.H.; Jeon, J.E.; Kim, S.H.; Lee, S.H.; Rho, B.J.; Oh, D.C.; Oh, K.B.; Shin, J. Brominated aromatic furanones and related esters from the ascidian synoicum sp. J. Nat. Prod. 2012, 75, 2055–2061. [Google Scholar] [CrossRef]
- Won, T.H.; Jeon, J.E.; Lee, S.H.; Rho, B.J.; Oh, K.B.; Shin, J. Beta-carboline alkaloids derived from the ascidian Synoicum sp. Bioorg. Med. Chem. 2012, 20, 4082–4087. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Bi, C.; Cai, H.; Liu, B.; Zhong, X.; Deng, X.; Wang, T.; Xiang, H.; Niu, X.; Wang, D. The therapeutic effect of chlorogenic acid against Staphylococcus aureus infection through sortase A inhibition. Front. Microbiol. 2015, 6, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.J.; Han, Y.R.; Park, W.; Nam, S.H.; Oh, K.B.; Lee, H.S. Synthetic analogs of indole-containing natural products as inhibitors of sortase A and isocitrate lyase. Bioorg. Med. Chem. Lett. 2010, 20, 6882–6885. [Google Scholar] [CrossRef]
- Wang, J.; Shi, Y.; Jing, S.; Dong, H.; Wang, D.; Wang, T. Astilbin inhibits the activity of sortase a from Streptococcus mutans. Molecules 2019, 24, 465. [Google Scholar] [CrossRef] [Green Version]
- Dong, J.; Zhang, L.; Xu, N.; Zhou, S.; Song, Y.; Yang, Q.; Liu, Y.; Yang, Y.; Ai, X. Rutin reduces the pathogenicity of Streptococcus agalactiae to tilapia by inhibiting the activity of sortase A. Aquaculture 2021, 530, 735743. [Google Scholar] [CrossRef]
- Oh, K.B.; Kim, S.H.; Lee, J.; Cho, W.J.; Lee, T.; Kim, S. Discovery of diarylacrylonitriles as a novel series of small molecule Sortase A inhibitors. J. Med. Chem. 2004, 47, 2418–2421. [Google Scholar] [CrossRef]
- Oh, K.B.; Nam, K.W.; Ahn, H.; Shin, J.; Kim, S.; Mar, W. Therapeutic effect of (Z)-3-(2,5-dimethoxyphenyl)-2-(4-methoxyphenyl) acrylonitrile (DMMA) against Staphylococcus aureus infection in a murine model. Biochem. Biophys. Res. Commun. 2010, 396, 440–444. [Google Scholar] [CrossRef]
- Oh, K.B.; Oh, M.N.; Kim, J.G.; Shin, D.S.; Shin, J. Inhibition of sortase-mediated Staphylococcus aureus adhesion to fibronectin via fibronectin-binding protein by sortase inhibitors. Appl. Microbiol. Biotechnol. 2006, 70, 102–106. [Google Scholar] [CrossRef]
- Maresso, A.W.; Wu, R.; Kern, J.W.; Zhang, R.; Janik, D.; Missiakas, D.M.; Duban, M.-E.; Joachimiak, A.; Schneewind, O. Activation of inhibitors by sortase triggers irreversible modification of the active site. J. Biol. Chem. 2007, 282, 23129–23139. [Google Scholar] [CrossRef] [Green Version]
- Zhulenkovs, D.; Rudevica, Z.; Jaudzems, K.; Turks, M.; Leonchiks, A. Discovery and structure-activity relationship studies of irreversible benzisothiazolinone-based inhibitors against Staphylococcus aureus sortase A transpeptidase. Bioorg. Med. Chem. 2014, 22, 5988–6003. [Google Scholar] [CrossRef]
- Wehrli, P.M.; Uzelac, I.; Olsson, T.; Jacso, T.; Tietze, D.; Gottfries, J. Discovery and development of substituted thiadiazoles as inhibitors of Staphylococcus aureus Sortase A. Bioorg. Med. Chem. 2019, 27, 115043. [Google Scholar] [CrossRef] [Green Version]
- Maggio, B.; Raffa, D.; Raimondi, M.V.; Cascioferro, S.; Plescia, F.; Schillaci, D.; Cusimano, M.G.; Leonchiks, A.; Zhulenkovs, D.; Basile, L.; et al. Discovery of a new class of sortase a transpeptidase inhibitors to tackle gram-positive pathogens: 2-(2-phenylhydrazinylidene)alkanoic acids and related derivatives. Molecules 2016, 21. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.; Zhang, T.; Guan, X.N.; Dong, Z.; Lan, L.; Yang, S.; Yang, C.G. Tideglusib and its analogues as inhibitors of Staphylococcus aureus SrtA. J. Med. Chem. 2020, 63, 8442–8457. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, P.; He, X.; Yuan, Z.W.; Yin, Z.Q.; Fu, H.; Lin, J.; He, C.; Liang, X.; Lv, C.; Shu, G.; et al. Erianin against staphylococcus aureus infection via inhibiting sortase A. Toxins 2018, 10, 385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Li, H.; Pan, J.; Dong, J.; Zhou, X.; Niu, X.; Deng, X. Oligopeptide targeting sortase a as potential anti-infective therapy for Staphylococcus aureus. Front. Microbiol. 2018, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Chenna, B.C.; Shinkre, B.A.; King, J.R.; Lucius, A.L.; Narayana, S.V.L.; Velu, S.E. Identification of novel inhibitors of bacterial surface enzyme Staphylococcus aureus Sortase A. Bioorg. Med. Chem. Lett. 2008, 18, 380–385. [Google Scholar] [CrossRef]
- Thappeta, K.R.V.; Zhao, L.N.; Nge, C.E.; Crasta, S.; Leong, C.Y.; Ng, V.; Kanagasundaram, Y.; Fan, H.; Ng, S.B. In-silico identified new natural sortase a inhibitors disrupt s. Aureus biofilm formation. Int. J. Mol. Sci. 2020, 21, 8601. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.H.; Wereszczynski, J.; Amer, B.R.; Yi, S.W.; Jung, M.E.; Mccammon, J.A.; Clubb, R.T. Discovery of staphylococcus aureus sortase a inhibitors using virtual screening and the relaxed complex scheme. Chem. Biol. Drug Des. 2013, 82, 418–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chenna, B.C.; King, J.R.; Shinkre, B.A.; Glover, A.L.; Lucius, A.L.; Velu, S.E. Synthesis and structure activity relationship studies of novel Staphylococcus aureus Sortase A inhibitors. Eur. J. Med. Chem. 2010, 45, 3752–3761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Liu, H.; Zhu, K.; Gong, S.; Dramsi, S.; Wang, Y.T.; Li, J.; Chen, F.; Zhang, R.; Zhou, L.; et al. Antiinfective therapy with a small molecule inhibitor of Staphylococcus aureus sortase. Proc. Natl. Acad. Sci. USA 2014, 111, 13517–13522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gill, S.R.; Fouts, D.E.; Archer, G.L.; Mongodin, E.F.; DeBoy, R.T.; Ravel, J.; Paulsen, I.T.; Kolonay, J.F.; Brinkac, L.; Beanan, M.; et al. Insights on evolution of virulence and resistance from the complete genome analysis of an early methicillin-resistant Staphylococcus aureus strain and a biofilm-producing methicillin-resistant Staphylococcus epidermidis strain. J. Bacteriol. 2005, 187, 2426–2438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misra, A.; Biswas, T.; Das, S.; Marathe, U.; Sehgal, D.; Roy, R.P.; Suryanarayanarao, R. Crystallization and preliminary X-ray diffraction studies of sortase A from Streptococcus pneumoniae. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2011, 67, 1195–1198. [Google Scholar] [CrossRef] [Green Version]
- Jaudzems, K.; Kurbatska, V.; Jekabsons, A.; Bobrovs, R.; Rudevica, Z.; Leonchiks, A. Targeting bacterial Sortase A with covalent inhibitors: 27 new starting points for structure-based hit-to-lead optimization. ACS Infect. Dis. 2020, 6, 186–194. [Google Scholar] [CrossRef]
- Chan, A.H.; Yi, S.W.; Weiner, E.M.; Amer, B.R.; Sue, C.K.; Wereszczynski, J.; Dillen, C.A.; Senese, S.; Torres, J.Z.; McCammon, J.A.; et al. NMR structure-based optimization of Staphylococcus aureus sortase A pyridazinone inhibitors. Chem. Biol. Drug Des. 2017, 90, 327–344. [Google Scholar] [CrossRef]
- Tonge, P.J. Quantifying the interactions between biomolecules: Guidelines for assay design and data analysis. ACS Infect. Dis. 2019, 5, 796–808. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zrelovs, N.; Kurbatska, V.; Rudevica, Z.; Leonchiks, A.; Fridmanis, D. Sorting out the Superbugs: Potential of Sortase A Inhibitors among Other Antimicrobial Strategies to Tackle the Problem of Antibiotic Resistance. Antibiotics 2021, 10, 164. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics10020164
Zrelovs N, Kurbatska V, Rudevica Z, Leonchiks A, Fridmanis D. Sorting out the Superbugs: Potential of Sortase A Inhibitors among Other Antimicrobial Strategies to Tackle the Problem of Antibiotic Resistance. Antibiotics. 2021; 10(2):164. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics10020164
Chicago/Turabian StyleZrelovs, Nikita, Viktorija Kurbatska, Zhanna Rudevica, Ainars Leonchiks, and Davids Fridmanis. 2021. "Sorting out the Superbugs: Potential of Sortase A Inhibitors among Other Antimicrobial Strategies to Tackle the Problem of Antibiotic Resistance" Antibiotics 10, no. 2: 164. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics10020164