
Colonization Dynamics of Multidrug-Resistant Klebsiella pneumoniae Are Dictated by Microbiota-Cluster Group Behavior over Individual Antibiotic Susceptibility: A Metataxonomic Analysis

 ,
,  , and
, and 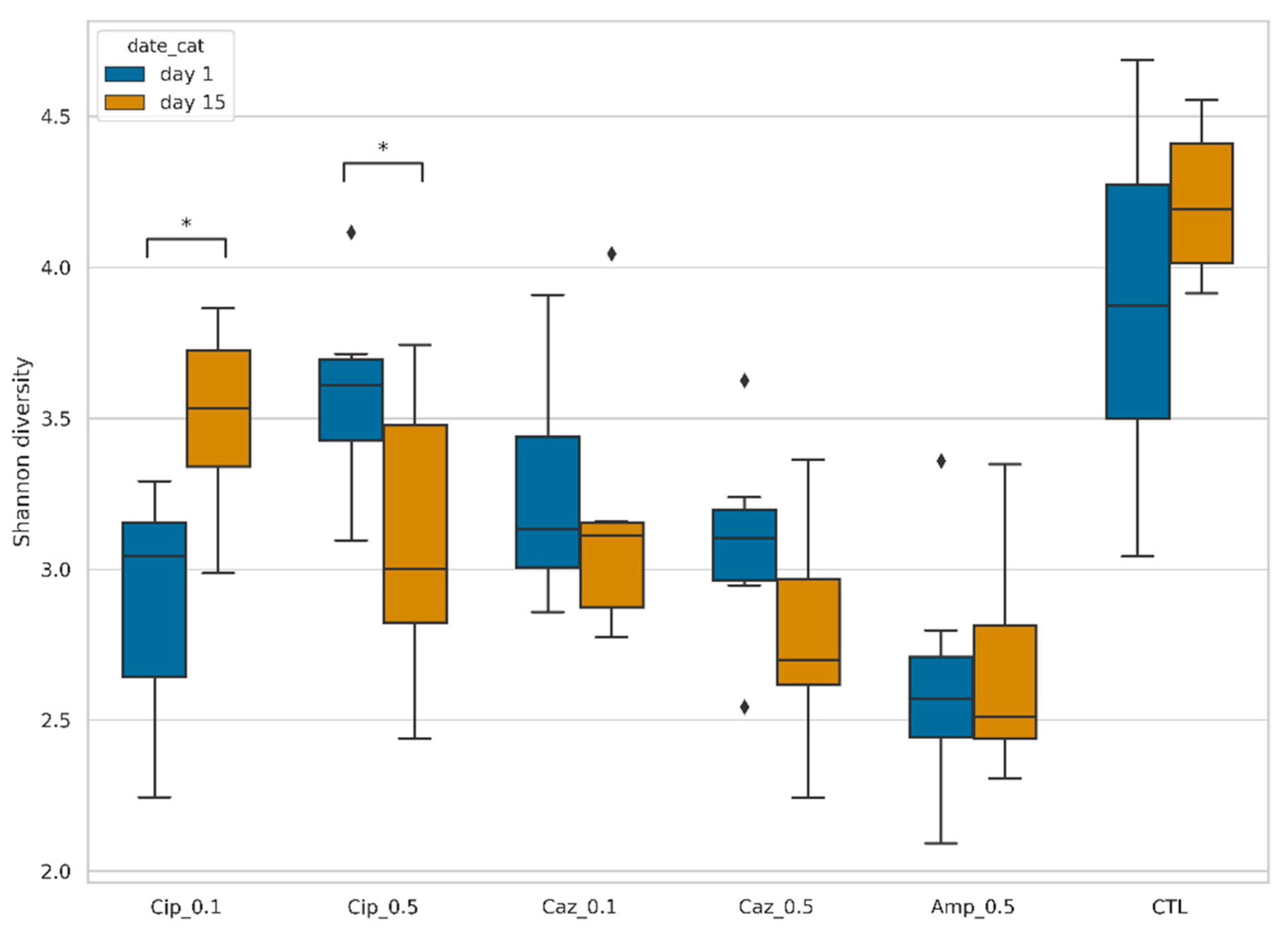{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
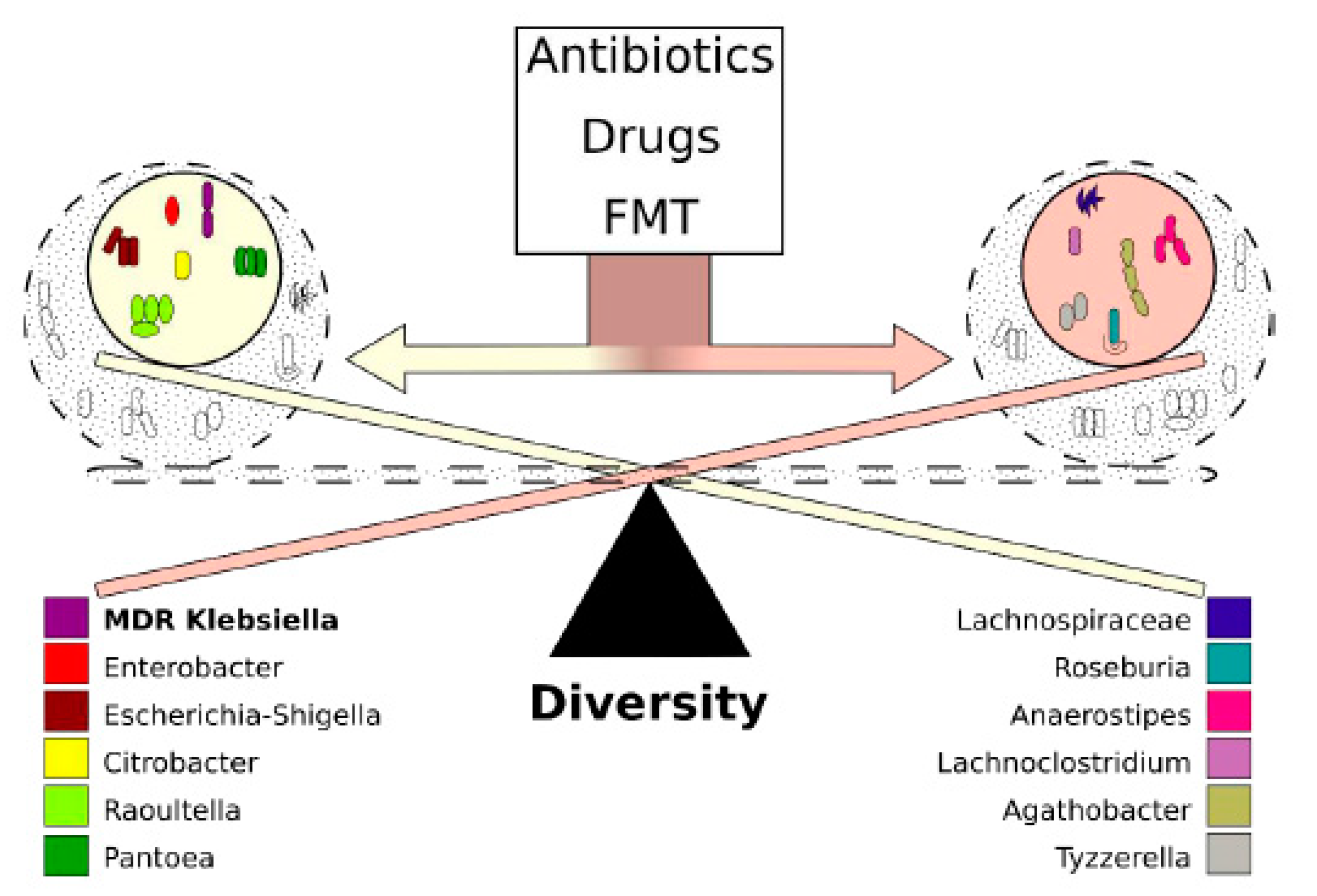
2.1. Fecal Microbiota Diversity during the Different Antibiotic Treatments after Colonization with ECKP
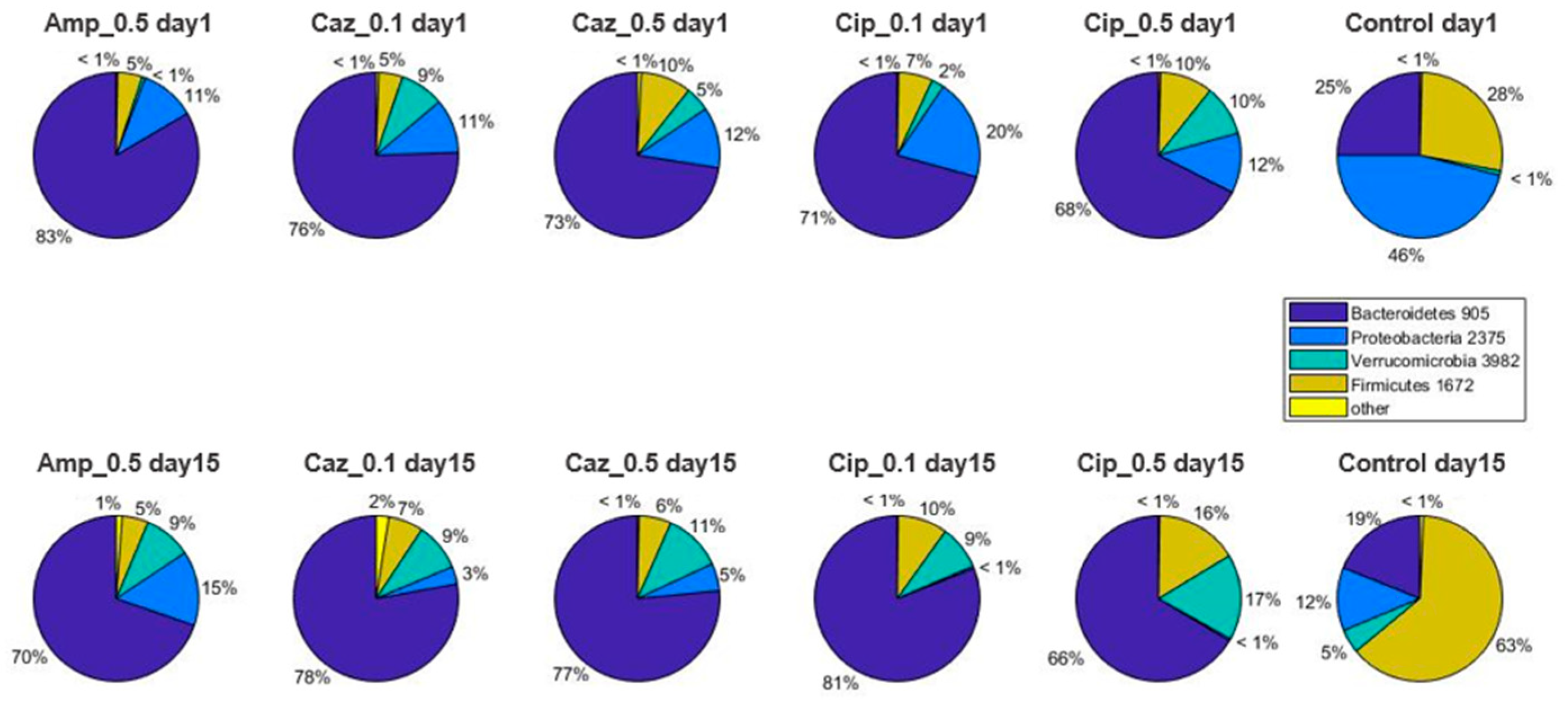
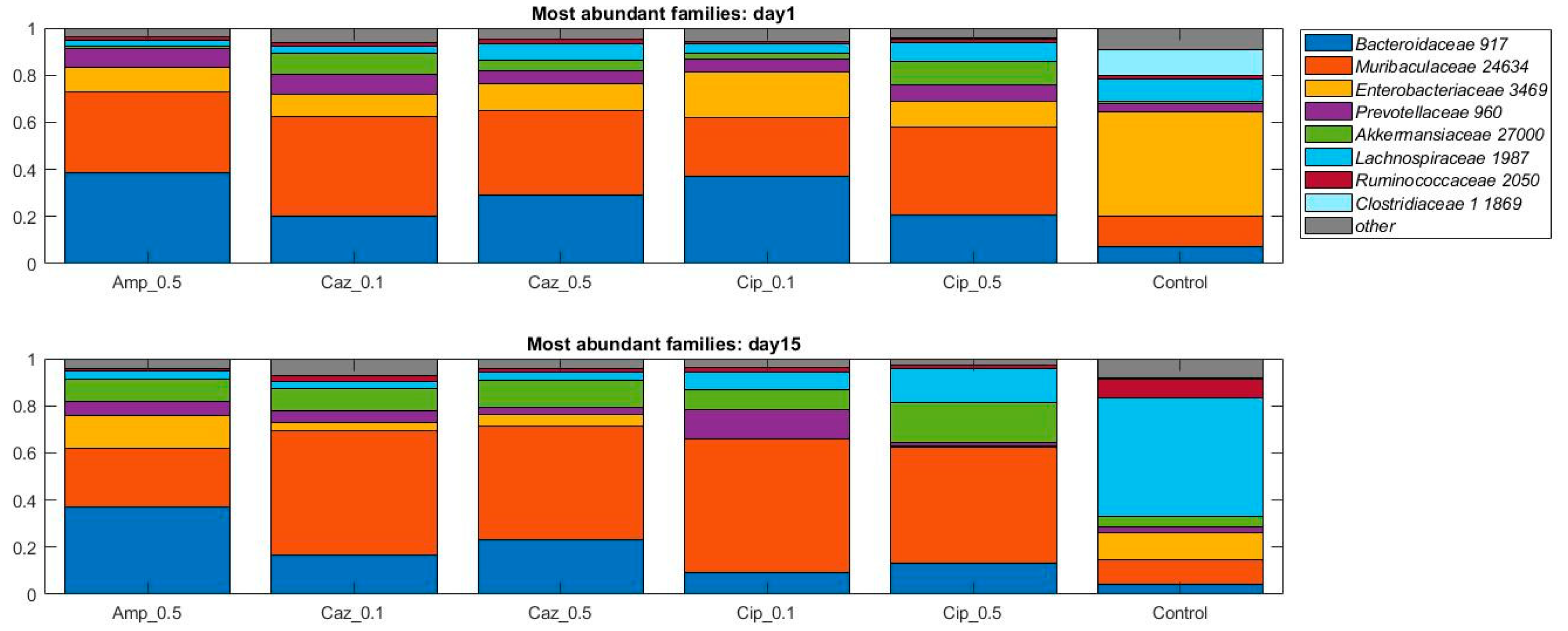
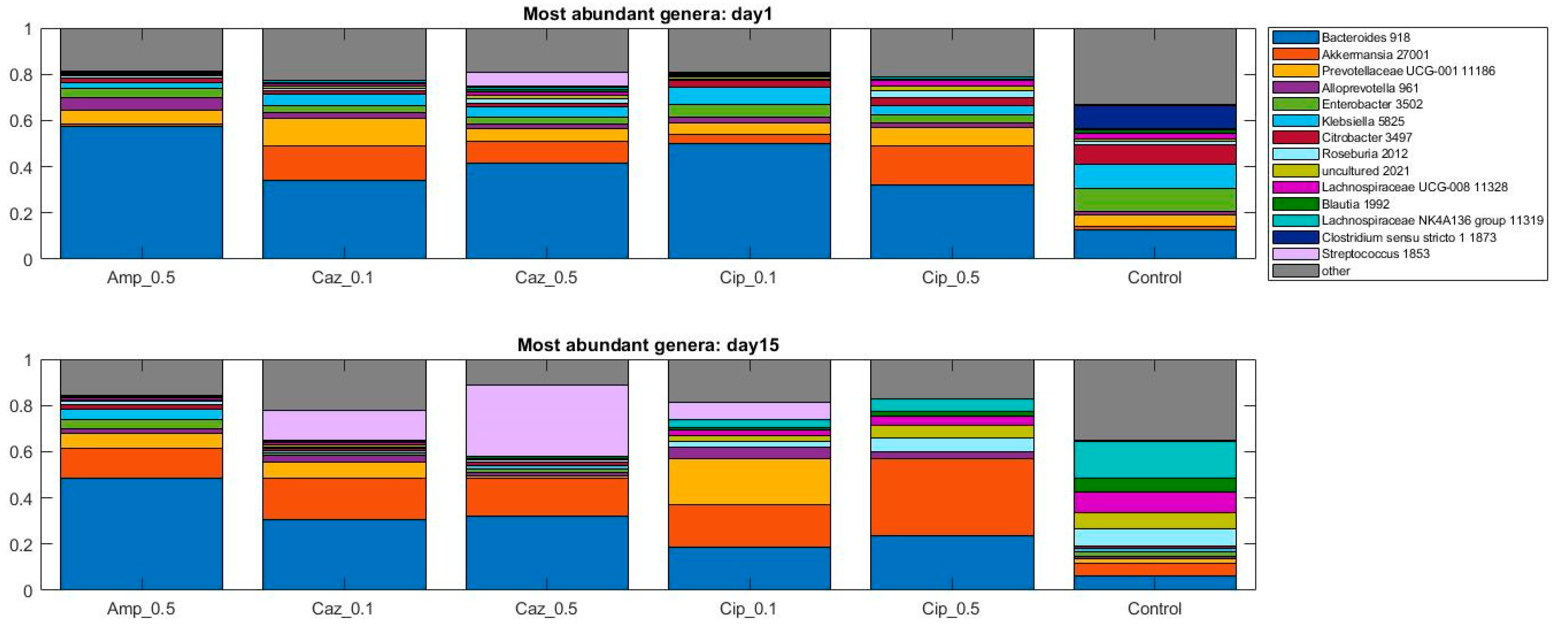
2.2. Alterations in Gut Microbiota Composition during the Different Antibiotic Treatments after Colonization with ECKP
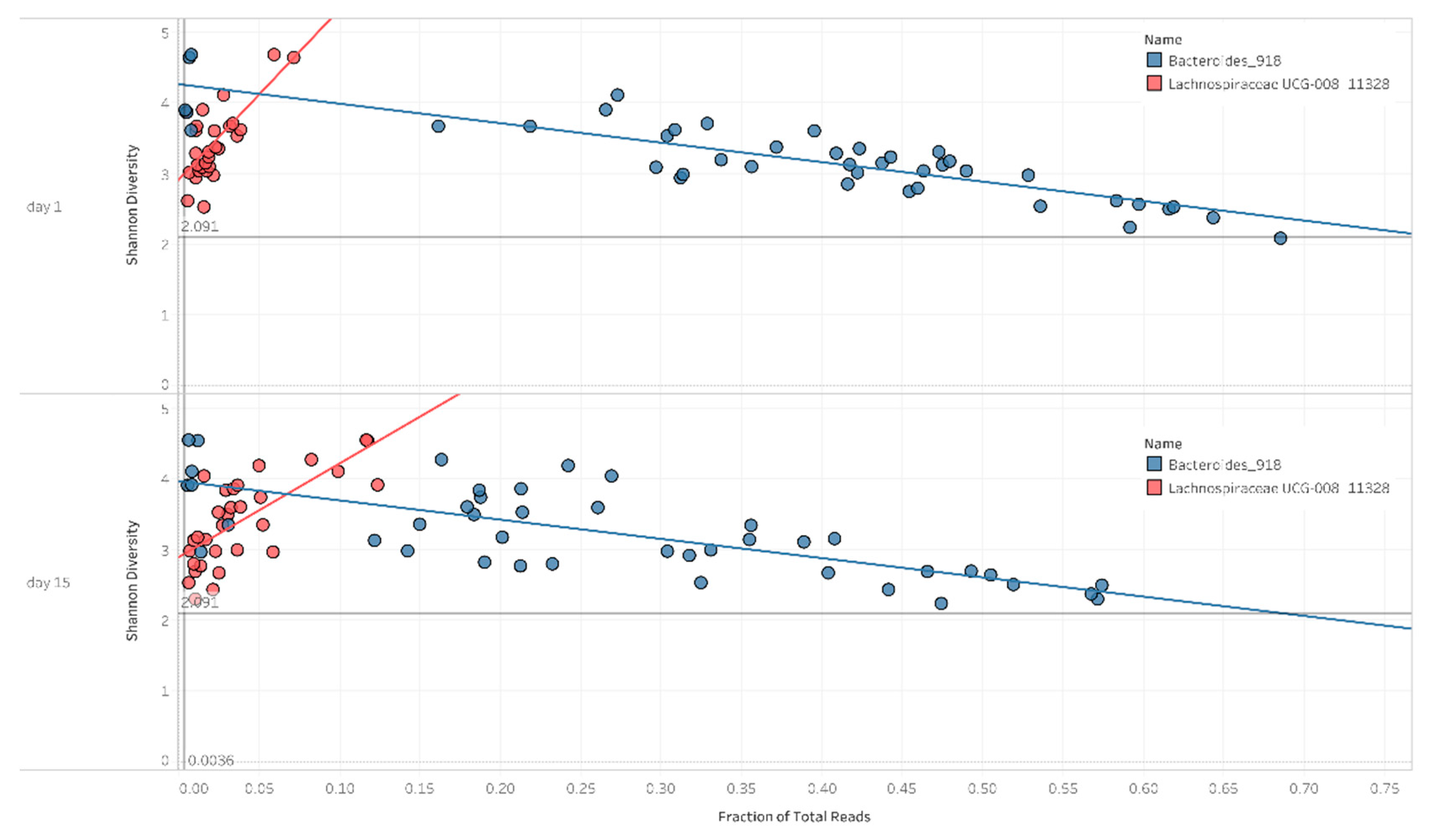
2.3. Correlation between Diversity and Taxonomy
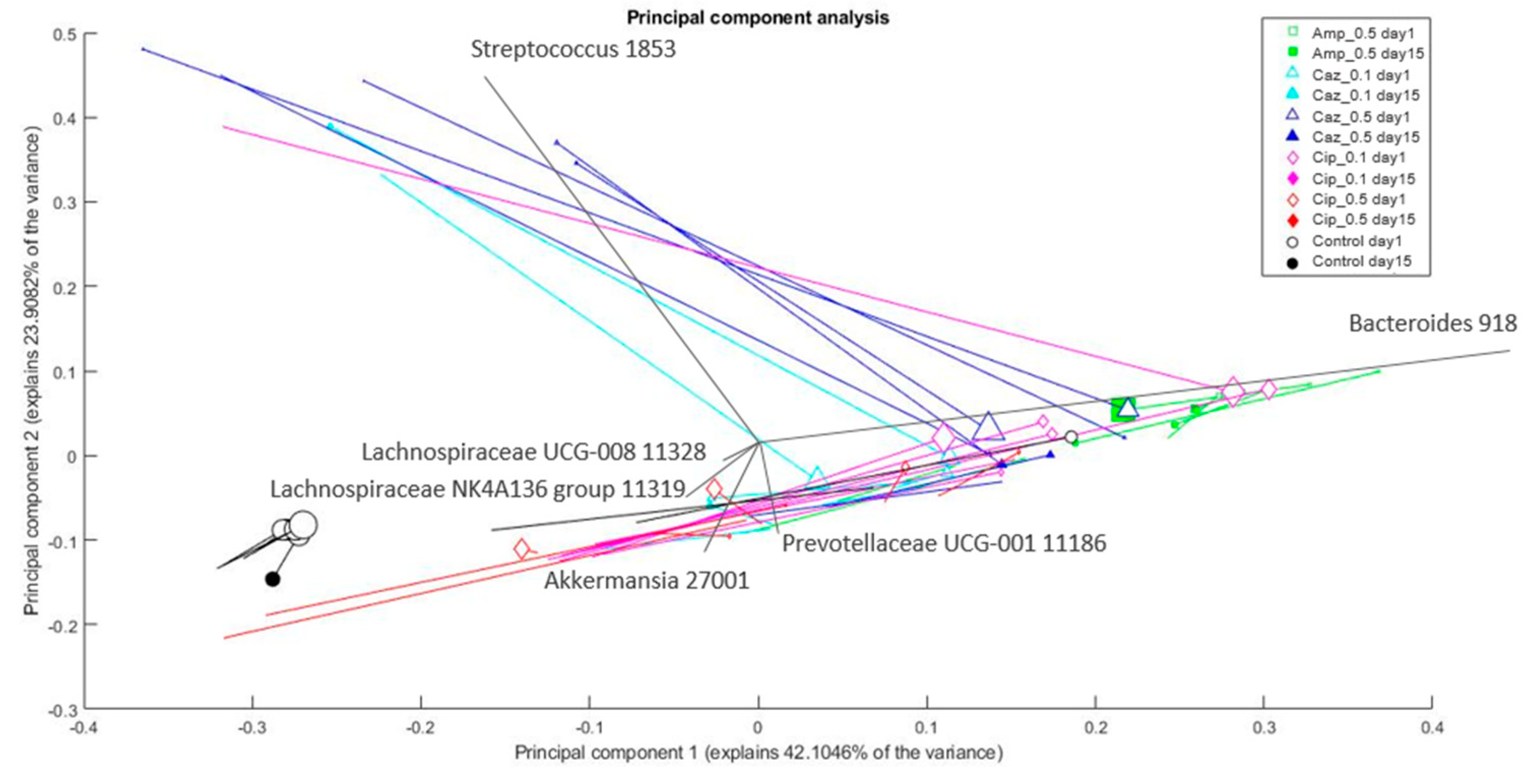
2.4. Microbiota Changes Corresponding to Individual Treatment during Colonization
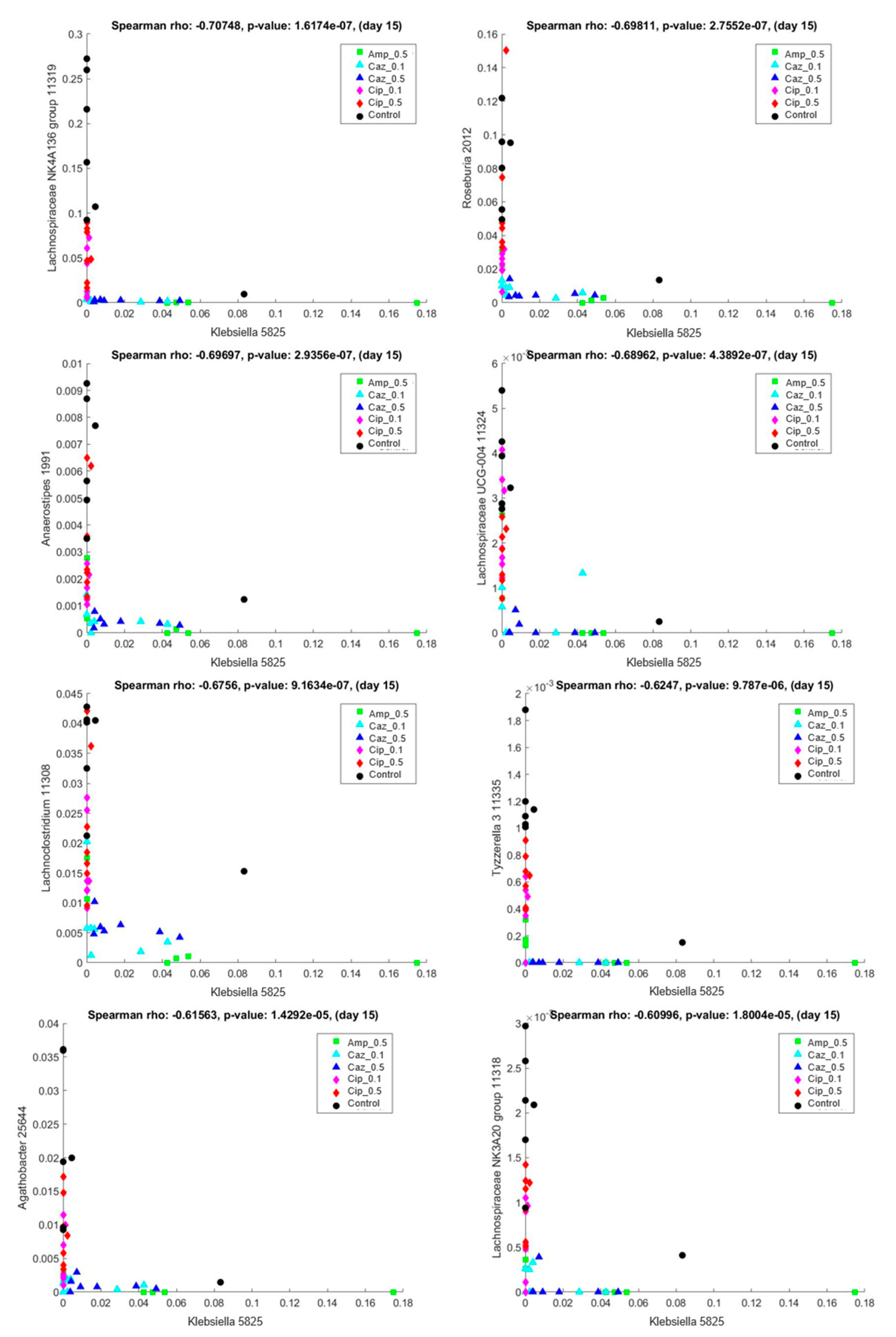
2.5. Interaction Analysis between the Colonizing ECKP and other Genera of the Microbiota
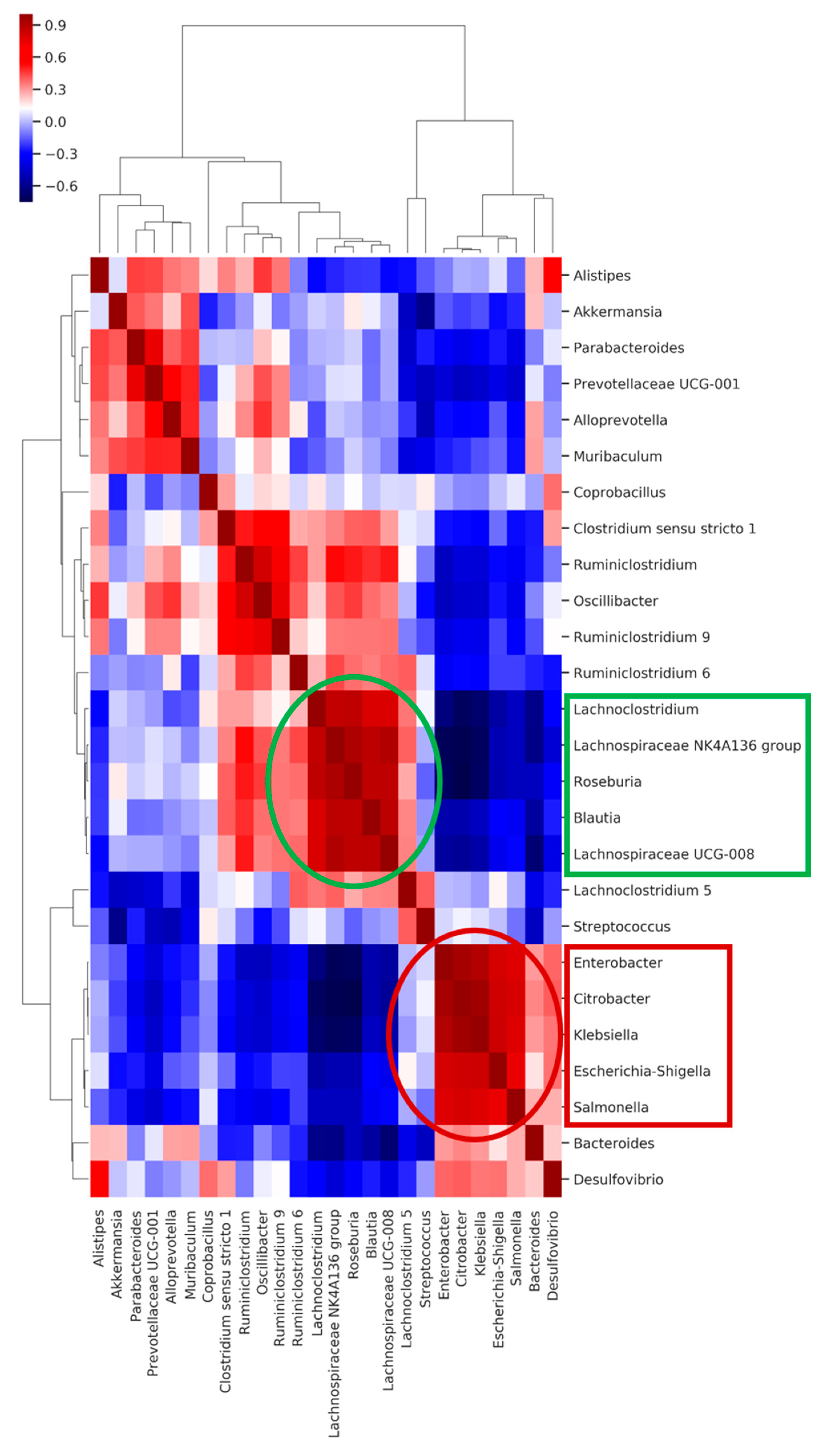
2.6. Co-Occurring Clusters of Bacteria during Dynamic Changes in the Microbiota Associated with ECKP Colonization
3. Discussion
4. Materials and Methods
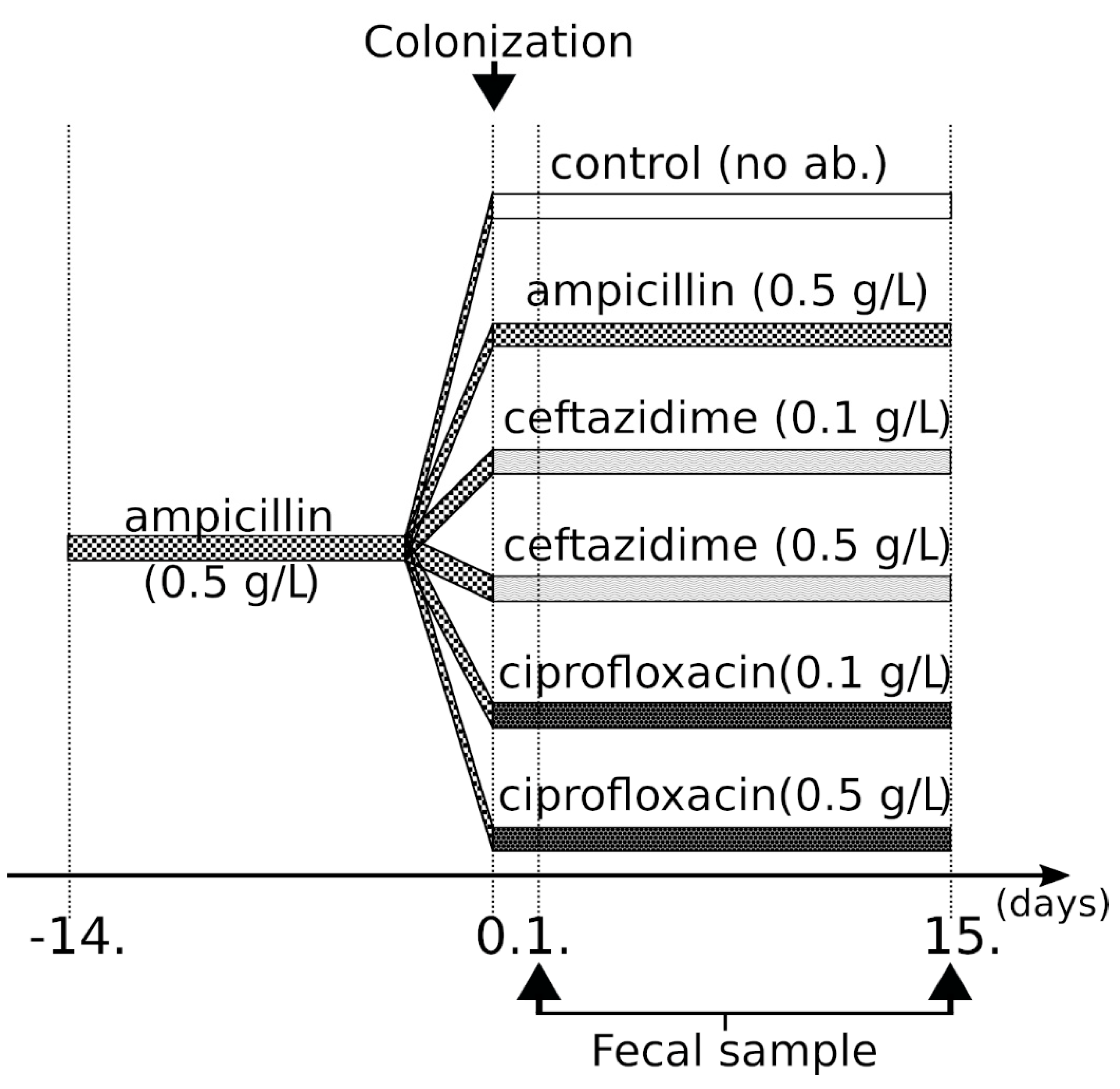
4.1. Study Design, Bacterial Strains
4.2. Microbiota Analysis
4.3. Metataxonomic Analysis
4.4. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Amp | ampicillin |
| Amp_0.5 | 0.5 g/L ampicillin |
| BSL | biosafety level |
| Caz | ceftazidime |
| Caz_0.1 | 0.1 g/L ceftazidime |
| Caz_0.5 | 0.5 g/L ceftazidime |
| CFU | colony-forming unit |
| Cip | ciprofloxacin |
| Cip_0.1 | 0.1 g/L ciprofloxacin |
| Cip_0.5 | 0.5 g/L ciprofloxacin |
| CRE | carbapenem-resistant Enterobacterales |
| CTL | control group |
| DALY | disability-adjusted life years |
| ECKP | ESBL- and carbapenemase-producing Klebsiella pneumoniae |
| EBSL | extended-spectrum β-lactamase |
| FMT | fecal microbiota transplantation |
| HGT | horizontal gene transfer |
| ICU | intensive care unit |
| iNOS | Nitric oxide synthase |
| KPC | Klebsiella pneumoniae carbapenemase |
| MDR | multidrug-resistant |
| NDM | New Delhi metallo-beta-lactamase |
| OUT | operational taxonomic unit |
| PCA | principal component analysis |
| PCR | polymerase chain reaction |
| QC | quality control |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 |
| SCFA | short-chain fatty acid |
| SSU | small subunit |
| T | thymine |
| U | uracil |
| WRE | vancomycin-resistant enterococci |
| WHO | World Health Organization |
References
- Lob, S.H.; Biedenbach, D.J.; Badal, R.E.; Kazmierczak, K.M.; Sahm, D.F. Antimicrobial resistance and resistance mechanisms of Enterobacteriaceae in ICU and non-ICU wards in Europe and North America: SMART 2011–2013. J. Glob. Antimicrob. Resist. 2015, 3, 190–197. [Google Scholar] [CrossRef]
- Jean, S.S.; Coombs, G.; Ling, T.; Balaji, V.; Rodrigues, C.; Mikamo, H.; Kim, M.J.; Rajasekaram, D.G.; Mendoza, M.; Tan, T.Y.; et al. Epidemiology and antimicrobial susceptibility profiles of pathogens causing urinary tract infections in the Asia-Pacific region: Results from the Study for Monitoring Antimicrobial Resistance Trends (SMART), 2010–2013. Int. J. Antimicrob. Agents 2016, 47, 328–334. [Google Scholar] [CrossRef] [PubMed]
- Grundmann, H.; Glasner, C.; Albiger, B.; Aanensen, D.M.; Tomlinson, C.T.; Andrasevic, A.T.; Cantón, R.; Carmeli, Y.; Friedrich, A.W.; Giske, C.G.; et al. Occurrence of car-bapenemase-producing Klebsiella pneumoniae and Escherichia coli in the European survey of carbapenemase-producing Enterobacteriaceae (EuSCAPE): A prospective, multinational study. Lancet Infect. Dis. 2017, 17, 153–163. [Google Scholar] [CrossRef] [Green Version]
- Karaiskos, I.; Giamarellou, H. Carbapenem-sparing strategies for ESBL producers: When and how. Antibiotics 2020, 9, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cano, A.; Gutierrez-Gutierrez, B.; Machuca, I.; Gracia-Ahufinger, I.; Perez-Nadales, E.; Causse, M.; Castón, J.J.; Guzman-Puche, J.; Torre-Giménez, J.; Kindelán, L.; et al. Risks of infection and mortality among patients colonized with Klebsiella pneumoniae carbapenemase-producing K. pneumoniae: Validation of scores and proposal for management. Clin. Infect. Dis. 2018, 66, 1204–1210. [Google Scholar] [CrossRef] [Green Version]
- Pitout, J.D.D.; Nordmann, P.; Poirel, L. Carbapenemase-producing Klebsiella pneumoniae, a key pathogen set for global nosocomial dominance. Antimicrob. Agents Chemother. 2015, 59, 5873–5884. [Google Scholar] [CrossRef] [Green Version]
- Forcina, A.; Baldan, R.; Marasco, V.; Cichero, P.; Bondanza, A.; Noviello, M.; Piemontese, S.; Soliman, C.; Greco, R.; Lorentino, F.; et al. Control of infectious mortality due to carbapenemase-producing Klebsiella pneumoniae in hematopoietic stem cell transplantation. Bone Marrow Transplant. 2017, 52, 114–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freire, M.P.; Abdala, E.; Moura, M.L.; De Paula, F.J.; Spadão, F.; Caiaffa-Filho, H.H.; David-Neto, E.; Nahas, W.C.; Pierrotti, L.C. Risk factors and outcome of infections with Klebsiella pneumoniae carbapenemase-producing K. pneumoniae in kidney transplant recipients. Infection 2015, 43, 315–323. [Google Scholar] [CrossRef]
- Freire, M.P.; Pierrotti, L.C.; Filho, H.H.; Ibrahim, K.Y.; Magri, A.S.; Bonazzi, P.R.; Hajar, L.; Diz, M.P.; Pereira, J.; Hoff, P.M.; et al. Infection with Klebsiella pneumoniae carbapenemase (KPC)-producing Klebsiella pneumoniae in cancer patients. Eur. J. Clin. Microbiol. Infect. Dis. 2015, 34, 277–286. [Google Scholar] [CrossRef]
- Rivera-Espinar, F.; Machuca, I.; Tejero, R.; Rodríguez, J.; Mula, A.; Marfil, E.; Cano, Á.; Gutiérrez-Gutiérrez, B.; Rodríguez, M.; Pozo, J.C.; et al. Impact of KPC Production and high-level meropenem resistance on all-cause mortality of ventilator-associated pneumonia in association with Klebsiella pneumoniae. Antimicrob. Agents Chemother. 2020, 64, 02164-19. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, Q.; Yin, Y.; Chen, H.; Jin, L.; Gu, B.; Xie, L.; Yang, C.; Ma, X.; Li, H.; et al. Epidemiology of carbapenem-resistant Enterobacteriaceae infections: Report from the China CRE Network. Antimicrob. Agents Chemother. 2017, 62, 01882-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, S.-K.; Ma, L.; Chan, M.-C.; Lin, Y.-T.; Fung, C.-P.; Wu, T.-L.; Chuang, Y.-C.; Lu, P.-L.; Wang, J.-T.; Lin, J.-C.; et al. Carbapenem nonsusceptible Klebsiella pneumoniae in Taiwan: Dissemination and increasing resistance of carbapenemase producers during 2012–2015. Sci. Rep. 2018, 8, e8468. [Google Scholar] [CrossRef] [Green Version]
- Tseng, W.-P.; Chen, Y.-C.; Chen, S.-Y.; Chen, S.-Y.; Chang, S.-C. Risk for subsequent infection and mortality after hospitalization among patients with multidrug-resistant gram-negative bacteria colonization or infection. Antimicrob. Resist. Infect. Control. 2018, 7, e93. [Google Scholar] [CrossRef] [Green Version]
- Han, R.; Shi, Q.; Wu, S.; Yin, D.; Peng, M.; Dong, D.; Zheng, Y.; Guo, Y.; Zhang, R.; Hu, F.; et al. Dissemination of Carbapenemases (KPC, NDM, OXA-48, IMP, and VIM) among carbapenem-resistant Enterobacteriaceae isolated from adult and children patients in China. Front. Cell. Infect. Microbiol. 2020, 10, 314. [Google Scholar] [CrossRef]
- World Health Organization. Prioritization of Pathogens to Guide Discovery, Research and Development of New Antibiotics for Drug Resistant Bacterial Infections, Including Tuberculosis; WHO: Geneva, Switzerland, 2017; p. 74. [Google Scholar]
- Kim, S.; Covington, A.; Pamer, E.G. The intestinal microbiota: Antibiotics, colonization resistance, and enteric pathogens. Immunol. Rev. 2017, 279, 90–105. [Google Scholar] [CrossRef] [PubMed]
- Khanna, S. Microbiota Replacement Therapies: Innovation in gastrointestinal care. Clin. Pharmacol. Ther. 2018, 103, 102–111. [Google Scholar] [CrossRef]
- Do, T.T.; Tamames, J.; Stedtfeld, R.D.; Guo, X.; Murphy, S.; Tiedje, J.M.; Walsh, F. Antibiotic resistance gene detection in the microbiome context. Microb. Drug Resist. 2018, 24, 542–546. [Google Scholar] [CrossRef]
- Karanika, S.; Karantanos, T.; Arvanitis, M.; Grigoras, C.; Mylonakis, E. Fecal colonization with extended-spectrum beta-lactamase-producing Enterobacteriaceae and risk factors among healthy individuals: A systematic review and metaanalysis. Clin. Infect. Dis. 2016, 63, 310–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, X.; Wu, S.; Hao, M.; Zhu, J.; Ding, B.; Yang, Y.; Xu, X.; Wang, M.; Yang, F.; Hu, F. The colonization of carbapenem-resistant Klebsiella pneumoniae: Epi-demiology, resistance mechanisms, and risk factors in patients admitted to intensive care units in China. J. Infect. Dis. 2020, 221, S206–S214. [Google Scholar] [CrossRef]
- Arcilla, M.S.; van Hattem, J.M.; Haverkate, M.R.; Bootsma, M.C.J.; van Genderen, P.J.J.; Goorhuis, A.; Grobusch, M.P.; Lashof, A.M.O.; Molhoek, N.; Schultsz, C.; et al. Import and spread of ex-tended-spectrum beta-lactamase-producing Enterobacteriaceae by international travellers (COMBAT study): A prospective, multicentre cohort study. Lancet Infect. Dis. 2017, 17, 78–85. [Google Scholar] [CrossRef]
- Woerther, P.L.; Andremont, A.; Kantele, A. Travel-acquired ESBL-producing Enterobacteriaceae: Impact of colonization at individual and community level. J. Travel Med. 2017, 24, S29–S34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimasaki, T.; Seekatz, A.; Bassis, C.; Rhee, Y.; Yelin, R.D.; Fogg, L.; Dangana, T.; Cisneros, E.C.; Weinstein, R.A.; Okamoto, K.; et al. Increased relative abundance of Klebsiella pneumoniae carbapenemase-producing Klebsiella pneumoniae within the gut microbiota is associated with risk of bloodstream infection in long-term acute care hospital patients. Clin. Infect. Dis. 2019, 68, 2053–2059. [Google Scholar] [CrossRef]
- Otter, J.A.; Natale, A.; Batra, R.; Tosas Auguet, O.; Dyakova, E.; Goldenberg, S.D.; Edgeworth, J.D. Individual- and community-level risk factors for ESBL Enterobacteriaceae colonization identified by universal admission screening in London. Clin. Microbiol. Infect. 2019, 25, 1259–1265. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-S.; Lai, L.-C.; Chen, Y.-A.; Lin, K.-Y.; Chou, Y.-H.; Chen, H.-C.; Wang, S.-S.; Wang, J.-T.; Chang, S.-C. Colonization with multidrug-770 resistant organisms among healthy adults in the community setting: Prevalence, risk factors, and composition of gut microbiome. Front. Microbiol. 2020, 11, 1402. [Google Scholar] [CrossRef]
- Schaumburg, F.; Sertic, S.M.; Correa-Martinez, C.; Mellmann, A.; Kock, R.; Becker, K. Acquisition and colonization dynamics of an-timicrobial-resistant bacteria during international travel: A prospective cohort study. Clin. Microbiol. Infect. 2019, 25, 1287 e1–1287 e7. [Google Scholar] [CrossRef]
- Meurs, L.; Lempp, F.S.; Lippmann, N.; Trawinski, H.; Rodloff, A.C.; Eckardt, M.; Klingeberg, A.; Eckmanns, T.; Walter, J.; Lübbert, C.; et al. Intestinal colonization with extended-spectrum beta-lactamase producing Enterobacterales (ESBL-PE) during long distance travel: A cohort study in a German travel clinic (2016–2017). Travel Med. Infect. Dis. 2020, 33, 101521. [Google Scholar] [CrossRef]
- Collingwood, A.; Blostein, F.; Seekatz, A.M.; Wobus, C.E.; Woods, R.J.; Foxman, B.; Bachman, M.A. Epidemiological and microbiome associations between Klebsiella pneumoniae and vancomycin-resistant Enterococcus colonization in intensive care unit patients. Open Forum Infect. Dis. 2020, 7, 012. [Google Scholar] [CrossRef] [PubMed]
- Modi, S.R.; Collins, J.J.; Relman, D.A. Antibiotics and the gut microbiota. J. Clin. Investig. 2014, 124, 4212–4218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stercz, B.; Tóth, Á.; Gajdács, M.; Domokos, J.; Horváth, V.; Ostorházi, E.; Makra, N.; Kocsis, B.; Juhász, J.; Ligeti, B.; et al. The influence of antibiotics on transitory resistome during gut colonization with CTX-M-15 and OXA-162 producing Klebsiella pneumoniae ST15. Sci. Rep 2021, in press. [Google Scholar]
- Cassini, A.; Hogberg, L.D.; Plachouras, D.; Quattrocchi, A.; Hoxha, A.; Simonsen, G.S.; Colomb-Cotinat, M.; Kretzschmar, M.E.; Devleesschauwer, B.; Cecchini, M.; et al. Attributable deaths and disability-adjusted life-years caused by infections with antibiotic-resistant bacteria in the EU and the European Economic Area in 2015: A population-level modelling analysis. Lancet Infect. Dis. 2019, 19, 56–66. [Google Scholar] [CrossRef] [Green Version]
- Woerther, P.L.; Burdet, C.; Chachaty, E.; Andremont, A. Trends in human fecal carriage of extended-spectrum beta-lactamases in the community: Toward the globalization of CTX-M. Clin. Microbiol. Rev. 2013, 26, 744–758. [Google Scholar] [CrossRef] [Green Version]
- Willems, R.P.J.; van Dijk, K.; Ket, J.C.F.; Vandenbroucke-Grauls, C. Evaluation of the association between gastric acid suppression and risk of intestinal colonization with multidrug-resistant microorganisms: A systematic review and meta-analysis. JAMA Intern. Med. 2020, 180, 561–571. [Google Scholar] [CrossRef] [Green Version]
- Pires, J.; Kraemer, J.G.; Kuenzli, E.; Kasraian, S.; Tinguely, R.; Hatz, C.; Endimiani, A.; Hilty, M. Gut microbiota dynamics in travelers returning from India colonized with extended-spectrum cephalosporin-resistant Enterobacteriaceae: A longitudinal study. Travel Med. Infect. Dis. 2019, 27, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Haverkate, M.R.; Derde, L.P.G.; Brun-Buisson, C.; Bonten, M.J.M.; Bootsma, M.C.J. Duration of colonization with antimicrobial-resistant bacteria after ICU discharge. Intensiv. Care Med. 2014, 40, 564–571. [Google Scholar] [CrossRef] [Green Version]
- Zimmerman, F.S.; Assous, M.V.; Bdolah-Abram, T.; Lachish, T.; Yinnon, A.M.; Wiener-Well, Y. Duration of carriage of car-bapenem-resistant Enterobacteriaceae following hospital discharge. Am. J. Infect. Control. 2013, 41, 190–194. [Google Scholar] [CrossRef]
- Doron, S.; Snydman, D.R. Risk and Safety of Probiotics. Clin. Infect. Dis. 2015, 60, S129–S134. [Google Scholar] [CrossRef] [Green Version]
- Jang, M.-O.; An, J.H.; Jung, S.-I.; Park, K.-H. RefractoryClostridium difficileInfection Cured With Fecal Microbiota Transplantation in Vancomycin-Resistant Enterococcus Colonized Patient. Intest. Res. 2015, 13, 80–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bilinski, J.; Grzesiowski, P.; Sorensen, N.; Madry, K.; Muszynski, J.; Robak, K.; Wroblewska, M.; Dzieciatkowski, T.; Dulny, G.; Dwilewicz-Trojaczek, J.; et al. Fecal Microbiota Transplantation in Patients With Blood Disorders Inhibits Gut Colonization With Antibiotic-Resistant Bacteria: Results of a Prospective, Single-Center Study. Clin. Infect. Dis. 2017, 65, 364–370. [Google Scholar] [CrossRef]
- Tavoukjian, V. Faecal microbiota transplantation for the decolonization of antibiotic-resistant bacteria in the gut: A systematic review and meta-analysis. J. Hosp. Infect. 2019, 102, 174–188. [Google Scholar] [CrossRef]
- Dinh, A.; Fessi, H.; Duran, C.; Batista, R.; Michelon, H.; Bouchand, F.; Lepeule, R.; Vittecoq, D.; Escaut, L.; Sobhani, I.; et al. Clearance of carbapenem-resistant Enterobacteriaceae vs vancomycin-resistant enterococci carriage after faecal microbiota transplant: A prospective comparative study. J. Hosp. Infect. 2018, 99, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; De Groot, P.F.; Geerlings, S.E.; Hodiamont, C.J.; Belzer, C.; Berge, I.J.M.T.; De Vos, W.M.; Bemelman, F.J.; Nieuwdorp, M. Fecal microbiota transplantation against intestinal colonization by extended spectrum beta-lactamase producing Enterobacteriaceae: A proof of principle study. BMC Res. Notes 2018, 11, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Jouhten, H.; Mattila, E.; Arkkila, P.; Satokari, R. Reduction of antibiotic resistance genes in intestinal microbiota of patients with recurrent Clostridium difficile infection after fecal microbiota transplantation. Clin. Infect. Dis. 2016, 63, 710–711. [Google Scholar] [CrossRef] [Green Version]
- Leung, V.; Vincent, C.; Edens, T.J.; Miller, M.; Manges, A.R. Antimicrobial resistance gene acquisition and depletion following fecal microbiota transplantation for recurrent Clostridium difficile Infection. Clin. Infect. Dis. 2018, 66, 456–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeFilipp, Z.; Bloom, P.P.; Soto, M.T.; Mansour, M.K.; Sater, M.R.; Huntley, M.H.; turbett, S.; Chung, R.T.; Chen, Y.-B.; Hohmann, E.L. Drug-resistant E. coli bacteremia transmitted by fecal microbiota Transplant. N. Engl. J. Med. 2019, 381, 2043–2050. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.C.; Chan, F.K.L.; Chan, P.K.S. Screening FMT donors during the COVID-19 pandemic: A protocol for stool SARS-CoV-2 viral quantification. Lancet Gastroenterol. Hepatol. 2020, 5, 642–643. [Google Scholar] [CrossRef]
- FDA. Fecal Microbiota for Transplantation: New Safety Information—Regarding Additional Protections for Screening Donors for COVID-19 and Exposure to SARS-CoV-2 and Testing for SARS-CoV-2. Available online: https://www.fda.gov/safety/medical-product-safety-information/fecal-microbiota-transplantation-new-safety-information-regarding-additional-protections-screening (accessed on 10 September 2020).
- Zao, T.; Zhang, F.; Lui, C.Y.G.; Yeoh, Y.K.; Li, A.Y.; Zhan, H.; Wan, Y.; Chung, A.C.K.; Cheung, C.P.; Chen, N.; et al. Alterations in gut micro-biota of patients with COVID-19 during time of hospitalization. Gastroenterology 2020, 159, 944–955.e8. [Google Scholar] [CrossRef]
- Corbellino, M.; Kieffer, N.; Kutateladze, M.; Balarjishvili, N.; Leshkasheli, L.; Askilashvili, L.; Tsertsvadze, G.; Rimoldi, S.G.; Nizharadze, D.; Hoyle, N.; et al. Eradication of a multidrug-resistant, carbapenemase-producing Klebsiella pneumoniae isolate following oral and intra-rectal therapy with a custom made, lytic bacteriophage preparation. Clin. Infect. Dis. 2019, 70, 1998–2001. [Google Scholar] [CrossRef]
- Caballero, S.; Carter, R.; Ke, X.; Susac, B.; Leiner, I.M.; Kim, G.J.; Miller, L.; Ling, L.; Manova, K.; Pamer, E.G. Distinct but spatially overlapping intestinal niches for vancomycin-resistant Enterococcus faecium and carbapenem-resistant Klebsiella pneumoniae. PLoS Pathog. 2015, 11, e1005132. [Google Scholar] [CrossRef] [PubMed]
- Ubeda, C.; Bucci, V.; Caballero, S.; Djukovic, A.; Toussaint, N.C.; Equinda, M.; Lipuma, L.; Ling, L.; Gobourne, A.; No, D.; et al. Intestinal microbiota containing Barnesiella species cures vancomycin-resistant Enterococcus faecium colonization. Infect. Immun. 2013, 81, 965–973. [Google Scholar] [CrossRef] [Green Version]
- Piewngam, P.; Quinones, M.; Thirakittiwatthana, W.; Yungyuen, T.; Otto, M.; Kiratisin, P. Composition of the intestinal microbiota in extended-spectrum beta-lactamase-producing Enterobacteriaceae carriers and non-carriers in Thailand. Int. J. Antimicrob. Agents 2019, 53, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, M.; Méndez-García, C.; Rojo, D.; Barbas, C.; Moya, A. Antibiotic use and microbiome function. Biochem. Pharmacol. 2017, 134, 114–126. [Google Scholar] [CrossRef]
- Penders, J.; Stobberingh, E.E.; Savelkoul, P.H.M.; Wolffs, P.F.G. The human microbiome as a reservoir of antimicrobial resistance. Front. Microbiol. 2013, 4, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, D.J.; Preston, T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes 2016, 7, 189–200. [Google Scholar] [CrossRef] [Green Version]
- Sorbara, M.T.; Dubin, K.; Littmann, E.R.; Moody, T.U.; Fontana, E.; Seok, R.; Leiner, I.M.; Taur, Y.; Peled, J.U.; Brink, M.R.V.D.; et al. Inhibiting antibiotic-resistant Enterobacteriaceae by microbiota-mediated intracellular acidification. J. Exp. Med. 2018, 216, 84–98. [Google Scholar] [CrossRef] [PubMed]
- Barcenilla, A.; Pryde, S.E.; Martin, J.C.; Duncan, S.H.; Stewart, C.S.; Henderson, C.; Flint, H.J. Phylogenetic relationships of butyr-ate-producing bacteria from the human gut. Appl. Environ. Microbiol. 2000, 66, 1654–1661. [Google Scholar] [CrossRef] [Green Version]
- Sorbara, M.T.; Littmann, E.R.; Fontana, E.; Moody, T.U.; Kohout, C.E.; Gjonbalaj, M.; Eaton, V.; Seok, R.; Leiner, I.M.; Pamer, E.G. Functional and genomic variation between human-derived isolates of Lachnospiraceae reveals inter- and intra-species diversity. Cell Host Microbe 2020, 28, 134–146.e4. [Google Scholar] [CrossRef] [PubMed]
- Ricke, S. Perspectives on the use of organic acids and short chain fatty acids as antimicrobials. Poult. Sci. 2003, 82, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Byndloss, M.X.; Olsan, E.E.; Rivera-Chavez, F.; Tiffany, C.R.; Cevallos, S.A.; Lokken, K.L.; Torres, T.P.; Byndloss, A.J.; Faber, F.; Gao, Y.; et al. Microbiota-activated PPAR-gamma signaling inhibits dysbiotic Enterobacteriaceae expansion. Science 2017, 357, 570–575. [Google Scholar] [CrossRef]
- Leo, S.; Lazarevic, V.; Girard, M.; Gaïa, N.; Schrenzel, J.; De Lastours, V.; Fantin, B.; Bonten, M.; Carmeli, Y.; Rondinaud, E.; et al. Metagenomic characterization of gut microbiota of carriers of extended-spectrum beta-lactamase or carbapenemase-producing Enterobacteriaceae following treatment with oral antibiotics and fecal microbiota transplantation: Results from a multicenter randomized trial. Microorganisms 2020, 8, 941. [Google Scholar] [CrossRef]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.; Salzberg, S. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Breitwieser, F.P.; Thielen, P.; Salzberg, S.L. Bracken: Estimating species abundance in metagenomics data. PeerJ Comput. Sci. 2017, 3, e104. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Juhász, J.; Ligeti, B.; Gajdács, M.; Makra, N.; Ostorházi, E.; Farkas, F.B.; Stercz, B.; Tóth, Á.; Domokos, J.; Pongor, S.; et al. Colonization Dynamics of Multidrug-Resistant Klebsiella pneumoniae Are Dictated by Microbiota-Cluster Group Behavior over Individual Antibiotic Susceptibility: A Metataxonomic Analysis. Antibiotics 2021, 10, 268. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics10030268
Juhász J, Ligeti B, Gajdács M, Makra N, Ostorházi E, Farkas FB, Stercz B, Tóth Á, Domokos J, Pongor S, et al. Colonization Dynamics of Multidrug-Resistant Klebsiella pneumoniae Are Dictated by Microbiota-Cluster Group Behavior over Individual Antibiotic Susceptibility: A Metataxonomic Analysis. Antibiotics. 2021; 10(3):268. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics10030268
Chicago/Turabian StyleJuhász, János, Balázs Ligeti, Márió Gajdács, Nóra Makra, Eszter Ostorházi, Ferenc Balázs Farkas, Balázs Stercz, Ákos Tóth, Judit Domokos, Sándor Pongor, and et al. 2021. "Colonization Dynamics of Multidrug-Resistant Klebsiella pneumoniae Are Dictated by Microbiota-Cluster Group Behavior over Individual Antibiotic Susceptibility: A Metataxonomic Analysis" Antibiotics 10, no. 3: 268. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics10030268