
Identification of Escherichia coli and Related Enterobacteriaceae and Examination of Their Phenotypic Antimicrobial Resistance Patterns: A Pilot Study at A Wildlife–Livestock Interface in Lusaka, Zambia

 ,
,  , , , , , , , ,
, , , , , , , ,
Abstract
:1. Introduction
2. Results
2.1. Microbiological, Biochemical and Molecular Analysis for Identification of Enterobacteriaceae Fecal Isolates
2.2. Phylogenetic Analysis
2.3. Antimicrobial Susceptibility Testing
3. Discussion
4. Materials and Methods
4.1. Ethical Consideration
4.2. Study Design
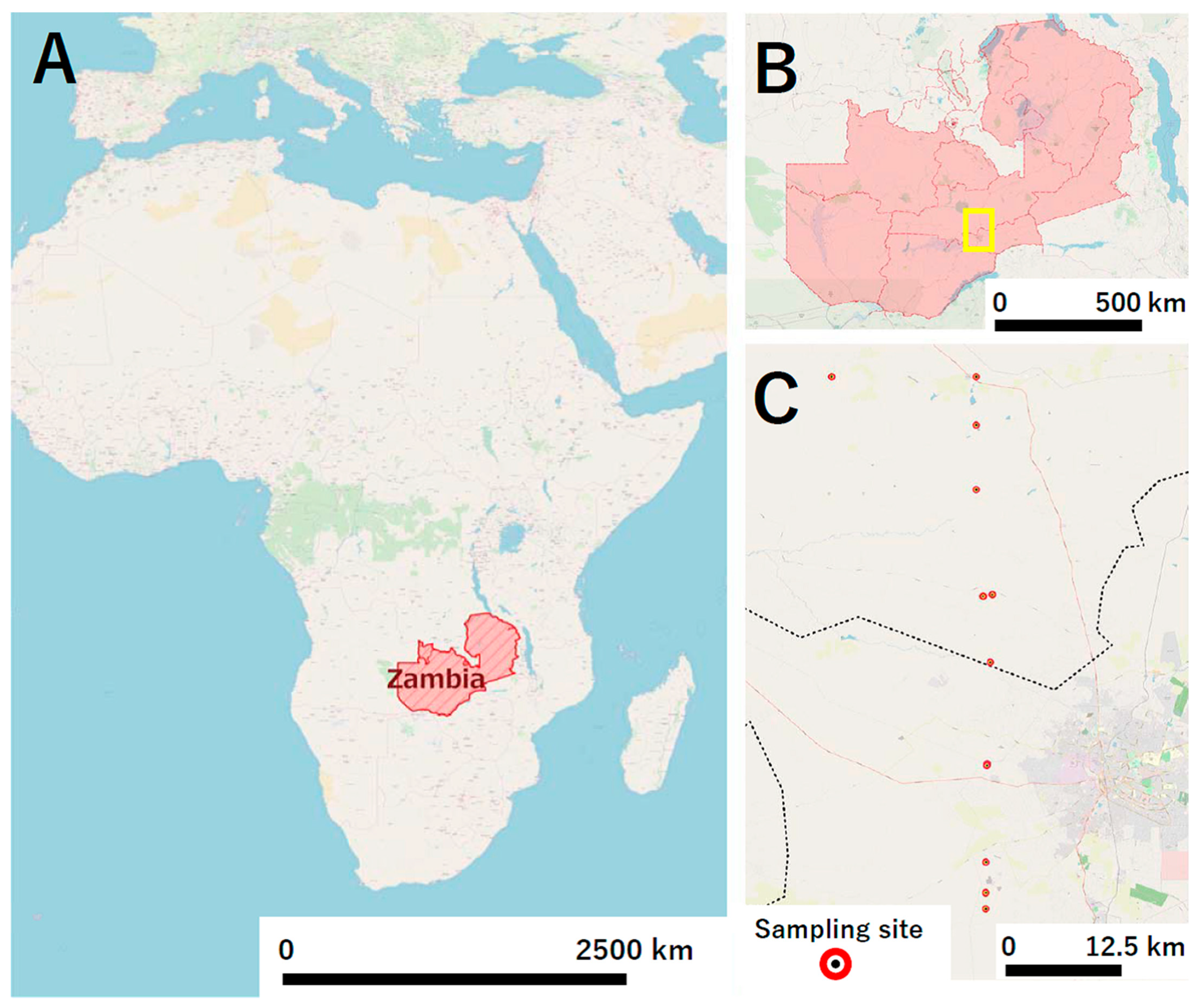
4.3. Sample Collection
4.4. Bacteria Isolation
4.5. Biochemical Identification
4.6. Molecular Identification
4.6.1. DNA Extraction and Polymerase Chain Reaction
4.6.2. Cycle Sequencing
4.6.3. Sequence Analysis
4.7. Antimicrobial Susceptibility Testing (AST)
4.8. Statistical Analysis
4.9. Study Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. Antimicrobial Resistance: Fact. Sheet. 2018. Available online: https://www.who.int/news-room/fact-sheets/detail/antimicrobial-resistance (accessed on 26 May 2019).
- WHO. World Health Assembly Addresses Antimicrobial Resistance, Immunization Gaps and Malnutrition. 2015. Available online: http://www.who.int/mediacentre/news/releases/2015/wha-25-may-2015/en/ (accessed on 12 February 2017).
- O’Neill, J. Review on Antimicrobial Resistance: Tackling Drug-Resistant Infections Globally. Tackling a Global Health Crisis: Initial Step. 2015. Available online: https://amr-review.org/sites/default/files/Report-52.15.pdf (accessed on 1 December 2018).
- Yang, Y.; Li, B.; Ju, F.; Zhang, T. Exploring variation of antibiotic resistance genes in activated sludge over a four-year period through a metagenomic approach. Environ. Sci. Technol. 2013, 47, 10197–10205. [Google Scholar] [CrossRef]
- Van Bijnen, E.M.E.; Paget, J.; de Lange-de Klerk, E.S.M.; den Heijer, C.D.J.; Versporten, A.; Stobberingh, E.E.; Goossens, H.; Schellevis, F.G. Antibiotic exposure and other risk factors for antimicrobial resistance in nasal commensal Staphylococcus aureus: An ecological study in 8 European countries. PLoS ONE 2015, 10, e0135094. [Google Scholar] [CrossRef] [Green Version]
- Pisano, E. Antimicrobial Resistance: What Does Medicine Quality Have to Do with it? For the Review on Antimicrobial Resistance. 2015. Available online: https://amr-review.org/sites/default/files/ElizabethPisaniMedicinesQualitypaper.pdf (accessed on 1 December 2018).
- Kabali, E.; Ward, K.; Bapoo, R. Review of the Effectiveness of Medicines Regulatory Systems in Zambia; Lambert Academic Publishing: Beau Bassin, Mauritius, 2018. [Google Scholar]
- FAO. Drivers, Dynamics and Epidemiology of Antimicrobial Resistance in Animal Production. 2016. Available online: http://www.fao.org/3/a-i6209e.pdf (accessed on 27 May 2019).
- Kho, Z.Y.; Lal, S.K. The human gut microbiome—A potential controller of wellness and disease. Front. Microbiol. 2018, 9, 1835. [Google Scholar] [CrossRef] [Green Version]
- Modi, S.R.; Collins, J.J.; Relman, D.A. Antibiotics and the gut microbiota. J. Clin. Investig. 2014, 124, 4212–4218. [Google Scholar] [CrossRef] [Green Version]
- Jaja, I.F.; Bhembe, N.L.; Green, E.; Oguttu, J.; Muchenje, V. Molecular characterization of antibiotic-resistant Salmonella enterica isolates recovered from meat in South Africa. Acta Tropica 2019, 190, 129–136. [Google Scholar] [CrossRef]
- Nikaido, H. Multidrug resistance in bacteria. Annu. Rev. Biochem. 2009, 78, 119–146. [Google Scholar] [CrossRef] [Green Version]
- Kabali, E.; Pandey, G.S.; Maboshe, M.; Munyeme, M. The wildlife-livestock-human interface, legislation and its impact on communities around Mosi-Oa-Tunya National Park in Zambia. J. Sustain. Dev. Africa 2015, 17, 125–145. [Google Scholar]
- Woods, R.; Reiss, A.; Cox-Witton, K.; Grillo, T.; Peters, A. The importance of wildlife disease monitoring as part of global surveillance for zoonotic diseases: The role of Australia. Trop Med. Inf. Dis. 2019, 4, 29. [Google Scholar] [CrossRef] [Green Version]
- UNEP Frontiers 2016 Report. Zoonoses: Blurred Lines of Emergent Disease and Ecosystem Health. 2016. Available online: http://uneplive.org/media/docs/early_warning/zoonoses.pdf (accessed on 26 May 2019).
- Wang, L.F.; Crameri, G. Emerging zoonotic viral diseases. Rev. Sci Tech. 2014, 33, 569–581. Available online: https://www.oie.int/doc/ged/D14089.PDF (accessed on 26 May 2019). [CrossRef]
- Kooriyama, T.; Okamoto, M.; Yoshida, T.; Nishida, T.; Tsubota, T.; Saito, A.; Tomonaga, M.; Matsuzawa, T.; Akari, T.; Nishimura, H.; et al. Epidemiological study of zoonoses derived from humans in captive chimpanzees. Primates 2013, 54, 89–98. [Google Scholar] [CrossRef]
- Alexander, K.A.; Lewis, B.L.; Marathe, M.; Eubank, S.; Blackburn, J.K. Modeling of wildlife-associated zoonoses: Applications and caveats. Vector Borne Zoonotic Dis. 2012, 12, 1005–1018. [Google Scholar] [CrossRef] [PubMed]
- OIE. Training Manual on Wildlife Diseases and Surveillance. 2010. Available online: https://www.oie.int/doc/ged/D12066.PDF (accessed on 27 May 2019).
- Mubita, C.; Syakalima, M.; Chisenga, C.; Munyeme, M.; Muma, B.; Chifumpa, G.; Hangombe, B.; Sinkala, P.; Simuunza, M.; Fukushi, H.; et al. Antimicrobial resistance of faecal Escherichia coli and Enterococci spp. from Apparently Health Pastoralist Cattle in the Interface Areas of the Kafue basin of Zambia-short communication. Veterinarski Arhiv 2008, 78, 179–185. [Google Scholar]
- Mubita, C.M.; Muma, B.J.; Nalubamba, K.; Pandey, G.S.; Samui, K.; Munyeme, M.; Masahiro, K.; Qui, Y.; Saasa, N.; Hang’ombe, B.M. Characterization of Salmonella isolated from domestic animals and wildlife from selected areas of Zambia. Sci. Afr. J. 2020. [Google Scholar] [CrossRef]
- Mshana, S.E.; Matee, M.; Rweyemamu, M. Antimicrobial resistance in human and animal pathogens in Zambia, Democratic Republic of Congo, Mozambique and Tanzania: An urgent need of a sustainable surveillance system. Ann. Clin. Microbiol. Antimicrob 2013, 12, 28. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.R. Shigella and Escherichia coli at the crossroads: Machiavellian masqueraders or taxonomic treachery? J. Med. Microbiol. 2000, 49, 583–585. [Google Scholar]
- Simulundu, E.; Lubaba, C.H.; Heerden, J.V.; Kajihara, M.; Matta, L.; Chambaro, H.M.; Sinkala, Y.; Munjita, S.M.; Munang’andu, H.M.; Nalubamba, K.S.; et al. The epidemiology of African swine fever in “nonendemic” regions of Zambia (1989–2015): Implications for disease prevention and control. Viruses 2017, 9, 236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nalubamba, K.S.; David, S.; Munyeme, M.; Kamboyi, H.K.; Saasa, N.; Mkandawire, E.; Munang’andu, H.M. Theileria spp. in free ranging giraffes (Giraffa camelopardalis) in Zambia. J. Vet. Med. Res. 2015, 2, 1039. Available online: https://www.researchgate.net/publication/311107984 (accessed on 26 May 2019).
- Kamboyi, H.K.; Garine-Wichatitsky, M.D.; Hang’ombe, B.M.; Munyeme, M. Risk mapping and eco-anthropogenic assessment of anthrax in the upper Zambezi basin. Vet. Med. Sci. 2019, 5, 419–427. [Google Scholar] [CrossRef]
- Wiethoelter, A.K.; Beltrán-Alcrudo, D.; Kock, R.; Mor, S.M. Global trends in infectious diseases at the wildlife livestock interface. Proc. Natl. Acad. Sci. USA 2015, 112, 9662–9667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gortázar, C.; Ferroglio, E.; Höfle, U.; Frölich, K.; Vicente, J. Diseases shared between wildlife and livestock: A European perspective. Eur, J. Wildl. Res. 2007, 53, 241–256. [Google Scholar] [CrossRef]
- Fischer, J.R.; Gerhold, R. Wildlife as a risk factor in animal health and zoonoses. Conf. OIE 2002, 273–280. Available online: https://pdfs.semanticscholar.org/1590/5c1e9c2e64edc7b0dd7422b99052ebfe74ba.pdf (accessed on 27 May 2019).
- Arnold, K.E.; Williams, N.J.; Bennett, M. ‘Disperse abroad in the land’: The role of wildlife in the dissemination of antimicrobial resistance. Biol. Lett. 2016, 12, pii:20160137. [Google Scholar] [CrossRef] [Green Version]
- Mpundu, P.; Mbewe, A.R.; Muma, J.B.; Zgambo, J.; Munyeme, M. Evaluation of bacterial contamination in dressed chickens in Lusaka abattoirs. Front. Public Health 2019, 7, 19. [Google Scholar] [CrossRef] [PubMed]
- Watkins, R.R.; Bonomo, R.A. β-Lactam Antibiotics. In Infectious Diseases, 4th ed.; Cohen, J., Powderly, W.G., Opal, S.M., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; Volume 2, pp. 1203–1216.e2. [Google Scholar] [CrossRef]
- Alcaide, B.; Aragoncillo, C.; Almendros, P. ‘Cephalosporins’ in Katritzky AR, Ramsden CA, Scrive EFV, Taylor RJK (eds.-in-chief) Comprehensive Heterocyclic Chemistry III. Elsevier Sci. 2008, 2, 111–171. [Google Scholar] [CrossRef]
- WHO Critically Important Antimicrobials for Human Medicine, 5th rev. 2016. Available online: http://who.int/foodsafety/publications/antimicrobials-fifth/en/ (accessed on 25 May 2019).
- OIE List of Antimicrobial Agents of Veterinary Importance. 2015. Available online: http://www.oie.int/fileadmin/Home/eng/Our_scientific_expertise/docs/pdf/Eng_OIE_List_antimicrobials_May2015.pdf (accessed on 25 May 2019).
- Kabwe, M.; Tembo, J.; Chilukutu, L.; Chilufya, M.; Ngulube, F.; Lukwesa, C.; Kapasa, M.; Enne, V.; Wexner, H.; Mwananyanda, L.; et al. Etiology, antibiotic resistance and risk factors for neonatal sepsis in a large referral center in Zambia. Pediatr Infect. Dis. J. 2016, 35, e191–e198. [Google Scholar] [CrossRef] [Green Version]
- Chame, M. Terrestrial mammal feces: A morphometric summary and description. Mem. Inst. Oswaldo Cruz 2003, 98, 71–94. [Google Scholar] [CrossRef]
- Tsen, H.Y.; Lin, C.K.; Chi, W.R. Development and use of 16S rRNA gene targeted PCR primers for the identification of Escherichia coli cells in water. J. Appl. Microbiol. 1998, 85, 554–560. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Paaterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [Green Version]
- Clinical and Laboratory Standards Institute. Performance Standards for Antimicrobial Susceptibility Testin, 28th ed.; CLSI supplementM100; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2018. [Google Scholar]
- Kozak, G.K.; Boerlin, P.; Janecko, N.; Reid-Smith, R.J.; Jardine, C. Antimicrobial resistance in Escherichia coli isolates from swine and wild small mammals in the proximity of swine farms and in natural environments in Ontario, Canada. Appl. Environ. Microbiol. 2009, 75, 559–566. [Google Scholar] [CrossRef] [Green Version]
- Magiorakos, A.P.; Srinivasan, A.; Carey, R.B.; Carmeli, Y.; Falagas, M.E.; Giske, C.G.; Harbarth, S.; Hindler, J.F.; Kahlmeter, G.; Olsson-Liljequist, B.; et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: An international expert proposal for interim standard definitions for acquired resistance. Clin. Microbiol. Infect. 2012, 18, 268–281. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Animal Species | Number of Samples Processed | Total Isolates (% Yield) | Identified Bacteria by Species | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Biochemical Identification Using API® 20E (n = 66) | Molecular Detection Using 16E1/16E2/16E3 and P3mod/P5 Primers (n = 65) | ||||||||||
| Escherichia coli Group 1 n (%) | Burkholderia cepacian (%) | Kluyvera spp. n (%) | Unidentified n (%) | Escherichia coli spp. n (%) | Escherichia fergusoniin (%) | Shigella spp. n (%) | Enterobacteriaceae bacteriumn (%) | ||||
| Impala | 16 | 15 (93.8) | 6 (40) | 0 | 1 (6.7) | 8 (53.3) | 11 (78.6) | 0 | 3 (21.4) | 0 | |
| Buffalo | 13 | 8 (61.5) | 5 (62.5) | 0 | 0 | 3 (37.5) | 6 (75) | 1 (12.5) | 1 (12.5) | 0 | |
| Ostrich | 7 | 6 (85.7) | 5 (83.3) | 0 | 0 | 1 (16.7) | 5 (83.3) | 0 | 1 (16.7) | 0 | |
| Cattle | 20 | 16 (80) | 11 (68.8) | 0 | 0 | 5 (31.3) | 13 (81.2) | 0 | 3 (18.8) | 0 | |
| Pig | 18 | 15 (83.3) | 11 (73.3) | 0 | 0 | 4 (26.7) | 10 (66.7) | 0 | 4 (26.7) | 1 (6.7) | |
| Goat | 10 | 6 (60) | 1 (16.7) | 1 (16.7) | 0 | 4 (66.7) | 3 (50) | 0 | 3 (50) | 0 | |
| Total | 84 | 66 (78.6) | 39 (59.1) | 1 (1.5) | 1 (1.5) | 25 (37.9) | 48 (72.7) | 1 (1.5) | 15 (22.7) | 1 (1.5) | |
| Sequence percentage homology (%) | Minimum | 90.00 | 1st Quantile | 98.10 | |||||||
| Mean | 98.29 | Median | 99.00 | ||||||||
| Maximum | 100.00 | 3rd Quantile | 99.30 | ||||||||
| Antimicrobial Agent | AMR Detected | All Isolates N = 63 | Impala N = 15 | Buffalo N = 8 | Ostrich N = 6 | Cattle N = 16 | Pig N = 15 | Goat N = 6 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| E. coli spp. (n = 48) n (%) | Shigella spp. (n = 15) n (%) | E. coli spp. (n = 11) n (%) | Shigella spp. (n = 3) n (%) | E. coli spp. (n = 6) n (%) | Shigella spp. (n = 1) n (%) | E. coli spp. (n = 5) n (%) | Shigella spp. (n = 1) n (%) | E. coli spp. (n = 13) n (%) | Shigella spp. (n = 3) n (%) | E. coli spp. (n = 10) n (%) | Shigella spp. (n = 4) n (%) | E. coli spp. (n = 3) n (%) | Shigella spp. (n = 3) n (%) | ||
| Ampicillin | R | 17 (27) | 3 (4.8) | 6 (42.9) | - | - | - | 3 (50) | 1 (16.7) | 7 (43.8) | - | 1 (6.7) | 1 (6.7) | - | 1 (16.7) |
| I | 10 (15.9) | 3 (4.8) | 2 (14.3) | - | - | 1 (12.5) | - | - | 2 (12.5) | 1 (6.2) | 5 (33.3) | - | 1 (16.7) | 1 (16.7) | |
| Amoxicillin + Clavulanic Acid | R | 5 (7.9) | - | 4 (28.6) | - | - | - | 1 (16.7) | - | - | - | - | - | - | - |
| I | 2 (3.2) | - | 1 (7.1) | - | - | - | 1 (16.7) | - | - | - | - | - | - | - | |
| Ceftazidime | R | 9 (14.3) | 1 (1.6) | 2 (14.3) | - | - | - | 1 (16.7) | - | 5 (31.2) | - | - | 1 (6.7) | 1 (16.7) | - |
| I | 4 (6.3) | - | - | - | - | - | - | - | 1(6.2) | - | 2 (13.3) | - | 1 (16.7) | - | |
| Cefotaxime | R | 6 (9.5) | 2 (3.2) | 2 (14.3) | - | - | - | 1(16.7) | 1(16.7) | 3 (18.8) | - | - | 1 (6.7) | - | - |
| I | 9 (14.3) | 1 (1.6) | 5 (35.7) | - | - | - | - | - | 2 (12.5) | - | 2(13.3) | - | - | 1 (16.7) | |
| Ceftriaxone | R | 4 (6.3) | - | 2 (14.3) | - | - | - | 1 (16.7) | - | 1 (6.2) | - | - | - | - | - |
| I | 7 (11.1) | 2 (3.2) | - | - | 1 (12.5) | - | - | 1 (16.7) | 4 (25) | 2 (12.5) | 1 (6.7) | - | - | - | |
| Kanamycin | R | 6 (9.5) | 3 (4.8) | 1 (7.1) | - | - | - | 2 (33.3) | 1 (16.7) | - | - | 1 (6.7) | 1 (6.7) | 2 (33.3) | 1 (16.7) |
| I | 20 (31.7) | 5 (7.9) | 5 (35.7) | 1 (7.1) | 3 (37.5) | 1 (12.5) | - | - | 6 (37.5) | 1 (6.2) | 4 (26.7) | 2 (13.3) | - | - | |
| Amikacin | R | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
| I | - | - | - | - | - | - | - | - | - | - | - | - | - | - | |
| Gentamicin | R | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
| I | 1 (1.6) | - | - | - | - | - | - | - | 1 (6.2) | - | - | - | - | - | |
| Imipenem | R | 1 (1.6) | - | 1 (7.1) | - | - | - | - | - | - | - | - | - | - | - |
| I | 2 (3.2) | - | 1 (7.1) | - | - | - | - | - | 2 (12.5) | - | - | - | - | - | |
| Nalidixic Acid | R | 3 (4.8) | 1(4) | 1 (7.1) | - | - | - | - | - | 2 (12.5) | - | 1 (6.7) | - | - | - |
| I | 1 (1.6) | - | 1 (7.1) | - | - | - | - | - | - | - | - | - | - | - | |
| Ciprofloxacin | R | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
| I | 1 (1.6) | - | - | - | - | - | - | - | 1 (6.2) | - | - | - | - | - | |
| Trimethoprim + Sulfamethoxazole | R | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
| I | - | - | - | - | - | - | - | - | - | - | - | - | - | - | |
| Chloramphenicol | R | 2 (3.2) | 1 (1.6) | - | - | - | - | - | - | - | 1 (6.2) | 1 (6.7) | - | 1 (16.7) | - |
| I | - | - | - | - | - | - | - | - | - | - | - | - | - | - | |
| Number of Antimicrobial Classes | All Isolates | Impala | Buffalo | Ostrich | Cattle | Pig | Goat | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| E. coli spp. (n = 48) n (%) | Shigella spp. (n = 15) n (%) | E. coli spp. (n = 11) n (%) | Shigella spp. (n = 3) n (%) | E. coli spp. (n = 6) n (%) | Shigella spp. (n = 1) n (%) | E. coli spp. (n = 5) n (%) | Shigella spp. (n = 1) n (%) | E. coli spp. (n = 13) n (%) | Shigella spp. (n = 3) n (%) | E. coli spp. (n = 10) n (%) | Shigella spp. (n = 4) n (%) | E. coli spp. (n = 3) n (%) | Shigella spp. (n = 3) n (%) | |
| 1 | 4 (8.4) | - | 2 (18.2) | - | - | - | - | - | - | - | 2 (20) | - | - | - |
| 2 | 10 (20.9) | 1 (6.7) | 1 (9.1) | - | - | - | 2 (40) | - | 6 (46.2) | - | 1 (10) | - | - | 1 (33.3) |
| 3 | 8 (16.7) | 1 (6.7) | 3 (27.3) | - | - | - | 1 (20) | 1 (100) | 2 (15.4) | - | 1 (10) | - | 1 (33.3) | - |
| 4 | 1 (2.1) | 1 (6.7) | - | - | - | - | - | - | - | - | - | 1 (25) | 1 (33.3) | - |
| 5 | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
| 6 | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
| 7 | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kabali, E.; Pandey, G.S.; Munyeme, M.; Kapila, P.; Mukubesa, A.N.; Ndebe, J.; Muma, J.B.; Mubita, C.; Muleya, W.; Muonga, E.M.; et al. Identification of Escherichia coli and Related Enterobacteriaceae and Examination of Their Phenotypic Antimicrobial Resistance Patterns: A Pilot Study at A Wildlife–Livestock Interface in Lusaka, Zambia. Antibiotics 2021, 10, 238. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics10030238
Kabali E, Pandey GS, Munyeme M, Kapila P, Mukubesa AN, Ndebe J, Muma JB, Mubita C, Muleya W, Muonga EM, et al. Identification of Escherichia coli and Related Enterobacteriaceae and Examination of Their Phenotypic Antimicrobial Resistance Patterns: A Pilot Study at A Wildlife–Livestock Interface in Lusaka, Zambia. Antibiotics. 2021; 10(3):238. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics10030238
Chicago/Turabian StyleKabali, Emmanuel, Girja Shanker Pandey, Musso Munyeme, Penjaninge Kapila, Andrew Nalishuwa Mukubesa, Joseph Ndebe, John Bwalya Muma, Charles Mubita, Walter Muleya, Elizabeth Muligisa Muonga, and et al. 2021. "Identification of Escherichia coli and Related Enterobacteriaceae and Examination of Their Phenotypic Antimicrobial Resistance Patterns: A Pilot Study at A Wildlife–Livestock Interface in Lusaka, Zambia" Antibiotics 10, no. 3: 238. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics10030238