
Cystic Fibrosis: Recent Insights into Inhaled Antibiotic Treatment and Future Perspectives

Abstract
:1. Introduction
2. Rationale for Inhaled Antibiotics in CF
3. Current Status of Inhaled Antibiotics in Cystic Fibrosis
3.1. Colistimethate Sodium
3.2. Tobramycin
3.3. Aztreonam Lysine
3.4. Levofloxacin
4. Antibiotic Strategies for Eradicating P. aeruginosa and Other Pathogenic Organisms
5. Inhaled Antibiotic Therapy for P. aeruginosa Chronic Pulmonary Infection
6. Inhaled Antibiotic Therapy for Preventing Pulmonary Exacerbations
7. Airway Conditions and Clinical Efficacy of Inhaled Antibiotics
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Goetz, D.; Ren, C.L. Review of cystic fibrosis. Pediatr. Ann. 2019, 48, e154–e161. [Google Scholar] [CrossRef]
- Hatziagorou, E.; Orenti, A.; Drevinek, P.; Kashirskaya, N.; Mei-Zahav, M.; De Boeck, K.; The European Cystic Fibrosis Society Patient Registry. Changing epidemiology of the respiratory bacteriology of patients with cystic fibrosis-data from the European cystic fibrosis society patient registry. J. Cyst. Fibros. 2020, 19, 376–383. [Google Scholar] [CrossRef] [PubMed]
- Hendricks, M.R.; Lashua, L.P.; Fischer, D.K.; Flitter, B.A.; Eichinger, K.M.; Durbin, J.E.; Sarkar, S.N.; Coyne, C.B.; Empey, K.M.; Bomberger, J.M. Respiratory syncytial virus infection enhances Pseudomonas aeruginosa biofilm growth through dysregulation of nutritional immunity. Proc. Natl. Acad. Sci. USA 2016, 113, 1642–1647. [Google Scholar] [CrossRef] [Green Version]
- Van Ewijk, B.E.; van der Zalm, M.M.; Wolfs, T.F.; Fleer, A.; Kimpen, J.L.; Wilbrink, B.; van der Ent, C.K. Prevalence and impact of respiratory viral infections in young children with cystic fibrosis: Prospective cohort study. Pediatrics 2008, 122, 1171–1176. [Google Scholar] [CrossRef]
- Dasenbrook, E.C.; Checkley, W.; Merlo, C.A.; Konstan, M.W.; Lechtzin, N.; Boyle, M.P. Association between respiratory tract methicillin-resistant Staphylococcus aureus and survival in cystic fibrosis. JAMA 2010, 303, 2386–2392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasenbrook, E.C. Update on methicillin-resistant Staphylococcus aureus in cystic fibrosis. Curr. Opin. Pulm. Med. 2011, 17, 437–441. [Google Scholar] [CrossRef]
- Hurley, M.N.; Smyth, A.R. Staphylococcus aureus in cystic fibrosis: Pivotal role or bit part actor? Curr. Opin. Pulm. Med. 2018, 24, 586–591. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.; Rowbotham, N.J.; Regan, K.H. Inhaled anti-pseudomonal antibiotics for long-term therapy in cystic fibrosis. Cochrane Database Syst. Rev. 2018, 3, CD001021. [Google Scholar] [CrossRef] [PubMed]
- Gajdács, M. The continuing threat of Methicillin-Resistant Staphylococcus aureus. Antibiotics 2019, 8, 52. [Google Scholar] [CrossRef] [Green Version]
- MacKenzie, T.; Gifford, A.H.; Sabadosa, K.A.; Quinton, H.B.; Knapp, E.A.; Goss, C.H.; Marshall, B.C. Longevity of patients with cystic fibrosis in 2000 to 2010 and beyond: Survival analysis of the Cystic Fibrosis Foundation patient registry. Ann. Intern. Med. 2014, 161, 233–241. [Google Scholar] [CrossRef]
- Cohen-Cymberknoh, M.; Shoseyov, D.; Kerem, E. Managing cystic fibrosis: Strategies that increase life expectancy and improve quality of life. Am. J. Respir. Crit. Care Med. 2011, 183, 1463–1471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zolin, A.; Orenti, A.; Naehrlich, L.; Jung, A.; van Rens, J. The European Cystic Fibrosis Society Patient Registry (ECFSPR), Annual Report 2018; European Cystic Fibrosis Society: Karup, Denmark, 2020; pp. 1–149. [Google Scholar]
- Cystic Fibrosis Foundation Patient Registry; 2019, Annual Data Report; Cystic Fibrosis Foundation: Bethesda, MD, USA, 2020; pp. 1–85.
- Castellani, C.; Duff, A.J.A.; Bell, S.C.; Heijerman, H.G.M.; Munck, A.; Ratjen, F.; Sermet-Gaudelus, I.; Southern, K.W.; Barben, J.; Flume, P.A.; et al. ECFS best practice guidelines: The 2018 revision. J. Cyst. Fibros. 2018, 17, 153–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flume, P.A.; Clancy, J.P.; Retsch-Bogart, G.Z.; Tullis, D.E.; Bresnik, M.; Derchak, P.A.; Lewis, S.A.; Ramsey, B.W. Continuous alternating inhaled antibiotics for chronic pseudomonal infection in cystic fibrosis. J. Cyst. Fibros. 2016, 15, 809–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van de Kerkhove, C.; Goeminne, P.C.; Kicinski, M.; Nawrot, T.S.; Lorent, N.; Van Bleyenbergh, P.; De Boeck, K.; Dupont, L.J. Continuous alternating inhaled antibiotic therapy in CF: A single centerretrospective analysis. J. Cyst. Fibros. 2016, 15, 802–808. [Google Scholar] [CrossRef] [Green Version]
- Middleton, P.G.; Taylor-Cousar, J.L. Development of elexacaftor-tezacaftor-ivacaftor: Highly effective CFTR modulation for the majority of people with cystic fibrosis. Expert Rev. Respir. Med. 2020, 1–13. [Google Scholar] [CrossRef]
- Paterson, S.L.; Barry, P.J.; Horsley, A.R. Tezacaftor and ivacaftor for the treatment of cystic fibrosis. Expert Rev. Respir. Med. 2020, 14, 15–30. [Google Scholar] [CrossRef]
- Davies, J.C.; Martin, I. New anti-pseudomonal agents for cystic fibrosis- still needed in the era of small molecule CFTR modulators? Rev. Expert Opin. Pharmacother. 2018, 19, 1327–1336. [Google Scholar] [CrossRef] [PubMed]
- Somayaji, R.; Nichols, D.P.; Bell, S.C. Cystic fibrosis—Ten promising therapeutic approaches in the current era of care. Expert Opin. Investig. Drugs 2020, 29, 1107–1124. [Google Scholar] [CrossRef] [PubMed]
- Szczesniak, R.; Heltshe, S.L.; Stanojevic, S.; Mayer-Hamblett, N. Use of FEV1 in cystic fibrosis epidemiologic studies and clinical trials: A statistical perspective for the clinical researcher. J. Cyst. Fibros. 2017, 16, 318–326. [Google Scholar] [CrossRef]
- VanDevanter, D.R.; Van Dalfsen, J.M. How much do Pseudomonas biofilms contribute to symptoms of pulmonary exacerbation in cystic fibrosis? Pediatr. Pulmonol. 2005, 39, 504–506. [Google Scholar] [CrossRef]
- Ciofu, O.; Tolker-Nielsen, T. Tolerance and resistance of Pseudomonas aeruginosa biofilms to antimicrobial agents-how P. aeruginosa can escape antibiotics. Front. Microbiol. 2019, 10, 913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behzadi, P.; Baráth, Z.; Gajdács, M. It’s not easy being green: A narrative review on the microbiology, virulence and therapeutic prospects of multidrug-resistant Pseudomonas aeruginosa. Antibiotics 2021, 10, 42. [Google Scholar] [CrossRef]
- Bacci, G.; Taccetti, G.; Dolce, D.; Armanini, F.; Segata, N.; Di Cesare, F.; Lucidi, V.; Fiscarelli, E.; Morelli, P.; Casciaro, R.; et al. Untargeted metagenomic investigation of the airway microbiome of cystic fibrosis patients with moderate-severe lung disease. Microorganisms 2020, 8, 1003. [Google Scholar] [CrossRef] [PubMed]
- Bevivino, A.; Bacci, G.; Drevinek, P.; Nelson, M.T.; Hoffman, L.; Mengoni, A. Deciphering the ecology of cystic fibrosis bacterial communities: Towards systems-level integration. Trends Mol. Med. 2019, 25, 1110–1122. [Google Scholar] [CrossRef] [Green Version]
- Di Sant’Agnese, P.E.A.; Andersen, D.H. Celiac syndrome; chemotherapy in infections of the respiratory tract associated with cystic fibrosis of the pancreas; observations with penicillin and drugs of the sulfonamide group, with special reference to penicillin aerosol. Am. J. Dis. Child. 1946, 72, 17–61. [Google Scholar] [CrossRef] [PubMed]
- Van der Ent, C.K.; Elborn, J.S. Improving inhaled antibiotic treatment—Practice defeats the proof. J. Cyst. Fibros. 2016, 15, 705–707. [Google Scholar] [CrossRef]
- Fiel, S.B. Aerosolized antibiotics in cystic fibrosis: Current and future trends. Expert. Rev. Respir. Med. 2008, 2, 479–487. [Google Scholar] [CrossRef]
- Scott, C.B.; Mall, M.A.; Gutierrez, H.; Macek, M.; Madge, S.; Davies, J.C.; Burgel, P.R.; Tullis, E.; Castaños, C.; Castellani, C.; et al. The future of cystic fibrosis care: A global perspective. Lancet Respir. Med. 2020, 8, 65–124. [Google Scholar] [CrossRef]
- Antoniu, S. Novel inhaled combined antibiotic formulations in the treatment of Pseudomonas aeruginosa airways infections in cystic fibrosis. Expert. Rev. Anti-Infect. Ther. 2015, 13, 897–905. [Google Scholar] [CrossRef]
- Geller, D.E.; Flume, P.A.; Griffith, D.C.; Morgan, E.; White, D.; Loutit, J.S.; Dudley, M.N. Pharmacokinetics and safety of MP-376 (levofloxacin inhalation solution) in cystic fibrosis subjects. Antimicrob. Agents Chemother. 2011, 55, 2636–2640. [Google Scholar] [CrossRef] [Green Version]
- Geller, D.E.; Flume, P.A.; Staab, D.; Fischer, R.; Loutit, J.S.; Conrad, D.J.; Mpex 204 Study Group. Levofloxacin inhalation solution (MP-376) in patients with cystic fibrosis with Pseudomonas aeruginosa. Am. J. Respir. Crit. Care Med. 2011, 183, 1510–1516. [Google Scholar] [CrossRef] [PubMed]
- Kirkby, S.; Novak, K.; McCoy, K. Aztreonam (for inhalation solution) for the treatment of chronic lung infection in patients with cystic fibrosis: An evidence-based review. Core Evid. 2011, 6, 59–66. [Google Scholar] [CrossRef] [Green Version]
- McCoy, K.S.; Quittner, A.L.; Oermann, C.M.; Gibson, R.L.; Retsch-Bogart, G.Z.; Montgomery, A.B. Inhaled aztreonam lysine for chronic airway Pseudomonas aeruginosa in cystic fibrosis. Am. J. Respir. Crit. Care Med. 2008, 178, 921–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Retsch-Bogart, G.Z.; Quittner, A.L.; Gibson, R.L.; Oermann, C.M.; McCoy, K.S.; Montgomery, A.B.; Cooper, P.J. Efficacy and safety of inhaled aztreonam lysine for airway Pseudomonas in cystic fibrosis. Chest 2009, 135, 1223–1232. [Google Scholar] [CrossRef] [Green Version]
- Elborn, J.S.; Geller, D.E.; Conrad, D.; Aaron, S.D.; Smyth, A.R.; Fischer, R.; Kerem, E.; Bell, S.C.; Loutit, J.S.; Dudley, M.N.; et al. A phase 3, open-label, randomized trial to evaluate the safety and efficacy of levofloxacin inhalation solution (APT-1026) versus tobramycin inhalation solutionin stable cystic fibrosis patients. J. Cyst. Fibros. 2015, 14, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Flume, P.A.; VanDevanter, D.R.; Morgan, E.E.; Dudley, M.N.; Loutit, J.S.; Bell, S.C.; Kerem, E.; Fischer, R.; Smyth, A.R.; Aaron, S.D.; et al. A phase 3, multi-center, multinational, randomized, double-blind, placebo-controlled study to evaluate the efficacy and safety of levofloxacin inhalation solution (APT-1026) in stable cystic fibrosis patients. J. Cyst Fibros. 2016, 15, 495–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, C.T.; McCaleb, R.; Manasco, K.B. New inhaled antimicrobial formulations for use in the cystic fibrosis patient population. Ann. Pharmacother. 2016, 50, 133–140. [Google Scholar] [CrossRef]
- Quon, B.S.; Goss, C.H.; Ramsey, B.W. Inhaled antibiotics for lower airway infections. Ann. Am. Thorac. Soc. 2014, 11, 425–434. [Google Scholar] [CrossRef]
- Bilton, D.; Pressler, T.; Fajac, I.; Clancy, J.P.; Sands, D.; Minic, P.; Cipolli, M.; Galeva, I.; Solé, A.; Quittner, A.L.; et al. Amikacin liposome inhalation suspension for chronic Pseudomonas aeruginosa infection in cystic fibrosis. J. Cyst. Fibros. 2020, 19, 284–291. [Google Scholar] [CrossRef]
- Flume, P.A.; Mogayzel, P.J., Jr.; Robinson, K.A.; Goss, C.H.; Rosenblatt, R.L.; Kuhn, R.J.; Marshall, B.C.; Clinical Practice Guidelines for Pulmonary Therapies Committee. Cystic fibrosis pulmonary guidelines: Treatment of pulmonary exacerbations. Am. J. Respir. Crit. Care Med. 2009, 180, 802–808. [Google Scholar] [CrossRef] [Green Version]
- Döring, G.; Flume, P.; Heijerman, H.; Elborn, J.S.; Consensus Study Group. Treatment of lung infection in patients with cystic fibrosis: Current and future strategies. J. Cyst. Fibros. 2012, 11, 461–479. [Google Scholar] [CrossRef] [Green Version]
- Zemanick, E.; Burgel, P.R.; Taccetti, G.; Holmes, A.; Ratjen, F.; Byrnes, C.A.; Waters, V.J.; Bell, S.C.; VanDevanter, D.R.; Stuart Elborn, J.; et al. Antimicrobial resistance in cystic fibrosis: A Delphi approach to defining best practices. J. Cyst. Fibros. 2020, 19, 370–375. [Google Scholar] [CrossRef]
- Sherrard, L.J.; Tunney, M.M.; Elborn, J.S. Antimicrobial resistance in the respiratory microbiota of people with cystic fibrosis. Lancet 2014, 384, 703–713. [Google Scholar] [CrossRef]
- Elborn, J.S.; Flume, P.A.; Cohen, F.; Loutit, J.; VanDevanter, D.R. Safety and efficacy of prolonged levofloxacin inhalation solution (APT-1026) treatment for cystic fibrosis and chronic Pseudomonas aeruginosa airway infection. J. Cyst. Fibros. 2016, 15, 634–640. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.E.L.; Powell, L.C.; Pritchard, M.F.; Thomas, D.W.; Jenkins, R.E. Anti-pseudomonal activity of manuka honey and antibiotics in a specialized ex vivo model simulating cystic fibrosis lung infection. Front. Microbiol. 2019, 10, 869. [Google Scholar] [CrossRef] [Green Version]
- Littlewood, J.M.; Koch, C.; Lambert, P.A.; Høiby, N.; Elborn, J.S.; Conway, S.P.; Dinwiddie, R.; Duncan-Skingle, F. A ten years review of colomycin. Respir. Med. 2000, 94, 632–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, T.; Pedersen, S.S.; Garne, S.; Heilmann, C.; Høiby, N.; Koch, C. Colistin inhalation therapy in cystic fibrosis patients with chronic Pseudomonas aeruginosa lung infection. J. Antimicrob. Chemother. 1987, 19, 831–838. [Google Scholar] [CrossRef]
- Hodson, M.E.; Gallagher, C.G.; Govan, J.R. A randomised clinical trial of nebulised tobramycin or colistin in cystic fibrosis. Eur. Respir. J. 2002, 20, 658–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCoy, K.S. Compounded colistimethate as possible cause of fatal acute respiratory distress syndrome. N. Engl. J. Med. 2007, 357, 2310–2311. [Google Scholar] [CrossRef]
- Schuster, A.; Haliburn, C.; Döring, G.; Goldman, M.H.; Freedom Study Group. Safety, efficacy and convenience of colistimethate sodium dry powder for inhalation (Colobreathe DPI) in patients with cystic fibrosis: A randomized study. Thorax 2013, 68, 344–350. [Google Scholar] [CrossRef] [Green Version]
- Konstan, M.W.; Flume, P.A.; Kappler, M.; Chiron, R.; Higgins, M.; Brockhaus, F.; Zhang, J.; Angyalosi, G.; He, E.; Geller, D.E. Safety, efficacy and convenience of tobramycin inhalation powder in cystic fibrosis patients: The EAGER trial. J. Cyst. Fibros. 2011, 10, 54–61. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, S.; Lee, A.; Caine, N.; Charman, S.C.; Bilton, D. Long-term safety study of colistimethate sodium (Colobreathe®): Findings from the UK Cystic Fibrosis Registry. J. Cyst. Fibros. 2020. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, B.W.; Pepe, M.S.; Quan, J.M.; Otto, K.L.; Montgomery, A.B.; Williams-Warren, J.; Vasiljev, K.M.; Borowitz, D.; Bowman, C.M.; Marshall, B.C.; et al. Intermittent administration of inhaled tobramycin in patients with cystic fibrosis. Cystic fibrosis inhaled tobramycin study group. N. Engl. J. Med. 1999, 340, 23–30. [Google Scholar] [CrossRef] [Green Version]
- Murphy, T.D.; Anbar, R.D.; Lester, L.A.; Nasr, S.Z.; Nickerson, B.; VanDevanter, D.R.; Colin, A.A. Treatment with tobramycin solution for inhalation reduces hospitalizations in young CF subjects with mild lung disease. Pediatr. Pulmonol. 2004, 38, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Moss, R.B. Long-term benefits of inhaled tobramycin in adolescent patients with cystic fibrosis. Chest 2002, 121, 55–63. [Google Scholar] [CrossRef]
- Hamed, K.; Debonnett, L. Tobramycin inhalation powder for the treatment of pulmonary Pseudomonas aeruginosa infection in patients with cystic fibrosis: A review based on clinical evidence. Ther. Adv. Respir. Dis. 2017, 11, 193–209. [Google Scholar] [CrossRef] [Green Version]
- Nichols, D.P.; Happoldt, C.L.; Bratcher, P.E.; Caceres, S.M.; Chmiel, J.F.; Malcolm, K.C.; Saavedra, M.T.; Saiman, L.; Taylor-Cousar, J.L.; Nick, J.A. Impact of azithromycin on the clinical and antimicrobial effectiveness of tobramycin in the treatment of cystic fibrosis. J. Cyst. Fibros. 2017, 16, 358–366. [Google Scholar] [CrossRef]
- Parkins, M.D.; Elborn, J.S. Aztreonam lysine: A novel inhalational antibiotic for cystic fibrosis. Expert Rev. Respir. Med. 2010, 4, 435–444. [Google Scholar] [CrossRef]
- Assael, B.M.; Pressler, T.; Bilton, D.; Fayon, M.; Fischer, R.; Chiron, R.; LaRosa, M.; Knoop, C.; McElvaney, N.; Lewis, S.A.; et al. AZLI Active Comparator Study Group. Inhaled aztreonam lysine vs. inhaled tobramycin in cystic fibrosis: A comparative efficacy trial. J. Cyst. Fibros. 2013, 12, 130–140. [Google Scholar] [CrossRef] [Green Version]
- Wainwright, C.E.; Quittner, A.L.; Geller, D.E.; Nakamura, C.; Wooldridge, J.L.; Gibson, R.L.; Lewis, S.; Montgomery, A.B. Aztreonam for inhalation solution (AZLI) in patients with cystic fibrosis, mild lung impairment, and P. aeruginosa. J. Cyst. Fibros. 2011, 10, 234–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tullis, D.E.; Burns, J.L.; Retsch-Bogart, G.Z.; Bresnik, M.; Henig, N.R.; Lewis, S.A.; Lipuma, J.J. Inhaled aztreonam for chronic Burkholderia infection in cystic fibrosis: A placebo-controlled trial. J. Cyst. Fibros. 2014, 13, 296–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, R.; Flume, P.A.; VanDevanter, D.R.; Polu, K.; Pecoraro, M.; Bhatt, N.; Elborn, J.S. Pulmonary exacerbations and changes in lung function in CF adults with P. aeruginosa treated with inhaled levofloxacin (Quinsair®) or tobramycin. The 30Th Annual North American Cystic Fibrosis Conference. Pediatr Pulmonol. 2016, 51 (Suppl. 45), 359. [Google Scholar] [CrossRef]
- Gajdács, M.; Urbán, E. Prevalence and antibiotic resistance of Stenotrophomonas maltophilia in respiratory tract samples: A 10-year epidemiological snapshot. Health Serv Res Manag Epidemiol. 2019, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elborn, J.S.; Vataire, A.L.; Fukushima, A.; Aballea, S.; Khemiri, A.; Moore, C.; Medic, G.; Hemels, M.E.H. Comparison of inhaled antibiotics for the treatment of chronic Pseudomonas aeruginosa lung infection in patients with cystic fibrosis: Systematic literature review and network meta-analysis. Clin. Ther. 2016, 38, 2204–2226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schelstraete, P.; Haerynck, F.; Van daele, S.; Deseyne, S.; De Baets, F. Eradication therapy for Pseudomonas aeruginosa colonization episodes in cystic fibrosis patients not chronically colonized by P. aeruginosa. J. Cyst. Fibros. 2013, 12, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratjen, F.; Munck, A.; Kho, P.; Angyalosi, G.; ELITE Study Group. Treatment of early Pseudomonas aeruginosa infection in patients with cystic fibrosis: The ELITE trial. Thorax 2010, 65, 286–291. [Google Scholar] [CrossRef] [Green Version]
- Langton Hewer, S.C.; Smyth, A.R. Antibiotic strategies for eradicating Pseudomonas aeruginosa in people with cystic fibrosis. Cochrane Database Syst. Rev. 2017, 4, Cd004197. [Google Scholar] [CrossRef]
- Treggiari, M.M.; Retsch-Bogart, G.; Mayer-Hamblett, N.; Khan, U.; Kulich, M.; Kronmal, R.; Williams, J.; Hiatt, P.; Gibson, R.L.; Spencer, T.; et al. Early Pseudomonas Infection Control (EPIC) Investigators. Comparative efficacy and safety of 4 randomized regimens to treat early Pseudomonas aeruginosa infection in children with cystic fibrosis. Arch. Pediatr. Adolesc. Med. 2011, 165, 847–856. [Google Scholar] [CrossRef] [Green Version]
- Taccetti, G.; Campana, S.; Festini, F.; Mascherini, M.; Döring, G. Early eradication therapy against Pseudomonas aeruginosa in cystic fibrosis patients. Eur. Respir. J. 2005, 26, 458–461. [Google Scholar] [CrossRef] [Green Version]
- Gibson, R.L.; Emerson, J.; McNamara, S.; Burns, J.L.; Rosenfeld, M.; Yunker, A.; Cystic Fibrosis Therapeutics Development Network Study Group. Significant microbiological effect of inhaled tobramycin in young children with cystic fibrosis. Am. J. Respir. Crit. Care Med. 2003, 167, 841–849. [Google Scholar] [CrossRef]
- Blanchard, A.C.; Horton, E.; Stanojevic, S.; Taylor, L.; Waters, V.; Ratjen, F. Effectiveness of a stepwise Pseudomonas aeruginosa eradication protocol in children with cystic fibrosis. J. Cyst. Fibros. 2017, 16, 395–400. [Google Scholar] [CrossRef]
- Cohen-Cymberknoh, M.; Gilead, N.; Gartner, S.; Rovira, S.; Blau, H.; Mussaffi, H.; Rivlin, J.; Gur, M.; Shteinberg, M.; Bentur, L.; et al. Eradication failure of newly acquired Pseudomonas aeruginosa isolates in cystic fibrosis. J. Cyst. Fibros. 2016, 15, 776–782. [Google Scholar] [CrossRef] [Green Version]
- Ratjen, F.; Moeller, A.; McKinney, M.L.; Asherova, I.; Alon, N.; Maykut, R.; Angyalosi, G.; EARLY Study Group. Eradication of early P. aeruginosa infection in children. J. Cyst. Fibros. 2019, 8, 78–85. [Google Scholar] [CrossRef]
- Ratjen, F. Treatment of early Pseudomonas aeruginosa infection in patients with cystic fibrosis. Curr. Opin. Pulm. Med. 2006, 12, 428–432. [Google Scholar] [CrossRef]
- Mayer-Hamblett, N.; Retsch-Bogart, G.; Kloster, M.; Accurso, F.; Rosenfeld, M.; Albers, G.; Black, P.; Brown, P.; Cairns, A.; Davis, S.D.; et al. Azithromycin for early Pseudomonas infection in cystic fibrosis. The OPTIMIZE Randomized Trial. Am. J. Respir. Crit. Care Med. 2018, 198, 1177–1187. [Google Scholar] [CrossRef]
- Stormbroek, B.V.; Zampoli, M.; Morrow, B.M. Nebulized gentamicin in combination with systemic antibiotics for eradicating early Pseudomonas aeruginosa infection in children with cystic fibrosis. Pediatr. Pulmonol. 2019, 54, 393–398. [Google Scholar] [CrossRef]
- Hewer, S.C.L.; Smyth, A.R.; Brown, M.; Jones, A.P.; Hickey, H.; Kenna, D.; Ashby, D.; Thompson, A.; Williamson, P.R.; TORPEDO-CF Study Group. Intravenous versus oral antibiotics for eradication of Pseudomonas aeruginosa in cystic fibrosis (TORPEDO-CF): A randomised controlled trial. Lancet Respir. Med. 2020, 8, 975–986. [Google Scholar] [CrossRef]
- Esposito, S.; Pennoni, G.; Mencarini, V.; Palladino, N.; Peccini, L.; Principi, N. Antimicrobial treatment of Staphylococcus aureus in patients with cystic fibrosis. Front. Pharmacol. 2019, 10, 849. [Google Scholar] [CrossRef]
- Lo, D.K.; Hurley, M.N.; Muhlebach, M.S.; Smyth, A.R. Interventions for the eradication of meticillin-resistant Staphylococcus aureus (MRSA) in people with cystic fibrosis. Cochrane Database Syst. Rev. 2015, 2, CD009650. [Google Scholar] [CrossRef] [Green Version]
- Kiefer, A.; Bogdan, C.; Melichar, V.O. Successful eradication of newly acquired MRSA in six of seven patients with cystic fibrosis applying a short-term local and systemic antibiotic scheme. BMC Pulm. Med. 2018, 18, 20. [Google Scholar] [CrossRef] [Green Version]
- Dolce, D.; Neri, S.; Grisotto, L.; Campana, S.; Ravenni, N.; Miselli, F.; Camera, E.; Zavataro, L.; Braggion, C.; Fiscarelli, E.V.; et al. Methicillin-resistant Staphylococcus aureus eradication in cystic fibrosis patients: A randomized multicentric study. PLoS ONE 2019, 14, e0213497. [Google Scholar] [CrossRef]
- Solis, A.; Brown, D.; Hughes, J.; Van Saene, H.K.; Heaf, D.P. Methicillin resistant Staphylococcus aureus in children with cystic fibrosis: An eradication protocol. Pediatr. Pulmonol. 2003, 36, 189–195. [Google Scholar] [CrossRef]
- Macfarlane, M.; Leavy, A.; McCaughan, J.; Fair, R.; Reid, A.J.M. Successful decolonization of methicillin-resistant Staphylococcus aureus in paediatric patients with cystic fibrosis (CF) using a three-step protocol. J. Hosp. Infect. 2007, 65, 231–236. [Google Scholar] [CrossRef]
- Vallières, E.; Rendall, J.C.; Moore, J.E.; McCaughan, J.; Hoeritzauer, A.I.; Tunney, M.M.; Elborn, J.S.; Downey, D.G. MRSA eradication of newly acquired lower respiratory tract infection in cystic fibrosis. ERJ Open Res. 2016, 2. [Google Scholar] [CrossRef] [Green Version]
- Kappler, M.; Nagel, F.; Feilcke, M.; Kröner, C.; Pawlita, I.; Naehrig, S.; Ripper, J.; Hengst, M.; Both, U.V.; Forstner, M.; et al. Eradication of methicillin resistant Staphylococcus aureus detected for the first time in cystic fibrosis: A single center observational study. Pediatr. Pulmonol. 2016, 51, 1010–1019. [Google Scholar] [CrossRef]
- Muhlebach, M.S.; Beckett, V.; Popowitch, E.; Miller, M.B.; Baines, A.; Mayer-Hamblett, N.; Zemanick, E.T.; Hoover, W.C.; VanDalfsen, J.M.; Campbell, P.; et al. Microbiological efficacy of early MRSA treatment in cystic fibrosis in a randomised controlled trial. Thorax 2017, 72, 318–326. [Google Scholar] [CrossRef] [Green Version]
- Dezube, R.; Jennings, M.T.; Rykiel, M.; Diener-West, M.; Boyle, M.P.; Chmiel, J.F.; Dasenbrook, E.C. Eradication of persistent methicillin-resistant Staphylococcus aureus infection in cystic fibrosis. J. Cyst. Fibros. 2019, 18, 357–363. [Google Scholar] [CrossRef]
- Garcia, B.A.; Carden, J.L.; Goodwin, D.L.; Smith, T.A.; Gaggar, A.; Leon, K.; Antony, V.B.; Rowe, S.M.; Solomon, G.M. Implementation of a successful eradication protocol for Burkholderia cepacia complex in cystic fibrosis patients. BMC Pulm. Med. 2018, 18, 35. [Google Scholar] [CrossRef]
- Regan, K.H.; Bhatt, J. Eradication therapy for Burkholderia cepacia complex in people with cystic fibrosis. Cochrane Database Syst Rev. 2019, 4, CD009876. [Google Scholar] [CrossRef]
- Ridderberg, W.; Bendstrup, K.E.; Olesen, H.V.; Jensen-Fangel, S.; NorskovLauritsen, N. Marked increase in incidence of Achromobacter xylosoxidans infections caused by sporadic acquisition from the environment. J. Cyst. Fibros. 2011, 10, 466–469. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Ridderberg, W.; Hansen, C.R.; Høiby, N.; Jensen-Fangel, S.; Olesen, H.V.; Skov, M.; Lemming, L.E.; Pressler, T.; Johansen, H.K.; et al. Early treatment with inhaled antibiotics postpones next occurrence of Achromobacter in cystic fibrosis J. Cyst. Fibros. 2013, 12, 638–643. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.L.; Ramsey, B.W.; Hedges, D.L.; Hack, B.; Williams-Warren, J.; Weber, A.; Gore, E.J.; Redding, G.J. Safety of aerosol tobramycin administration for 3 months to patients with cystic fibrosis. Pediatr. Pulmonol. 1989, 7, 265–271. [Google Scholar] [CrossRef]
- Konstan, M.W.; Geller, D.E.; Minić, P.; Brockhaus, F.; Zhang, J.; Angyalosi, G. Tobramycin inhalation powder for P. aeruginosa infection in cystic fibrosis: The EVOLVE trial. Pediatr. Pulmonol. 2011, 46, 230–238. [Google Scholar] [CrossRef] [Green Version]
- Oermann, C.M.; Retsch-Bogart, G.Z.; Quittner, A.L.; Gibson, R.L.; McCoy, K.S.; Montgomery, A.B.; Cooper, P.J. An 18-month study of the safety and efficacy of repeated courses of inhaled aztreonam lysine in cystic fibrosis. Pediatr. Pulmonol. 2010, 45, 1121–1134. [Google Scholar] [CrossRef] [Green Version]
- Dasenbrook, E.C.; Konstan, M.W.; VanDevanter, D.R. Association between the introduction of a new cystic fibrosis inhaled antibiotic class and change in prevalence of patients receiving multiple inhaled antibiotic classes. J. Cyst. Fibros. 2015, 14, 370–375. [Google Scholar] [CrossRef] [Green Version]
- Nichols, D.P.; Durmowicz, A.G.; Field, A.; Flume, P.A.; VanDevanter, D.R.; Mayer-Hamblett, N. Developing inhaled antibiotics in cystic fibrosis: Current challenges and opportunities. Ann. Am. Thorac. Soc. 2019, 16, 534–539. [Google Scholar] [CrossRef]
- Rojo-Molinero, E.; D Macià, M.; Rubio, R.; Moyà, B.; Cabot, G.; López-Causapé, C.; Pérez, J.L.; Cantón, R.; Oliver, O. Sequential treatment of biofilms with aztreonam and tobramycin is a novel strategy for combating Pseudomonas aeruginosa chronic respiratory infections. Antimicrob. Agents Chemother. 2016, 60, 2912–2922. [Google Scholar] [CrossRef] [Green Version]
- Stanford, G.E.; Dave, K.; Simmonds, N.J. Pulmonary exacerbations in adults with cystic fibrosis: A grown-up issue in a changing cystic fibrosis landscape. Chest 2021, 159, 93–102. [Google Scholar] [CrossRef]
- Goss, C.H. Acute pulmonary exacerbation in cystic fibrosis. Semin. Respir. Crit. Care Med. 2019, 40, 792–803. [Google Scholar] [CrossRef]
- Waters, V.; Ratjen, F. Pulmonary exacerbations in children with cystic fibrosis. Ann. Am. Thorac. Soc. 2015, 12, S200–S206. [Google Scholar] [CrossRef]
- Cogen, J.D.; Faino, A.V.; Onchiri, F.; Hoffman, L.R.; Kronman, M.P.; Nelson, M.; Nichols, D.P.; Rosenfeld, M.; VanDevanter, D.R.; Gibson, R.L. Association of inhaled antibiotics in addition to standard IV therapy and outcomes of pediatric inpatient pulmonary exacerbations. Ann. Am. Thorac. Soc. 2020. [Google Scholar] [CrossRef]
- Frost, F.; Young, G.R.; Wright, L.; Miah, N.; Smith, D.L.; Winstanley, C.; Walshaw, M.J.; Fothergill, J.L.; Nazareth, D. The clinical and microbiological utility of inhaled aztreonam lysine for the treatment of acute pulmonary exacerbations of cystic fibrosis: An open-label randomised crossover study (AZTEC-CF). J. Cyst. Fibros. 2020. [Google Scholar] [CrossRef]
- Bhatt, J.M. Treatment of pulmonary exacerbations in cystic fibrosis. Eur. Respir. Rev. 2013, 22, 205–216. [Google Scholar] [CrossRef] [Green Version]
- Conway, S.P.; Pond, M.N.; Hamnett, T.; Watson, A. Compliance with treatment in adult patients with cystic fibrosis. Thorax 1996, 51, 29–33. [Google Scholar] [CrossRef] [Green Version]
- Bodnár, R.; Mészáros, A.; Oláh, M.; Ágh, T. Inhaled antibiotics for the treatment of chronic Pseudomonas aeruginosa infection in cystic fibrosis patients: Challenges to treatment adherence and strategies to improve outcomes. Patient Prefer. Adherence 2016, 10, 183–193. [Google Scholar] [CrossRef] [Green Version]
- Deacon, J.; Abdelghany, S.M.; Quinn, D.J.; Schmid, D.; Megaw, J.; Donnelly, R.F.; Jones, D.S.; Kissenpfennig, A.; Elborn, J.S.; Gilmore, B.F.; et al. Antimicrobial efficacy of tobramycin polymeric nanoparticles for Pseudomonas aeruginosa infections in cystic fibrosis: Formulation, characterisation and functionalisation with dornase alfa (DNase). J. Control. Release 2015, 198, 55–61. [Google Scholar] [CrossRef] [Green Version]
- Gillis, J.R.; Iglewski, B.H. Azithromycin retards Pseudomonas aeruginosa biofilm formation. J. Clin. Microbiol. 2004, 42, 5842–5845. [Google Scholar] [CrossRef] [Green Version]
- Høiby, N. Understanding bacterial biofilms in patients with cystic fibrosis: Current and innovative approaches to potential therapies. J. Cyst. Fibros. 2002, 1, 249–254. [Google Scholar] [CrossRef] [Green Version]
- Southern, K.W.; Barker, P.M. Azithromycin for cystic fibrosis. Eur. Respir. J. 2004, 24, 834–838. [Google Scholar] [CrossRef] [Green Version]
- Gordon, C.A.; Hodges, N.A.; Marriott, C. Antibiotic interaction and diffusion through alginate and exopolysaccharide of cystic fibrosis-derived Pseudomonas aeruginosa. J. Antimicrob. Chemother. 1988, 22, 667–674. [Google Scholar] [CrossRef]
- Huang, J.X.; Blaskovich, M.A.; Pelingon, R.; Ramu, S.; Kavanagh, A.; Elliott, A.G.; Butler, M.S.; Montgomery, A.B.; Cooper, M.A. Mucin binding reduces colistin antimicrobial activity. Antimicrob. Agents Chemother. 2015, 59, 5925–5931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bos, A.C.; Passé, K.M.; Mouton, J.W.; Janssens, H.M.; Tiddens, H.A. The fate of inhaled antibiotics after deposition in cystic fibrosis: How to get drug to the bug? J. Cyst. Fibros. 2017, 16, 13–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, L.; Murgia, X.; Siebenbürger, L.; Börger, C.; Schwarzkopf, K.; Sewald, K.; Häussler, S.; Braun, A.; Lehr, C.M.; Hittinger, M.; et al. Human airway mucus alters susceptibility of Pseudomonas aeruginosa biofilms totobramycin, but not colistin. J. Antimicrob. Chemother. 2018, 73, 2762–2769. [Google Scholar] [CrossRef]
- King, P.; Lomovskaya, O.; Griffith, D.C.; Burns, J.L.; Dudley, M.N. In vitro pharmacodynamics of levofloxacin and other aerosolized antibiotics under multiple conditions relevant to chronic pulmonary infection in cystic fibrosis. Antimicrob. Agents Chemother. 2010, 54, 143–148. [Google Scholar] [CrossRef] [Green Version]
- Bahamondez-Canas, T.F.; Zhang, H.; Tewes, F.; Leal, J.; Smyth, H.D.C. PEGylation of tobramycin improves mucus penetration and antimicrobial activity against Pseudomonas aeruginosa biofilms in vitro. Mol. Pharm. 2018, 15, 1643–1652. [Google Scholar] [CrossRef]
- Beaudoin, T.; Yau, Y.C.W.; Stapleton, P.J.; Gong, Y.; Wang, P.W.; Guttman, D.S.; Waters, V. Staphylococcus aureus interaction with Pseudomonas aeruginosa biofilm enhances tobramycin resistance. NPJ Biofilms Microbiomes 2017, 3, 25. [Google Scholar] [CrossRef]
- Carmody, L.A.; Zhao, J.; Kalikin, L.M.; LeBar, W.; Simon, R.H.; Venkataraman, A.; Schmidt, T.M.; Abdo, Z.; Schloss, P.D.; LiPuma, J.J. The daily dynamics of cystic fibrosis airway microbiota during clinical stability and at exacerbation. Microbiome 2015, 3, 12. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.J.; LiPuma, J.J. The microbiome in cystic fibrosis. Clin. Chest. Med. 2016, 37, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Sherrard, L.J.; McGrath, S.J.; McIlreavey, L.; Hatch, J.; Wolfgang, M.C.; Muhlebach, M.S.; Gilpin, D.F.; Elborn, J.S.; Tunney, M.M. Production of extended-spectrum β-lactamases and the potential indirect pathogenic role of Prevotella isolates from the cystic fibrosis respiratory microbiota. Int. J. Antimicrob. Agents 2016, 47, 140–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherrard, L.J.; Schaible, B.; Graham, K.A.; McGrath, S.J.; McIlreavey, L.; Hatch, J.; Wolfgang, M.C.; Muhlebach, M.S.; Gilpin, D.F.; Schneiders, T.; et al. Mechanisms of reduced susceptibility and genotypic prediction of antibiotic resistance in Prevotella isolated from cystic fibrosis (CF) and non-CF patients. J. Antimicrob. Chemother. 2014, 69, 2690–2698. [Google Scholar] [CrossRef] [Green Version]
- Carmody, L.A.; Caverly, L.J.; Foster, B.K.; Rogers, M.A.M.; Kalikin, L.M.; Simon, R.H.; VanDevanter, D.R.; LiPuma, J.J. Fluctuations in airway bacterial communities associated with clinical states and disease stages in cystic fibrosis. PLoS ONE 2018, 13, e0194060. [Google Scholar] [CrossRef]
- Smith, D.J.; Badrick, A.C.; Zakrzewski, M.; Krause, L.; Bell, S.C.; Anderson, G.J.; Reid, D.W. Pyrosequencing reveals transient cystic fibrosis lung microbiome changes with intravenous antibiotics. Eur. Respir. J. 2014, 44, 922–930. [Google Scholar] [CrossRef] [Green Version]
- Heirali, A.A.; Workentine, M.L.; Acosta, N.; Poonja, A.; Storey, D.G.; Somayaji, R.; Rabin, H.R.; Whelan, F.J.; Surette, M.G.; Parkins, M.D. The effects of inhaled aztreonam on the cystic fibrosis lung microbiome. Microbiome 2017, 5, 51. [Google Scholar] [CrossRef] [Green Version]
- Heirali, A.A.; Acosta, N.; Storey, D.G.; Workentine, M.L.; Somayaji, R.; Laforest-Lapointe, I.; Leung, W.; Quon, B.S.; Berthiaume, Y.; Rabin, H.R.; et al. The effects of cycled inhaled aztreonam on the cystic fibrosis (CF) lung microbiome. J. Cyst. Fibros. 2019, 18, 829–837. [Google Scholar] [CrossRef]



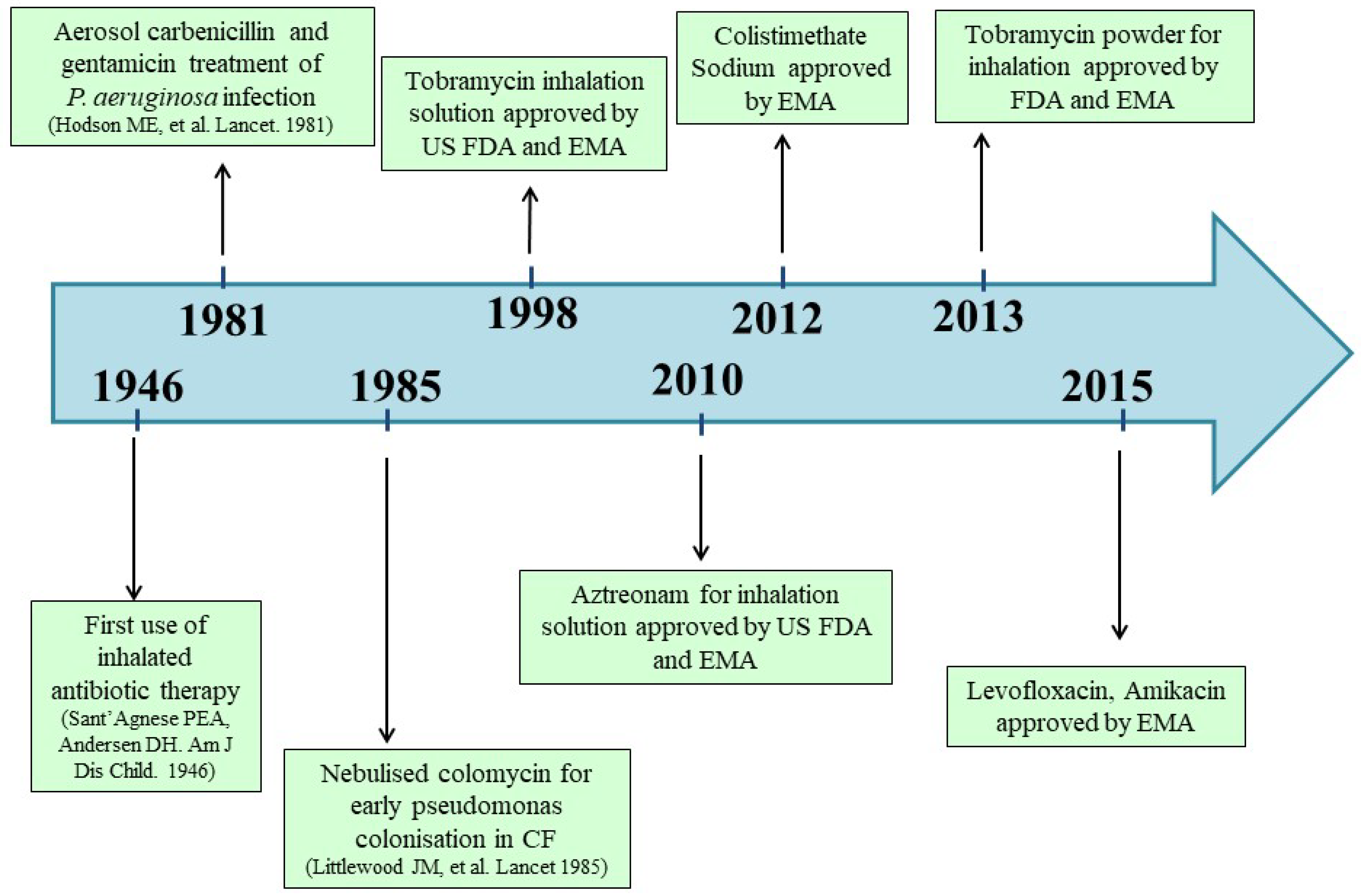{kind=link}
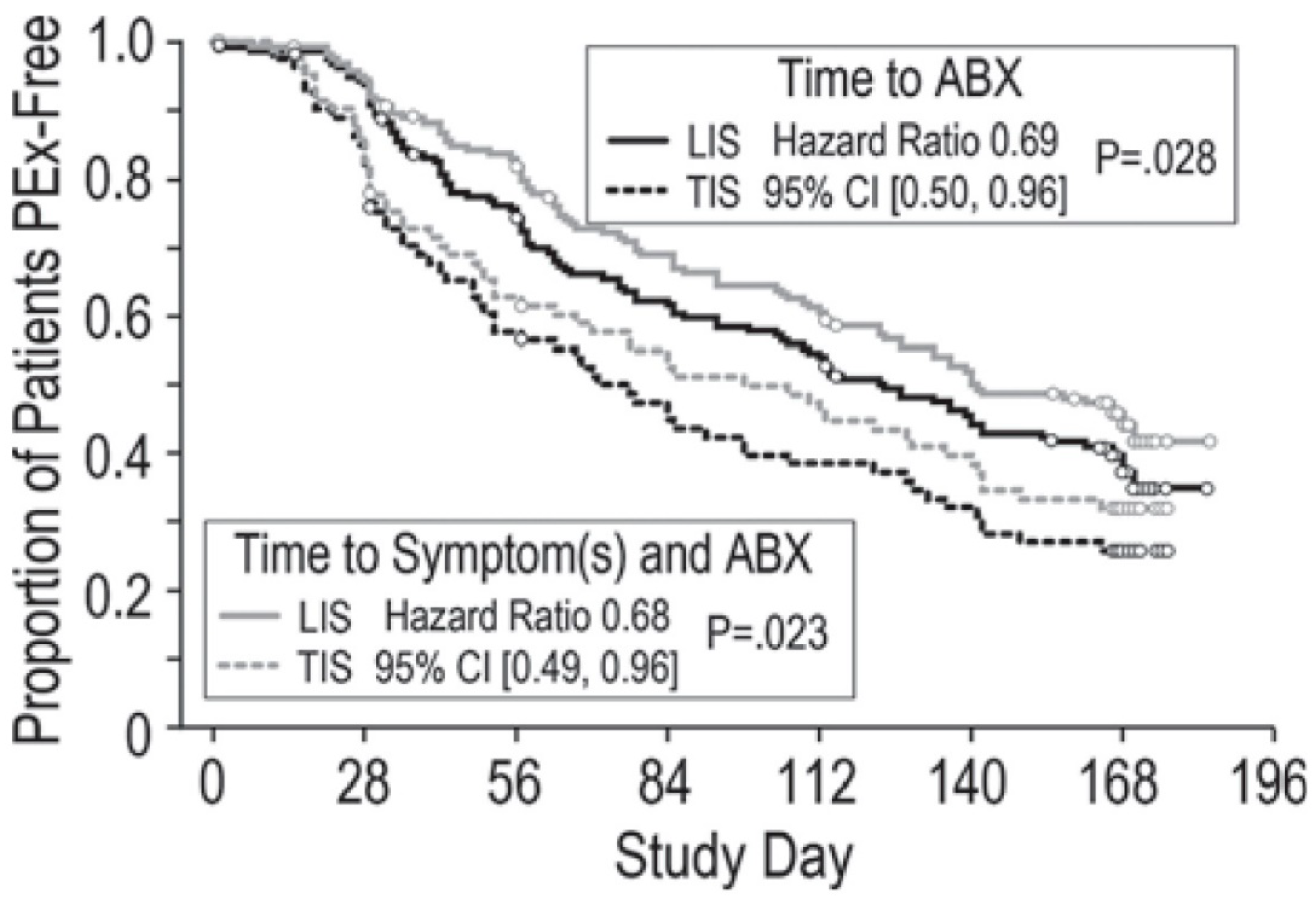{kind=link}
| Antibiotics | Type of Antibiotic | Mechanism of Action | Formulations | Trade Name | Nebulization Time | Dosage | Frequency |
|---|---|---|---|---|---|---|---|
| Tobramycin | Aminoglycosides | Inhibition of protein synthesis | Solution for nebulization | Tobramycin | 15 min | 300 mg/5 mL | Twice daily |
| Tobi | 15 min | 300 mg/5 mL | Twice daily | ||||
| Bramitob | 15 min | 300 mg/4 mL | Twice daily | ||||
| Vantobra | 4 min | 170 mg/1.7 mL | Twice daily | ||||
| Aztreonam lysine | Monobactams | Inhibition of bacterial cell wall synthesis | Solution for nebulization | Cayston | 2–3 min | 75 mg/1 mL | Three times daily |
| Levofloxacin | Fluoroquinolones | DNA gyrase and topoisomerase IV | Solution for nebulization | Quinsair | 5 min | 240 mg/3 mL | Twice daily |
| Colistimethate sodium * | Polymyxins | Disruption of bacterial cell membrane | Solution for nebulization | Promixin | 3 min | 80 mg/3 mL | Twice/Three times daily |
| Colfinair | 3–4 min | 80 mg/3 mL | Twice/Three times daily |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taccetti, G.; Francalanci, M.; Pizzamiglio, G.; Messore, B.; Carnovale, V.; Cimino, G.; Cipolli, M. Cystic Fibrosis: Recent Insights into Inhaled Antibiotic Treatment and Future Perspectives. Antibiotics 2021, 10, 338. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics10030338
Taccetti G, Francalanci M, Pizzamiglio G, Messore B, Carnovale V, Cimino G, Cipolli M. Cystic Fibrosis: Recent Insights into Inhaled Antibiotic Treatment and Future Perspectives. Antibiotics. 2021; 10(3):338. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics10030338
Chicago/Turabian StyleTaccetti, Giovanni, Michela Francalanci, Giovanna Pizzamiglio, Barbara Messore, Vincenzo Carnovale, Giuseppe Cimino, and Marco Cipolli. 2021. "Cystic Fibrosis: Recent Insights into Inhaled Antibiotic Treatment and Future Perspectives" Antibiotics 10, no. 3: 338. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics10030338