
The Phosin PptA Plays a Negative Role in the Regulation of Antibiotic Production in Streptomyces lividans

 ,
,  , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
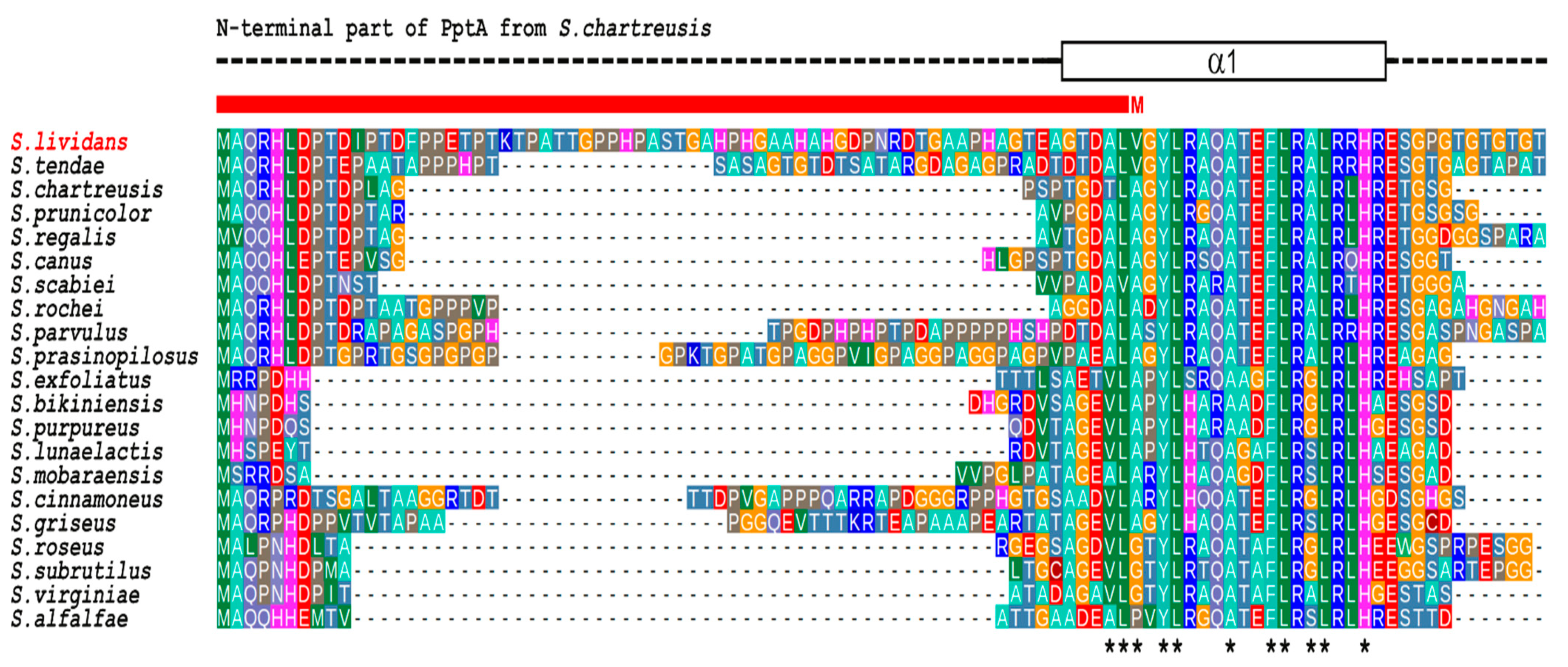
2.1. SLI_4382 and SLI_4383 Amino Acid Sequence Features
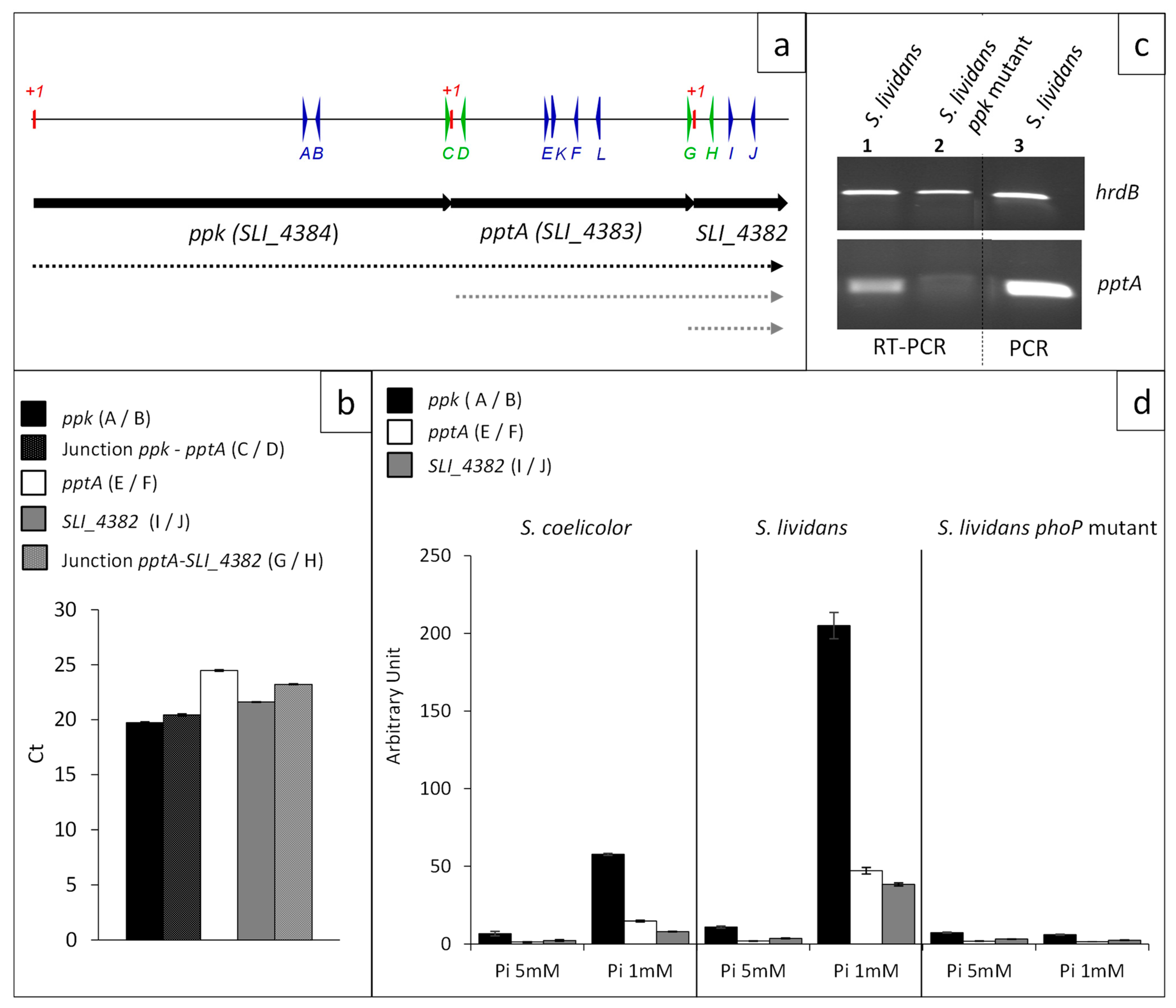
2.2. Ppk, pptA and SLI_4382 Are Co-Transcribed But pptA and SLI_4382 Can Also Be Transcribed Alone from Their Own Promoter
2.3. The ppk/pptA/SLI_4382 Operon Belongs to the Pho Regulon
2.4. The Disruption of SLI_4383/pptA Confers an Actinorhodin Over-Producing Phenotype to S. lividans TK24
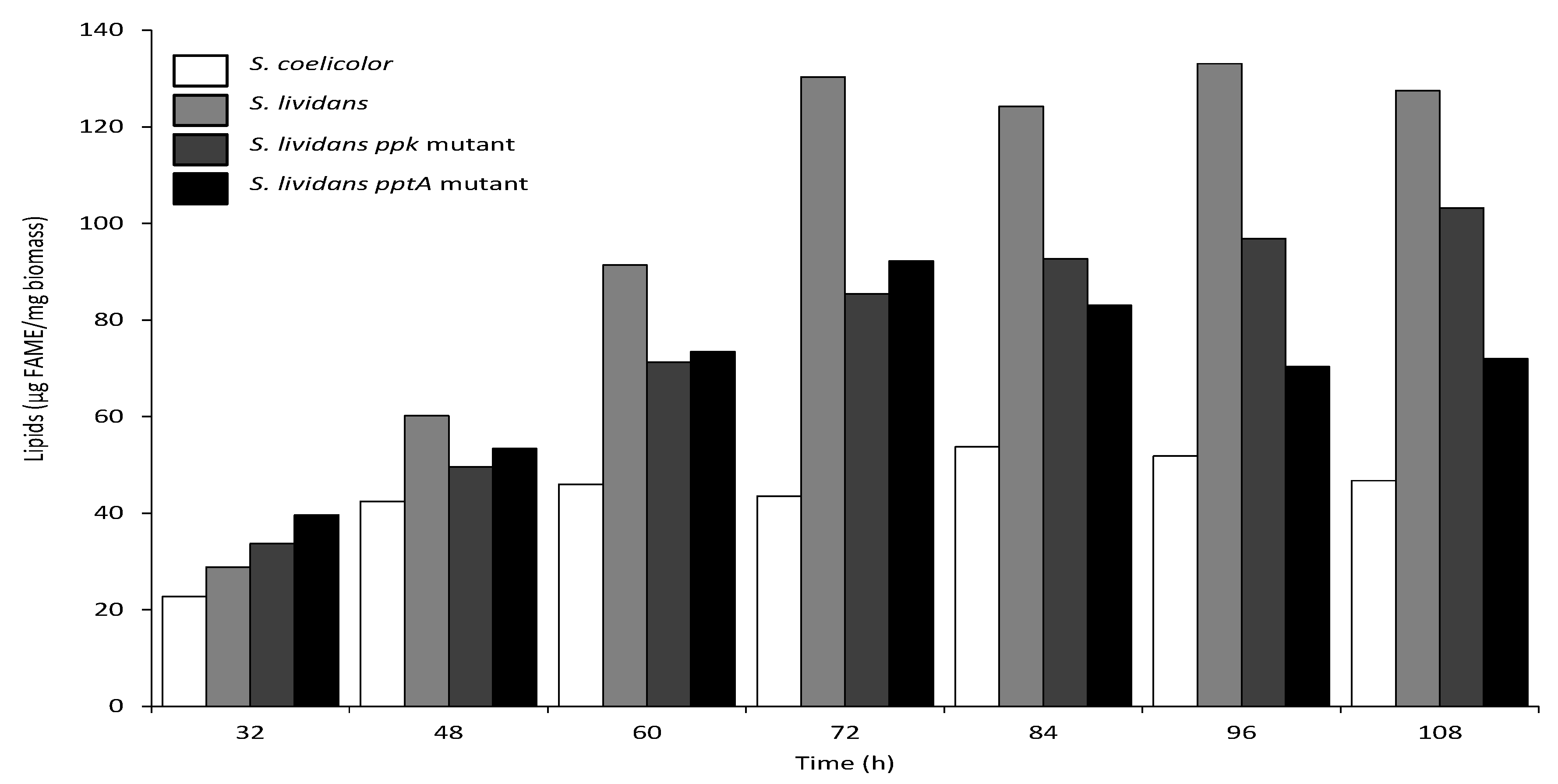
2.5. The Total Lipid Content of the SLI_4383/pptA Deletion Mutant Was Lower Than That of the Wild Type and ppk Mutant Strains of S. lividans
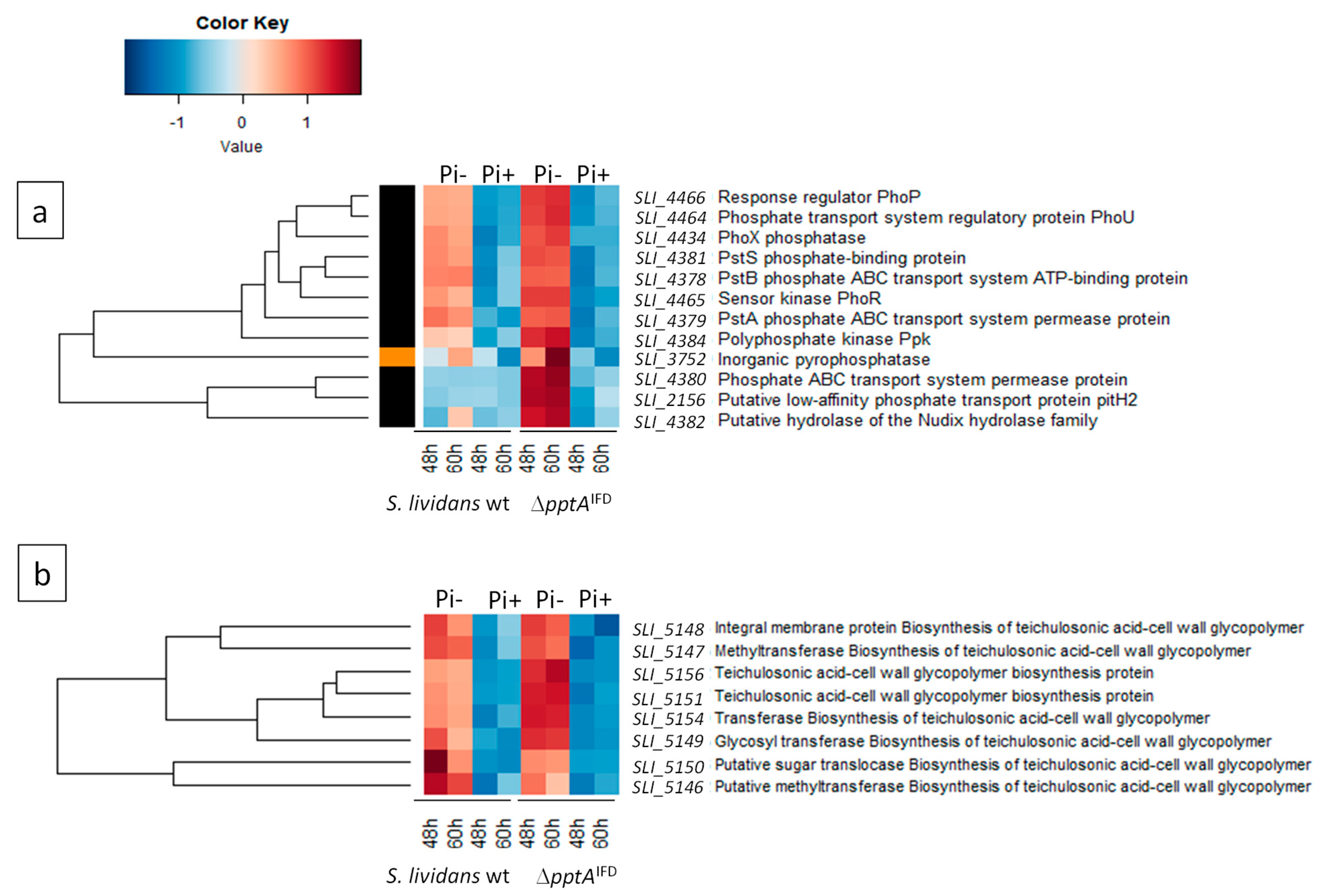
2.6. Proteins of the Pho Regulon Involved in Pi Supply and Saving Are Up-Regulated in the SLI_4383/pptA Mutant
2.7. The ppk-pptA-SLI_4382 Region Is Highly Conserved in Streptomyces Species But Not in Other Prokaryotes
3. Discussion
4. Materials and Methods
4.1. Strains, Plasmids, and Culture Conditions
4.2. DNA Manipulations and Transformation of Streptomyces and E. coli Strains
4.3. Construction of KO Mutant Strains and Complementation of the SLI_4383/pptA Mutant Strain
4.4. Southern Blot Analysis
4.5. RNA Preparation
4.6. RT-qPCR Experiments
4.7. 5′. Rapid-Amplification-of-cDNA-Ends (5′ RACE) PCR
4.8. Assay of Extracellular and Intracellular Actinorhodin (ACT) Production
4.9. Determination of Total Lipid Content Using Attenuated Total Reflectance-Fourier Transform Infra Red Spectroscopy (ATR-FTIRS) Measurements
4.10. Comparative Proteomic Analysis of the Wild Type Strain of S. lividans and of the ∆SLI_4383 Mutant
4.11. Computer Analysis of Protein Sequences
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pham, J.V.; Yilma, M.A.; Feliz, A.; Majid, M.T.; Maffetone, N.; Walker, J.R.; Kim, E.; Cho, H.J.; Reynolds, J.M.; Song, M.C.; et al. A Review of the Microbial Production of Bioactive Natural Products and Biologics. Front. Microbiol. 2019, 10, 1404. [Google Scholar] [CrossRef] [Green Version]
- Barka, E.A.; Vatsa, P.; Sanchez, L.; Gaveau-Vaillant, N.; Jacquard, C.; Meier-Kolthoff, J.P.; Klenk, H.P.; Clement, C.; Ouhdouch, Y.; van Wezel, G.P. Taxonomy, Physiology, and Natural Products of Actinobacteria. Microbiol. Mol. Biol. Rev. 2015, 80, 1–43. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Chater, K.F.; Chandra, G.; Niu, G.; Tan, H. Molecular regulation of antibiotic biosynthesis in Streptomyces. Microbiol. Mol. Biol. Rev. 2013, 77, 112–143. [Google Scholar] [CrossRef] [Green Version]
- Martin, J.F. Phosphate control of the biosynthesis of antibiotics and other secondary metabolites is mediated by the PhoR-PhoP system: An unfinished story. J. Bacteriol. 2004, 186, 5197–5201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esnault, C.; Dulermo, T.; Smirnov, A.; Askora, A.; David, M.; Deniset-Besseau, A.; Holland, I.B.; Virolle, M.J. Strong antibiotic production is correlated with highly active oxidative metabolism in Streptomyces coelicolor M145. Sci. Rep. 2017, 7, 200. [Google Scholar] [CrossRef] [Green Version]
- Albi, T.; Serrano, A. Inorganic polyphosphate in the microbial world. Emerging roles for a multifaceted biopolymer. World J. Microbiol. Biotechnol. 2016, 32, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, M.J. Interactions between DksA and Stress-Responsive Alternative Sigma Factors Control Inorganic Polyphosphate Accumulation in Escherichia coli. J. Bacteriol. 2020, 202, e00133-20. [Google Scholar] [CrossRef] [PubMed]
- Rudat, A.K.; Pokhrel, A.; Green, T.J.; Gray, M.J. Mutations in Escherichia coli Polyphosphate Kinase That Lead to Dramatically Increased In Vivo Polyphosphate Levels. J. Bacteriol. 2018, 200, e00697-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crooke, E.; Akiyama, M.; Rao, N.N.; Kornberg, A. Genetically altered levels of inorganic polyphosphate in Escherichia coli. J. Biol. Chem. 1994, 269, 6290–6295. [Google Scholar] [CrossRef]
- Morohoshi, T.; Maruo, T.; Shirai, Y.; Kato, J.; Ikeda, T.; Takiguchi, N.; Ohtake, H.; Kuroda, A. Accumulation of inorganic polyphosphate in phoU mutants of Escherichia coli and Synechocystis sp. strain PCC6803. Appl. Environ. Microbiol. 2002, 68, 4107–4110. [Google Scholar] [CrossRef] [Green Version]
- Gray, M.J.; Jakob, U. Oxidative stress protection by polyphosphate—New roles for an old player. Curr. Opin. Microbiol. 2015, 24, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Kuroda, A.; Tanaka, S.; Ikeda, T.; Kato, J.; Takiguchi, N.; Ohtake, H. Inorganic polyphosphate kinase is required to stimulate protein degradation and for adaptation to amino acid starvation in Escherichia coli. Proc. Natl. Acad. Sci. USA 1999, 96, 14264–14269. [Google Scholar] [CrossRef] [Green Version]
- Kulakovskaya, T. Inorganic polyphosphates and heavy metal resistance in microorganisms. World J. Microbiol. Biotechnol. 2018, 34, 139. [Google Scholar] [CrossRef] [PubMed]
- Rao, N.N.; Kornberg, A. Inorganic polyphosphate regulates responses of Escherichia coli to nutritional stringencies, environmental stresses and survival in the stationary phase. Prog. Mol. Subcell. Biol. 1999, 23, 183–195. [Google Scholar]
- Chouayekh, H.; Virolle, M.J. The polyphosphate kinase plays a negative role in the control of antibiotic production in Streptomyces lividans. Mol. Microbiol. 2002, 43, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Ghorbel, S.; Smirnov, A.; Chouayekh, H.; Sperandio, B.; Esnault, C.; Kormanec, J.; Virolle, M.J. Regulation of ppk expression and in vivo function of Ppk in Streptomyces lividans TK24. J. Bacteriol. 2006, 188, 6269–6276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghorbel, S.; Kormanec, J.; Artus, A.; Virolle, M.J. Transcriptional studies and regulatory interactions between the phoR-phoP operon and the phoU, mtpA, and ppk genes of Streptomyces lividans TK24. J. Bacteriol. 2006, 188, 677–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tumlirsch, T.; Jendrossek, D. Proteins with CHADs (Conserved Histidine alpha-Helical Domains) Are Attached to Polyphosphate Granules In Vivo and Constitute a Novel Family of Polyphosphate-Associated Proteins (Phosins). Appl. Environ. Microbiol. 2017, 83, e03399-16. [Google Scholar] [CrossRef] [Green Version]
- Lorenzo-Orts, L.; Hohmann, U.; Zhu, J.; Hothorn, M. Molecular characterization of CHAD domains as inorganic polyphosphate-binding modules. Life Sci. Alliance 2019, 2, e201900385. [Google Scholar] [CrossRef] [Green Version]
- Werten, S.; Rustmeier, N.H.; Gemmer, M.; Virolle, M.J.; Hinrichs, W. Structural and biochemical analysis of a phosin from Streptomyces chartreusis reveals a combined polyphosphate- and metal-binding fold. FEBS Lett. 2019, 593, 2019–2029. [Google Scholar] [CrossRef] [PubMed]
- Chater, K.F.; Chandra, G. The use of the rare UUA codon to define “expression space” for genes involved in secondary metabolism, development and environmental adaptation in Streptomyces. J. Microbiol. 2008, 46, 1–11. [Google Scholar] [CrossRef]
- Millan-Oropeza, A.; Henry, C.; Lejeune, C.; David, M.; Virolle, M.J. Expression of genes of the Pho regulon is altered in Streptomyces coelicolor. Sci. Rep. 2020, 10, 8492. [Google Scholar] [CrossRef]
- de Smit, M.H.; Verlaan, P.W.; van Duin, J.; Pleij, C.W. Intracistronic transcriptional polarity enhances translational repression: A new role for Rho. Mol. Microbiol. 2008, 69, 1278–1289. [Google Scholar] [CrossRef]
- David, M.; Lejeune, C.; Abreu, S.; Thibessard, A.; Leblond, P.; Chaminade, P.; Virolle, M.J. Negative Correlation between Lipid Content and Antibiotic Activity in Streptomyces: General Rule and Exceptions. Antibiotics 2020, 9, 280. [Google Scholar] [CrossRef] [PubMed]
- Virolle, M.J. A Challenging View: Antibiotics Play a Role in the Regulation of the Energetic Metabolism of the Producing Bacteria. Antibiotics 2020, 9, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lejeune, C.; Abreu, S.; Dulermo, T.; Chaminade, P.; Werten, S.; David, M.; Virolle, M.J. Impact of phosphate availability on membrane lipid content of the model strains, Streptomyces lividans and Streptomyces coelicolor. Frontiers 2021, in press. [Google Scholar] [CrossRef]
- Santos-Beneit, F.; Rodriguez-Garcia, A.; Franco-Dominguez, E.; Martin, J.F. Phosphate-dependent regulation of the low- and high-affinity transport systems in the model actinomycete Streptomyces coelicolor. Microbiology 2008, 154, 2356–2370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostash, B.; Shashkov, A.; Streshinskaya, G.; Tul’skaya, E.; Baryshnikova, L.; Dmitrenok, A.; Dacyuk, Y.; Fedorenko, V. Identification of Streptomyces coelicolor M145 genomic region involved in biosynthesis of teichulosonic acid-cell wall glycopolymer. Folia Microbiol. 2014, 59, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Le Marechal, P.; Decottignies, P.; Marchand, C.H.; Degrouard, J.; Jaillard, D.; Dulermo, T.; Froissard, M.; Smirnov, A.; Chapuis, V.; Virolle, M.J. Comparative proteomic analysis of Streptomyces lividans Wild-Type and ppk mutant strains reveals the importance of storage lipids for antibiotic biosynthesis. Appl. Environ. Microbiol. 2013, 79, 5907–5917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatusova, T.; Ciufo, S.; Fedorov, B.; O’Neill, K.; Tolstoy, I. RefSeq microbial genomes database: New representation and annotation strategy. Nucleic Acids Res. 2014, 42, D553–D559. [Google Scholar] [CrossRef] [Green Version]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. NCBI BLAST: A better web interface. Nucleic Acids Res. 2008, 36, W5–W9. [Google Scholar] [CrossRef]
- Klimchuk, O.I.; Konovalov, K.A.; Perekhvatov, V.V.; Skulachev, K.V.; Dibrova, D.V.; Mulkidjanian, A.Y. COGNAT: A web server for comparative analysis of genomic neighborhoods. Biol. Direct 2017, 12, 26. [Google Scholar] [CrossRef] [Green Version]
- Fong, C.; Rohmer, L.; Radey, M.; Wasnick, M.; Brittnacher, M.J. PSAT: A web tool to compare genomic neighborhoods of multiple prokaryotic genomes. BMC Bioinform. 2008, 9, 170. [Google Scholar] [CrossRef] [Green Version]
- Dehal, P.S.; Joachimiak, M.P.; Price, M.N.; Bates, J.T.; Baumohl, J.K.; Chivian, D.; Friedland, G.D.; Huang, K.H.; Keller, K.; Novichkov, P.S.; et al. MicrobesOnline: An integrated portal for comparative and functional genomics. Nucleic Acids Res. 2010, 38, D396–D400. [Google Scholar] [CrossRef] [Green Version]
- Song, H.; Dharmasena, M.N.; Wang, C.; Shaw, G.X.; Cherry, S.; Tropea, J.E.; Jin, D.J.; Ji, X. Structure and activity of PPX/GppA homologs from Escherichia coli and Helicobacter pylori. FEBS J. 2020, 287, 1865–1885. [Google Scholar] [CrossRef]
- McLennan, A.G. The Nudix hydrolase superfamily. Cell. Mol. Life Sci. 2006, 63, 123–143. [Google Scholar] [CrossRef] [PubMed]
- Mildvan, A.S.; Xia, Z.; Azurmendi, H.F.; Saraswat, V.; Legler, P.M.; Massiah, M.A.; Gabelli, S.B.; Bianchet, M.A.; Kang, L.W.; Amzel, L.M. Structures and mechanisms of Nudix hydrolases. Arch. Biochem. Biophys. 2005, 433, 129–143. [Google Scholar] [CrossRef]
- McLennan, A.G. The MutT motif family of nucleotide phosphohydrolases in man and human pathogens (review). Int. J. Mol. Med. 1999, 4, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Rigden, D.J. The histidine phosphatase superfamily: Structure and function. Biochem. J. 2008, 409, 333–348. [Google Scholar] [CrossRef] [Green Version]
- Millan-Oropeza, A.; Henry, C.; Blein-Nicolas, M.; Aubert-Frambourg, A.; Moussa, F.; Bleton, J.; Virolle, M.J. Quantitative Proteomics Analysis Confirmed Oxidative Metabolism Predominates in Streptomyces coelicolor versus Glycolytic Metabolism in Streptomyces lividans. J. Proteome Res. 2017, 16, 2597–2613. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Ellis, H.M.; Lee, E.C.; Jenkins, N.A.; Copeland, N.G.; Court, D.L. An efficient recombination system for chromosome engineering in Escherichia coli. Proc. Natl. Acad. Sci. USA 2000, 97, 5978–5983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, R.C.; Schimke, R.T. Preparation of electro competent E. coli using salt-free growth medium. Biotechniques 1996, 20, 42–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kieser, T.; Bibb, M.J.; Buttner, M.J.; Chater, K.F.; Hopwood, D.A. Practical Streptomyces Genetics; John Innes Foundation: Norwich, UK, 2000. [Google Scholar]
- Gust, B.; Challis, G.L.; Fowler, K.; Kieser, T.; Chater, K.F. PCR-targeted Streptomyces gene replacement identifies a protein domain needed for biosynthesis of the sesquiterpene soil odor geosmin. Proc. Natl. Acad. Sci. USA 2003, 100, 1541–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bentley, S.D.; Chater, K.F.; Cerdeno-Tarraga, A.M.; Challis, G.L.; Thomson, N.R.; James, K.D.; Harris, D.E.; Quail, M.A.; Kieser, H.; Harper, D.; et al. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3 (2). Nature 2002, 417, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Redenbach, M.; Kieser, H.M.; Denapaite, D.; Eichner, A.; Cullum, J.; Kinashi, H.; Hopwood, D.A. A set of ordered cosmids and a detailed genetic and physical map for the 8 Mb Streptomyces coelicolor A3 (2) chromosome. Mol. Microbiol. 1996, 21, 77–96. [Google Scholar] [CrossRef]
- Raynal, A.; Karray, F.; Tuphile, K.; Darbon-Rongere, E.; Pernodet, J.L. Excisable cassettes: New tools for functional analysis of Streptomyces genomes. Appl. Environ. Microbiol. 2006, 72, 4839–4844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Willems, E.; Leyns, L.; Vandesompele, J. Standardization of real-time PCR gene expression data from independent biological replicates. Anal. Biochem. 2008, 379, 127–129. [Google Scholar] [CrossRef]
- Xu, D.; Seghezzi, N.; Esnault, C.; Virolle, M.J. Repression of antibiotic production and sporulation in Streptomyces coelicolor by overexpression of a TetR family transcriptional regulator. Appl. Environ. Microbiol. 2010, 76, 7741–7753. [Google Scholar] [CrossRef] [Green Version]
- Deniset-Besseau, A.; Prater, C.B.; Virolle, M.J.; Dazzi, A. Monitoring TriAcylGlycerols Accumulation by Atomic Force Microscopy Based Infrared Spectroscopy in Streptomyces Species for Biodiesel Applications. J. Phys. Chem. Lett. 2014, 5, 654–658. [Google Scholar] [CrossRef]
- Millan-Oropeza, A.; Rebois, R.; David, M.; Moussa, F.; Dazzi, A.; Bleton, J.; Virolle, M.J.; Deniset-Besseau, A. Attenuated Total Reflection Fourier Transform Infrared (ATR FT-IR) for Rapid Determination of Microbial Cell Lipid Content: Correlation with Gas Chromatography-Mass Spectrometry (GC-MS). Appl. Spectrosc. 2017, 71, 2344–2352. [Google Scholar] [CrossRef] [PubMed]
- Vitry, P.; Bourillot, E.; Tetard, L.; Plassard, C.; Lacroute, Y.; Lesniewska, E. Mode-synthesizing atomic force microscopy for volume characterization of mixed metal nanoparticles. J. Microsc. 2016, 263, 307–311. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukashin, A.V.; Borodovsky, M. GeneMark.hmm: New solutions for gene finding. Nucleic Acids Res. 1998, 26, 1107–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Besemer, J.; Lomsadze, A.; Borodovsky, M. GeneMarkS: A self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001, 29, 2607–2618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, P.; Krogh, A. Large-scale prokaryotic gene prediction and comparison to genome annotation. Bioinformatics 2005, 21, 4322–4329. [Google Scholar] [CrossRef] [Green Version]
- Larsen, T.S.; Krogh, A. EasyGene—A prokaryotic gene finder that ranks ORFs by statistical significance. BMC Bioinform. 2003, 4, 21. [Google Scholar] [CrossRef] [Green Version]
- Bocs, S.; Cruveiller, S.; Vallenet, D.; Nuel, G.; Medigue, C. AMIGene: Annotation of MIcrobial Genes. Nucleic Acids Res. 2003, 31, 3723–3726. [Google Scholar] [CrossRef] [Green Version]
- Marchler-Bauer, A.; Anderson, J.B.; Cherukuri, P.F.; DeWeese-Scott, C.; Geer, L.Y.; Gwadz, M.; He, S.; Hurwitz, D.I.; Jackson, J.D.; Ke, Z.; et al. CDD: A Conserved Domain Database for protein classification. Nucleic Acids Res. 2005, 33, D192–D196. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shikura, N.; Darbon, E.; Esnault, C.; Deniset-Besseau, A.; Xu, D.; Lejeune, C.; Jacquet, E.; Nhiri, N.; Sago, L.; Cornu, D.; et al. The Phosin PptA Plays a Negative Role in the Regulation of Antibiotic Production in Streptomyces lividans. Antibiotics 2021, 10, 325. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics10030325
Shikura N, Darbon E, Esnault C, Deniset-Besseau A, Xu D, Lejeune C, Jacquet E, Nhiri N, Sago L, Cornu D, et al. The Phosin PptA Plays a Negative Role in the Regulation of Antibiotic Production in Streptomyces lividans. Antibiotics. 2021; 10(3):325. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics10030325
Chicago/Turabian StyleShikura, Noriyasu, Emmanuelle Darbon, Catherine Esnault, Ariane Deniset-Besseau, Delin Xu, Clara Lejeune, Eric Jacquet, Naima Nhiri, Laila Sago, David Cornu, and et al. 2021. "The Phosin PptA Plays a Negative Role in the Regulation of Antibiotic Production in Streptomyces lividans" Antibiotics 10, no. 3: 325. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics10030325