
Insight into the AcrAB-TolC Complex Assembly Process Learned from Competition Studies

 , ,
, ,
Abstract
:1. Introduction
2. Results
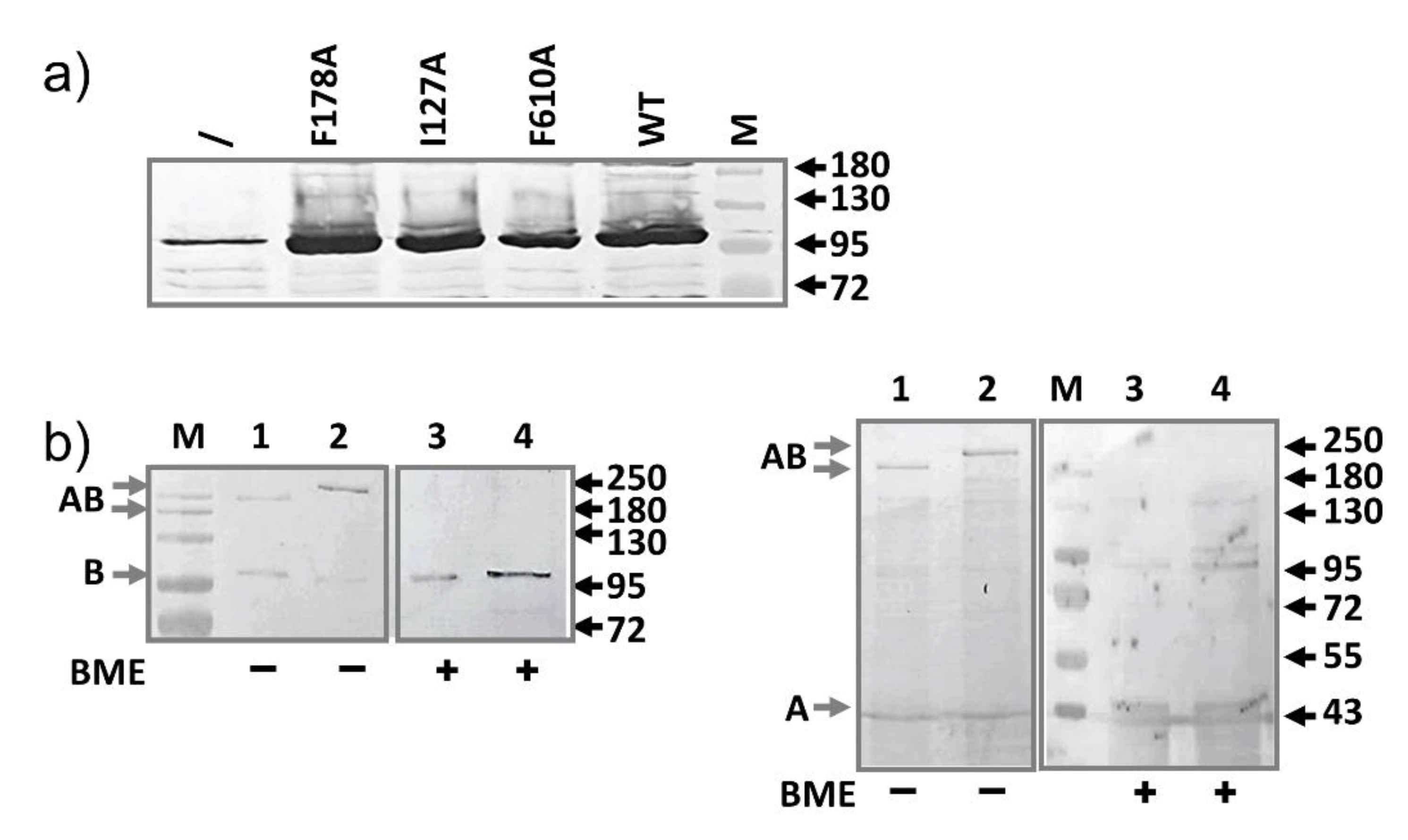
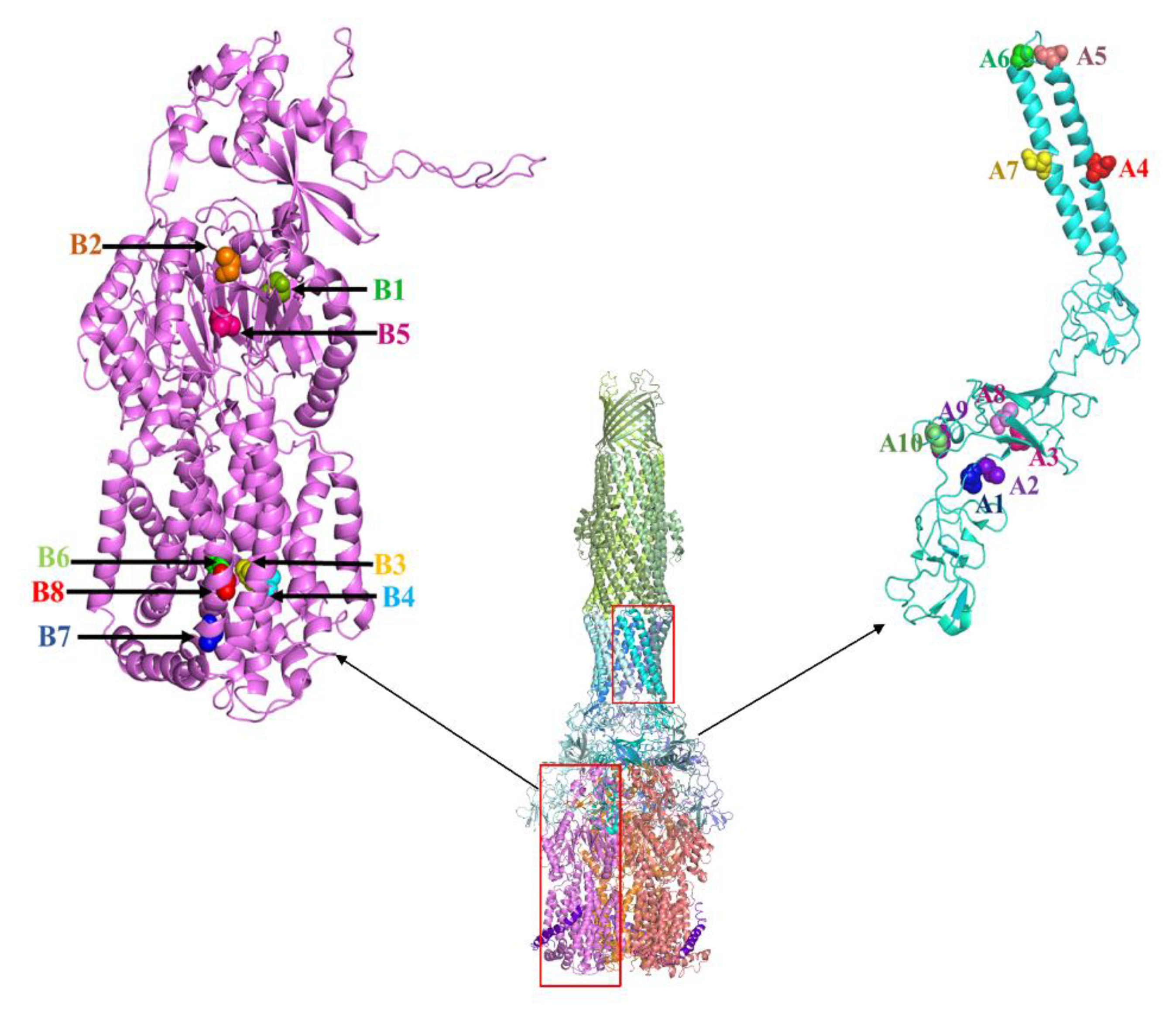
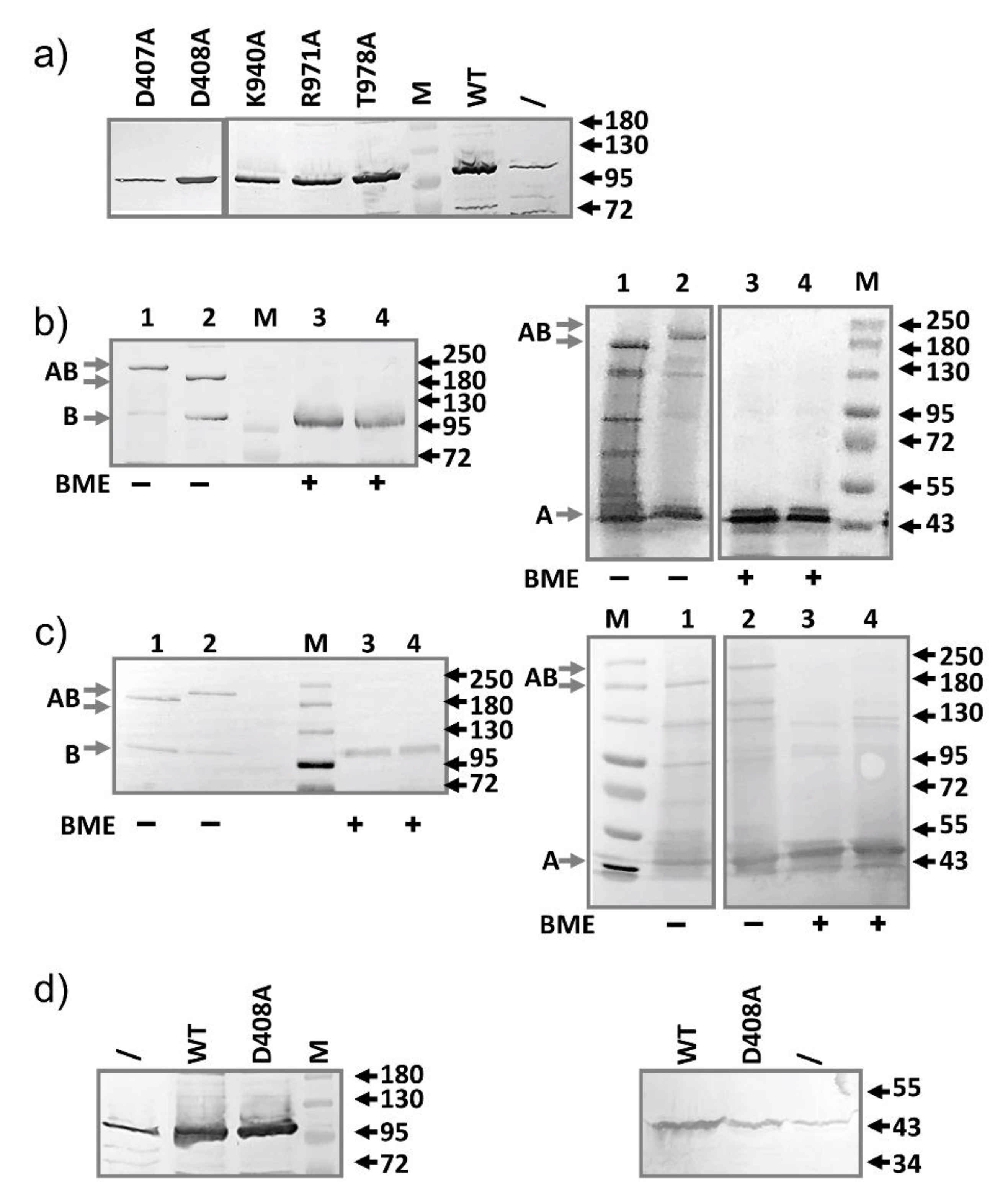
2.1. AcrB Mutants Defective in Proton Transport
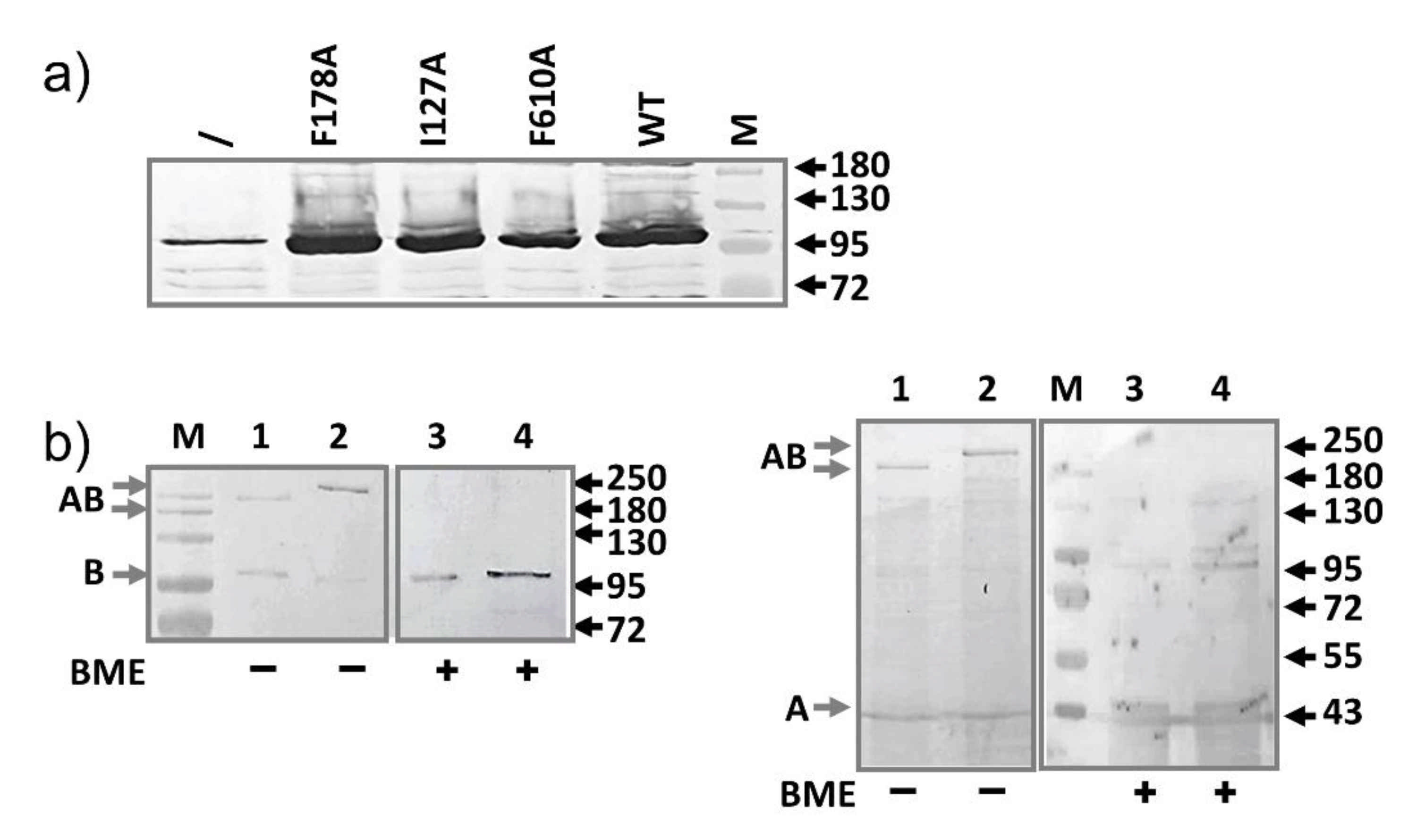
2.2. AcrB Mutants Defective in Substrate Binding
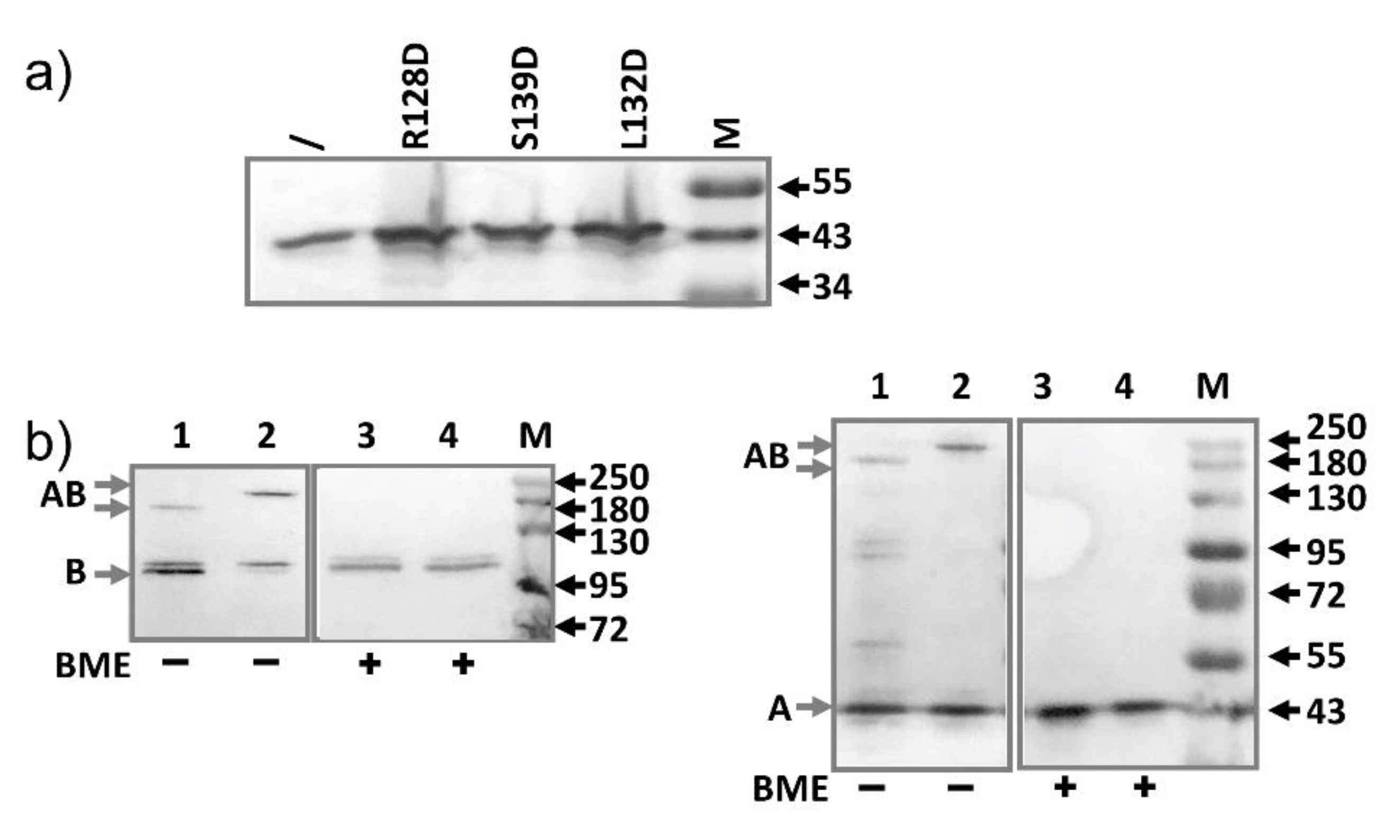
2.3. AcrA Mutant Defective in TolC Interaction
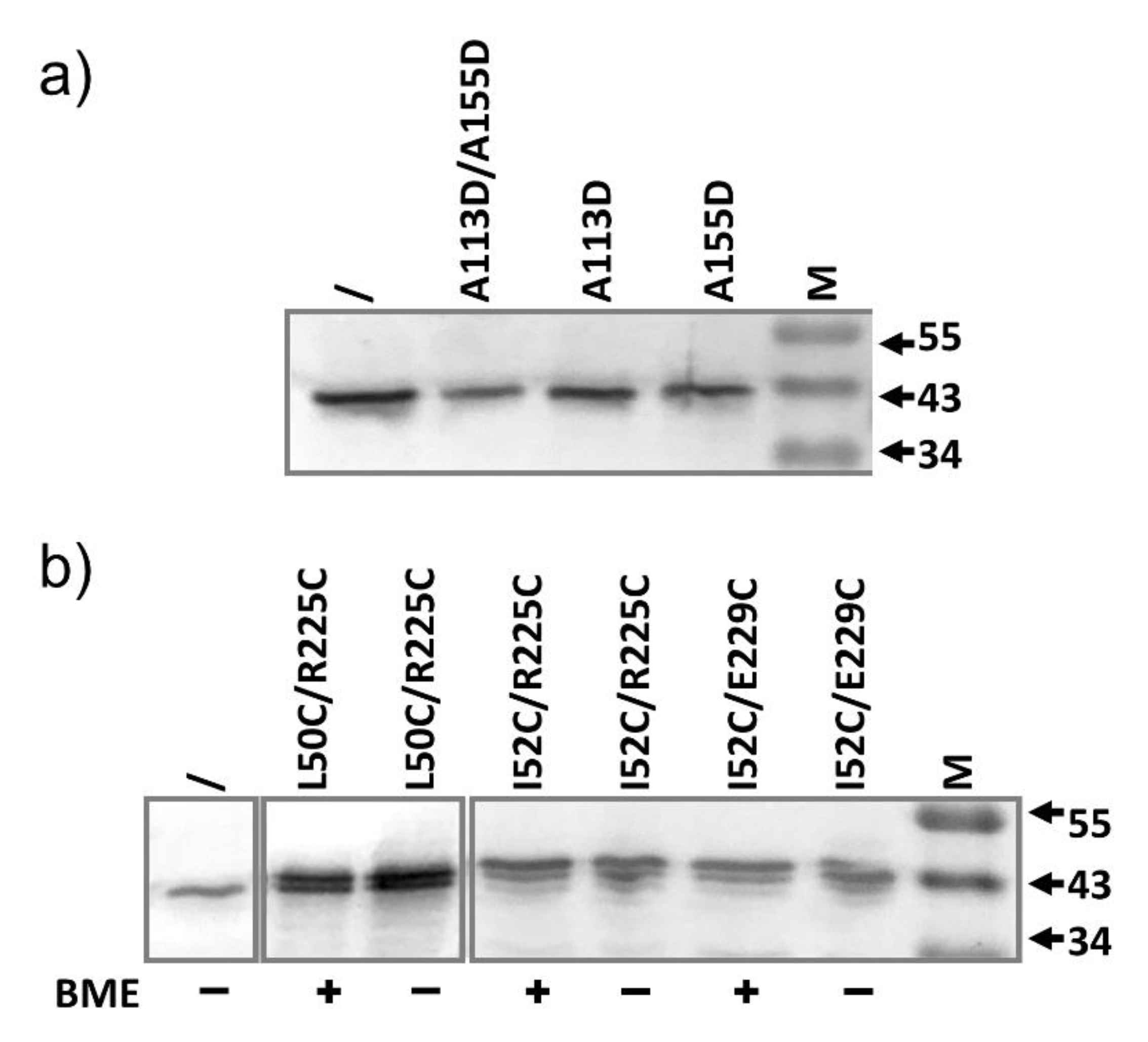
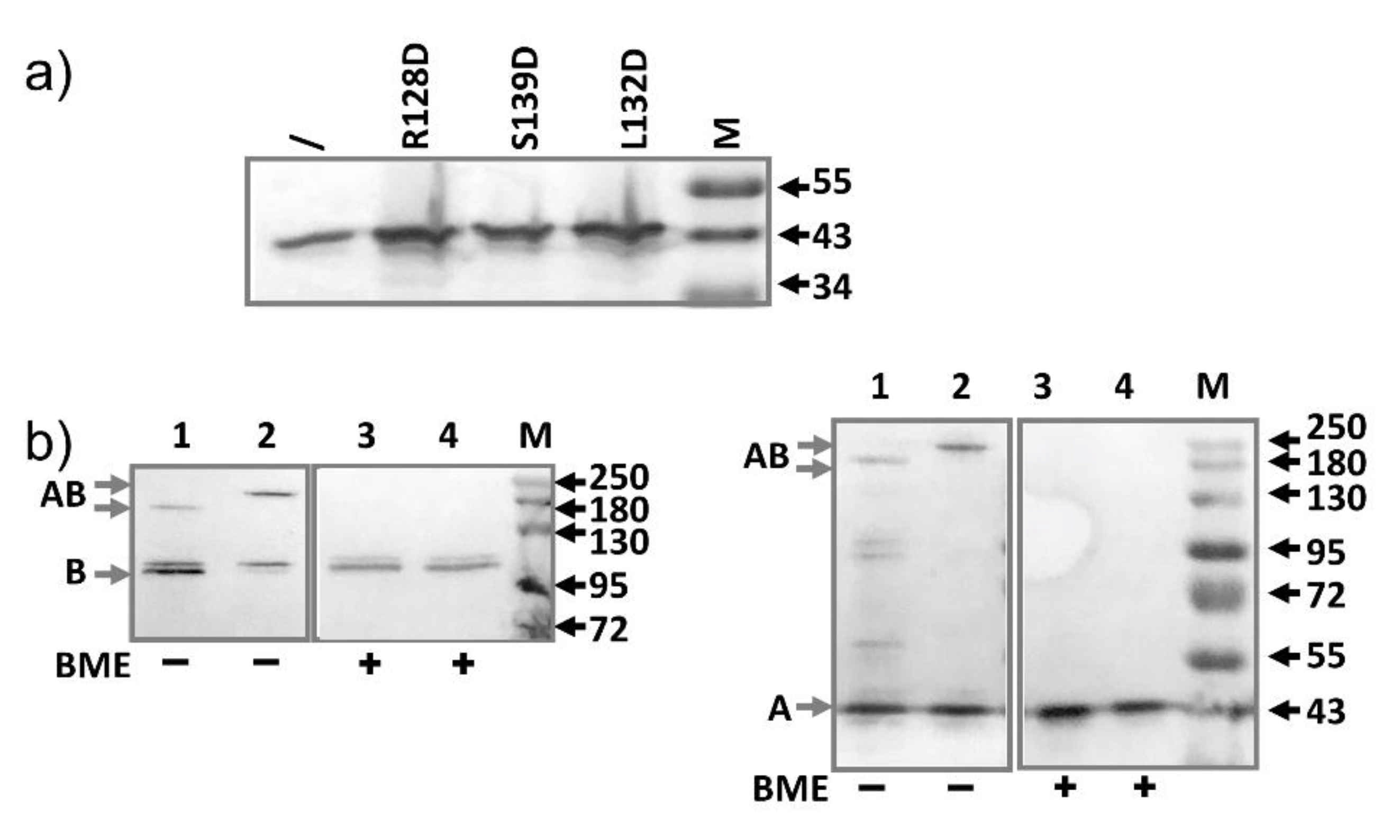
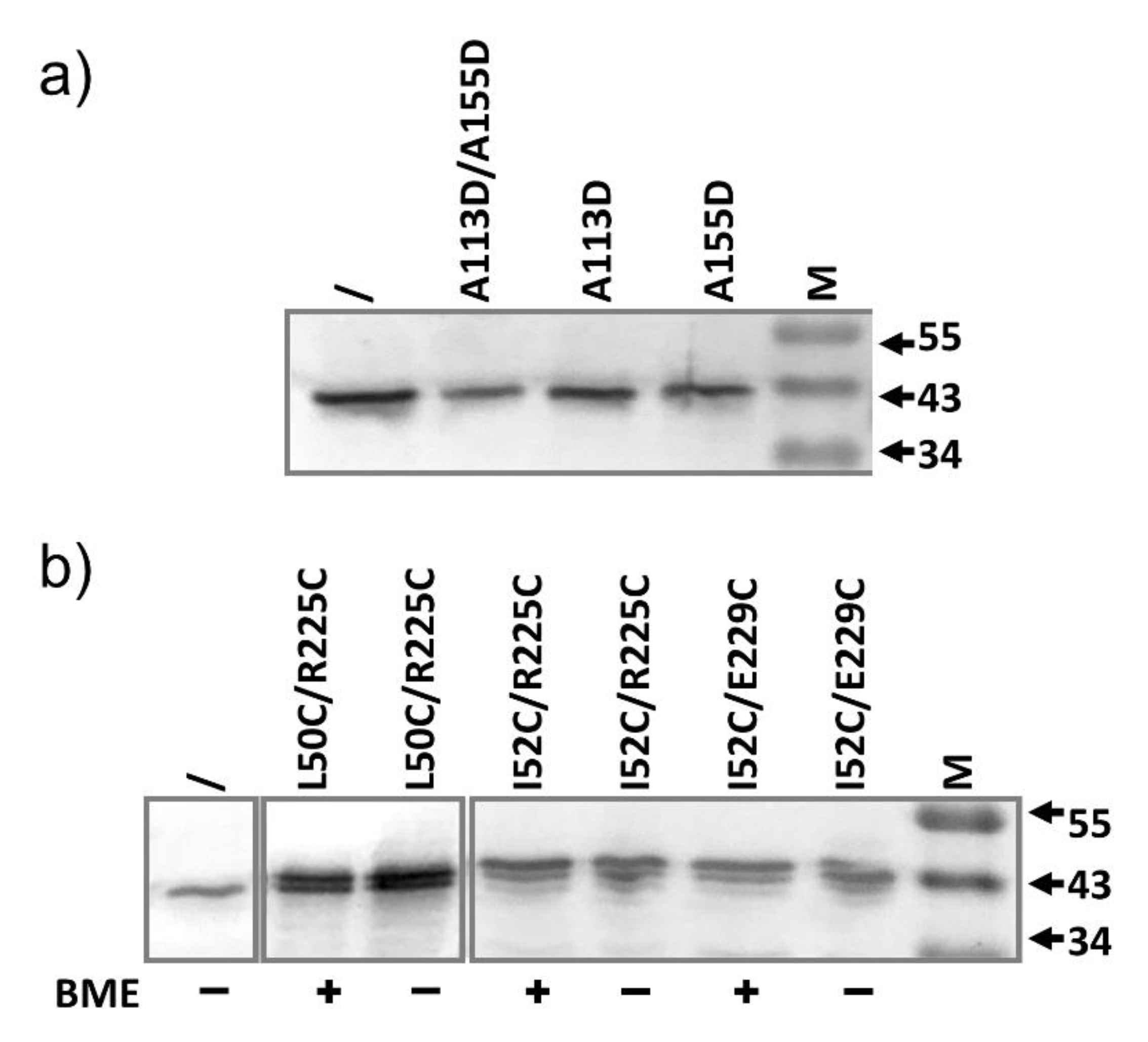
2.4. AcrA Mutant Defective in AcrA Assembly
2.5. AcrA Mutant Defective in Conformational Change
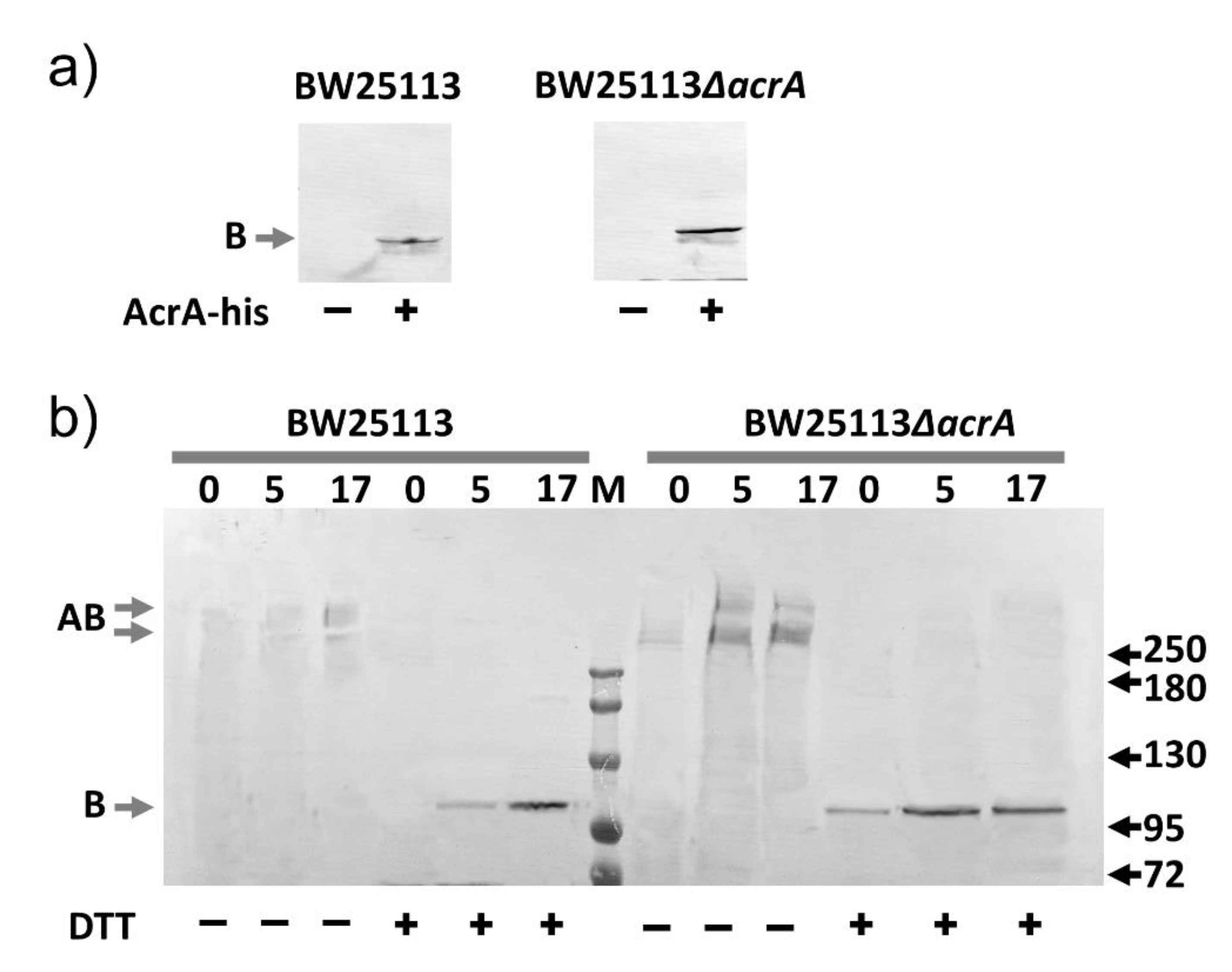
2.6. AcrA Mutants in a Strain Containing Anchor-Free AcrA
2.7. Slow Dissociation of the AcrAB Complex
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains, Plasmids, and Growth Conditions
4.2. Drug Susceptibility Assay
4.3. Protein Expression, SDS-PAGE and Western Blot Analysis
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- WHO. Antimicrobial resistance. Wkly. Epidemiol. Rec. Relev. Épidémiol. Hebd. 2000, 75, 336. [Google Scholar]
- Krishnamoorthy, G.; Tikhonova, E.B.; Dhamdhere, G.; Zgurskaya, H.I. On the role of TolC in multidrug efflux: The function and assembly of AcrAB-TolC tolerate significant depletion of intracellular TolC protein. Mol. Microbiol. 2013, 87, 982–997. [Google Scholar] [CrossRef] [Green Version]
- Nikaido, H. Structure and mechanism of RND-type multidrug efflux pumps. Adv. Enzym. Relat. Areas Mol. Biol. 2011, 77, 1–60. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, A.; Nakashima, R.; Sakurai, K. Structural basis of RND-type multidrug exporters. Front. Microbiol. 2015, 6, 327. [Google Scholar] [CrossRef] [Green Version]
- Du, D.; van Veen, H.W.; Murakami, S.; Pos, K.M.; Luisi, B.F. Structure, mechanism and cooperation of bacterial multidrug transporters. Curr. Opin. Struct. Biol. 2015, 33, 76–91. [Google Scholar] [CrossRef] [Green Version]
- Nishino, K.; Nikaido, E.; Yamaguchi, A. Regulation and physiological function of multidrug efflux pumps in Escherichia coli and Salmonella. Biochim. Et Biophys. Acta Proteins Proteom. 2009, 1794, 834–843. [Google Scholar] [CrossRef]
- Nikaido, H. Multidrug efflux pumps of gram-negative bacteria. J. Bacteriol. 1996, 178, 5853. [Google Scholar] [CrossRef] [Green Version]
- Eswaran, J.; Koronakis, E.; Higgins, M.K.; Hughes, C.K.; Oronakis, V. Three’s company: Component structures bring a closer view of tripartite drug efflux pumps. Curr. Opin. Struct. Biol. 2004, 14, 741–747. [Google Scholar] [CrossRef]
- Eicher, T.; Seeger, M.A.; Anselmi, C.; Zhou, W.; Brandstätter, L.; Verrey, F.; Diederichs, K.; Faraldo-Gómez, J.D.; Pos, K.M. Coupling of remote alternating-access transport mechanisms for protons and substrates in the multidrug efflux pump AcrB. Elife 2014, 3, e03145. [Google Scholar] [CrossRef] [Green Version]
- Daury, L.; Orange, F.; Taveau, J.-C.; Verchère, A.; Monlezun, L.; Gounou, C.; Marreddy, R.K.; Picard, M.; Broutin, I.; Pos, K.M.; et al. Tripartite assembly of RND multidrug efflux pumps. Nat. Commun. 2016, 7, 10731. [Google Scholar] [CrossRef] [Green Version]
- Du, D.; Wang, Z.; James, N.R.; Voss, J.E.; Klimont, E.; Ohene-Agyei, T.; Venter, H.; Chiu, W.; Luisi, B.F. Structure of the AcrAB–TolC multidrug efflux pump. Nature 2014, 509, 512–515. [Google Scholar] [CrossRef] [Green Version]
- Kobylka, J.; Kuth, M.S.; Müller, R.T.; Geertsma, E.R.; Pos, K.M. AcrB: A mean, keen, drug efflux machine. Ann. N. Y. Acad. Sci. 2020, 1459, 38–68. [Google Scholar] [CrossRef] [Green Version]
- Zgurskaya, H.I.; Nikaido, H. AcrA is a highly asymmetric protein capable of spanning the periplasm. J. Mol. Biol. 1999, 285, 409–420. [Google Scholar] [CrossRef]
- Zgurskaya, H.I.; Weeks, J.W.; Ntreh, A.T.; Nickels, L.M.; Wolloscheck, D. Mechanism of coupling drug transport reactions located in two different membranes. Front. Microbiol. 2015, 6, 100. [Google Scholar] [CrossRef] [Green Version]
- Higgins, C.F. Multiple molecular mechanisms for multidrug resistance transporters. Nature 2007, 446, 749–757. [Google Scholar] [CrossRef]
- Yen, M.R.; Peabody, C.R.; Partovi, S.M.; Zhai, Y.; Tseng, Y.H.; Saier, M.H. Protein-translocating outer membrane porins of Gram-negative bacteria. Biochim. Biophys. Acta 2002, 1562, 6–31. [Google Scholar] [CrossRef] [Green Version]
- Lobedanz, S.; Bokma, E.; Symmons, M.F.; Koronakis, E.; Hughes, C.; Koronakis, V. A periplasmic coiled-coil interface underlying TolC recruitment and the assembly of bacterial drug efflux pumps. Proc. Natl. Acad. Sci. USA 2007, 104, 4612–4617. [Google Scholar] [CrossRef] [Green Version]
- Koronakis, V.; Sharff, A.; Koronakis, E.; Luisi, B.; Hughes, C. Crystal structure of the bacterial membrane protein TolC central to multidrug efflux and protein export. Nature 2000, 405, 914–919. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Chen, M.; Yu, Z.; Bell, J.M.; Wang, H.; Forrester, I.; Villarreal, H.; Jakana, J.; Du, D.; Luisi, B.F.; et al. In situ structure and assembly of the multidrug efflux pump AcrAB-TolC. Nat. Commun. 2019, 10, 2635. [Google Scholar] [CrossRef] [PubMed]
- Murakami, S.; Nakashima, R.; Yamashita, E.; Yamaguchi, A. Crystal structure of bacterial multidrug efflux transporter AcrB. Nature 2002, 419, 587. [Google Scholar] [CrossRef] [PubMed]
- Schulz, R.; Vargiu, A.V.; Collu, F.; Kleinekathöfer, U.; Ruggerone, P. Functional rotation of the transporter AcrB: Insights into drug extrusion from simulations. PLoS Comput. Biol. 2010, 6, e1000806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, S.; Nakashima, R.; Yamashita, E.; Matsumoto, T.; Yamaguchi, A. Crystal structures of a multidrug transporter reveal a functionally rotating mechanism. Nature 2006, 443, 173–179. [Google Scholar] [CrossRef]
- Mikolosko, J.; Bobyk, K.; Zgurskaya, H.I.; Ghosh, P. Conformational flexibility in the multidrug efflux system protein AcrA. Structure 2006, 14, 577–587. [Google Scholar] [CrossRef] [Green Version]
- Symmons, M.F.; Bokma, E.; Koronakis, E.; Hughes, C.; Koronakis, V. The assembled structure of a complete tripartite bacterial multidrug efflux pump. Proc. Natl. Acad. Sci. USA 2009, 106, 7173–7178. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Fan, G.; Hryc, C.F.; Blaza, J.N.; Serysheva, I.I.; Schmid, M.F.; Chiu, W.; Luisi, B.F.; Du, D. An allosteric transport mechanism for the AcrAB-TolC multidrug efflux pump. Elife 2017, 6, e24905. [Google Scholar] [CrossRef]
- Marshall, R.L.; Bavro, V.N. Mutations in the TolC periplasmic domain affect substrate specificity of the AcrAB-TolC pump. Front. Mol. Biosci. 2020, 7, 166. [Google Scholar] [CrossRef] [PubMed]
- Murakami, S. Multidrug efflux transporter, AcrB—the pumping mechanism. Curr. Opin. Struct. Biol. 2008, 18, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, R.; Sakurai, K.; Yamasaki, S.; Nishino, K.; Yamaguchi, A. Structures of the multidrug exporter AcrB reveal a proximal multisite drug-binding pocket. Nature 2011, 480, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Pos, K.M.; Schiefner, A.; Seeger, M.A.; Diederichs, K. Crystallographic analysis of AcrB. FEBS Lett. 2004, 564, 333–339. [Google Scholar] [CrossRef] [Green Version]
- Seeger, M.A.; Schiefner, A.; Eicher, T.; Verrey, F.; Diederichs, K.; Pos, K.M. Structural asymmetry of AcrB trimer suggests a peristaltic pump mechanism. Science 2006, 313, 1295–1298. [Google Scholar] [CrossRef] [Green Version]
- Tikhonova, E.B.; Zgurskaya, H.I. AcrA, AcrB, and TolC of Escherichia coli form a stable intermembrane multidrug efflux complex. J. Biol. Chem. 2004, 279, 32116–32124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.C.; Liu, M.; Han, L. Energy coupling mechanisms of AcrB-like RND transporters. Biophys. Rep. 2017, 3, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Eicher, T.; Cha, H.-J.; Seeger, M.A.; Brandstätter, L.; El-Delik, J.; Bohnert, J.A.; Kern, W.V.; Verrey, F.; Grütter, M.G.; Diederichs, K.; et al. Transport of drugs by the multidrug transporter AcrB involves an access and a deep binding pocket that are separated by a switch-loop. Proc. Natl. Acad. Sci. USA 2012, 109, 5687–5692. [Google Scholar] [CrossRef] [Green Version]
- Su, C.-C.; Li, M.; Gu, R.; Takatsuka, Y.; McDermott, G.; Nikaido, H.; Edward, W.Y. Conformation of the AcrB multidrug efflux pump in mutants of the putative proton relay pathway. J. Bacteriol. 2006, 188, 7290–7296. [Google Scholar] [CrossRef] [Green Version]
- Li, X.-Z.; Plésiat, P.; Nikaido, H. The challenge of efflux-mediated antibiotic resistance in Gram-negative bacteria. Clin. Microbiol. Rev. 2015, 28, 337–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, S.; Yamaguchi, A. Multidrug-exporting secondary transporters. Curr. Opin. Struct. Biol. 2003, 13, 443–452. [Google Scholar] [CrossRef]
- Guan, L.; Nakae, T. Identification of Essential Charged Residues in Transmembrane Segments of the Multidrug Transporter MexB ofPseudomonas aeruginosa. J. Bacteriol. 2001, 183, 1734–1739. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Zhang, X.C. Energy-coupling mechanism of the multidrug resistance transporter AcrB: Evidence for membrane potential-driving hypothesis through mutagenic analysis. Protein Cell 2017, 8, 623–627. [Google Scholar] [CrossRef] [Green Version]
- Lu, W.; Zhong, M.; Wei, Y. Folding of AcrB subunit precedes trimerization. J. Mol. Biol. 2011, 411, 264–274. [Google Scholar] [CrossRef]
- Matsunaga, Y.; Yamane, T.; Terada, T.; Moritsugu, K.; Fujisaki, H.; Murakami, S.; Ikeguchi, M.; Kidera, A. Energetics and conformational pathways of functional rotation in the multidrug transporter AcrB. Elife 2018, 7, e31715. [Google Scholar] [CrossRef] [Green Version]
- Craig, D.B.; Dombkowski, A.A. Disulfide by Design 2.0: A web-based tool for disulfide engineering in proteins. BMC Bioinform. 2013, 14, 346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohnert, J.A.; Schuster, S.; Seeger, M.A.; Fähnrich, E.; Pos, K.M.; Kern, W.V. Site-directed mutagenesis reveals putative substrate binding residues in the Escherichia coli RND efflux pump AcrB. J. Bacteriol. 2008, 190, 8225–8229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinana, A.D.; Vargiu, A.V.; Nikaido, H. Effect of site-directed mutations in multidrug efflux pump AcrB examined by quantitative efflux assays. Biochem. Biophys. Res. Commun. 2016, 480, 552–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.-M.; Xu, Y.; Lee, M.; Piao, S.; Sim, S.-H.; Ha, N.-C.; Lee, K. Functional relationships between the AcrA hairpin tip region and the TolC aperture tip region for the formation of the bacterial tripartite efflux pump AcrAB-TolC. J. Bacteriol. 2010, 192, 4498–4503. [Google Scholar] [CrossRef] [Green Version]
- Hazel, A.J.; Abdali, N.; Leus, I.V.; Parks, J.M.; Smith, J.C.; Zgurskaya, H.I.; Gumbart, J.C. Conformational dynamics of AcrA govern multidrug efflux pump assembly. ACS Infect. Dis. 2019, 5, 1926–1935. [Google Scholar] [CrossRef]
- Torres, V.J.; McClain, M.S.; Cover, T.L. Mapping of a domain required for protein-protein interactions and inhibitory activity of a Helicobacter pylori dominant-negative VacA mutant protein. Infect. Immun. 2006, 74, 2093–2101. [Google Scholar] [CrossRef] [Green Version]
- Valente, L.; Nishikura, K. RNA binding-independent dimerization of adenosine deaminases acting on RNA and dominant negative effects of nonfunctional subunits on dimer functions. J. Biol. Chem. 2007, 282, 16054–16061. [Google Scholar] [CrossRef] [Green Version]
- Chevrier, L.; De Brevern, A.; Hernandez, E.; Leprince, J.; Vaudry, H.; Guedj, A.M.; De Roux, N. PRR repeats in the intracellular domain of KISS1R are important for its export to cell membrane. Mol. Endocrinol. 2013, 27, 1004–1014. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, R.; Li, X.-Y.; Pessara, U.; Van Huisjduijnen, R.H.; Benoist, C.; Mathis, D. Dominant negative analogs of NF-YA. J. Biol. Chem. 1994, 269, 20340–20346. [Google Scholar] [CrossRef]
- Zgurskaya, H.I.; Nikaido, H. Cross-linked complex between oligomeric periplasmic lipoprotein AcrA and the inner-membrane-associated multidrug efflux pump AcrB from Escherichia coli. J. Bacteriol. 2000, 182, 4264–4267. [Google Scholar] [CrossRef] [Green Version]
- Soparkar, K.; Kinana, A.D.; Weeks, J.W.; Morrison, K.D.; Nikaido, H.; Misra, R. Reversal of the drug binding pocket defects of the AcrB multidrug efflux pump protein of Escherichia coli. J. Bacteriol. 2015, 197, 3255–3264. [Google Scholar] [CrossRef] [Green Version]
- Lu, W.; Chai, Q.; Zhong, M.; Yu, L.; Fang, J.; Wang, T.; Li, H.; Zhu, H.; Wei, Y. Assembling of AcrB trimer in cell membrane. J. Mol. Biol. 2012, 423, 123–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Wen, A.; Shen, B.; Lu, J.; Huang, Y.; Chang, Y. FastCloning: A highly simplified, purification-free, sequence-and ligation-independent PCR cloning method. BMC Biotechnol. 2011, 11, 92. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Chen, B.; Duan, C.; Sun, B.; Yang, J.; Yang, S. Multigene editing in the Escherichia coli genome via the CRISPR-Cas9 system. Appl. Environ. Microbiol. 2015, 81, 2506–2514. [Google Scholar] [CrossRef] [Green Version]
- Humphries, R.M.; Ambler, J.; Mitchell, S.L.; Castanheira, M.; Dingle, T.; Hindler, J.A.; Koeth, L.; Sei, K. CLSI methods development and standardization working group best practices for evaluation of antimicrobial susceptibility tests. J. Clin. Microbiol. 2018, 56, e01934-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrate 1 | NOV | ERY | TPP | EtBr | R6G | NA |
|---|---|---|---|---|---|---|
| BW25113∆acrB containing | ||||||
| / | 4 | 4 | 4 | 8 | 8 | 1 |
| WT | 128 | 64 | 256 | 128 | 256 | 4 |
| D407A | 4 | 4 | 4 | 8 | 8 | ND |
| D408A | 4 | 4 | 4 | 8 | 8 | ND |
| K940A | 8 | 4 | 4 | 8 | 8 | ND |
| R971A | 8 | 4 | 4 | 8 | 16 | ND |
| T978A | 8 | 8 | 16 | 32 | 128 | ND |
| BW25113 containing | ||||||
| / | 256 | 64 | 1024 | 512 | 1024 | 4 |
| WT | 512 | 128 | 1024 | 512 | 1024 | 4 |
| D407A | 128 | 64 | 1024 | 256 | 512 | 4 |
| D408A | 128 | 64 | 1024 | 512 | 1024 | 4 |
| K940A | 256 | 64 | 1024 | 512 | 1024 | 4 |
| R971A | 128 | 32 | 1024 | 512 | 1024 | 4 |
| T978A | 256 | 64 | 1024 | 512 | 1024 | 2 |
| Substrate | NOV | ERY | TPP | EtBr | R6G |
|---|---|---|---|---|---|
| BW25113∆acrAB containing | |||||
| / | 4 | 4 | 8 | 4 | 16 |
| WT | 32 | 32 | 128 | 128 | 32 |
| AcrA/AcrB-D408A | 4 | 2 | 8 | 8 | 8 |
| AcrAP57C/AcrBN191C | 32 | 16 | 64 | 64 | 32 |
| AcrA-T217C/AcrB-S258C | 64 | 16 | 128 | 128 | 32 |
| BW25113 containing | |||||
| / | 256 | 32 | 1024 | 512 | 512 |
| AcrAB-WT | 256 | 64 | 1024 | 512 | 1024 |
| AcrAB-D408A | 256 | 32 | 1024 | 512 | 512 |
| Substrate | NOV | ERY | TPP | EtBr | R6G | NA |
|---|---|---|---|---|---|---|
| BW25113∆acrB containing | ||||||
| / | 4 | 4 | 4 | 8 | 8 | 1 |
| WT | 128 | 64 | 256 | 128 | 256 | 4 |
| F610A | 8 | 8 | 64 | 32 | 128 | ND |
| F178A | 64 | 8 | 128 | 128 | 256 | ND |
| I278A | 32 | 32 | 128 | 128 | 128 | ND |
| BW25113 containing | ||||||
| / | 256 | 64 | 1024 | 512 | 1024 | 4 |
| WT | 512 | 128 | 1024 | 512 | 1024 | 4 |
| F610A | 512 | 64 | 1024 | 512 | 1024 | 2 |
| F178A | 512 | 64 | 1024 | 512 | 1024 | 4 |
| I278A | 512 | 64 | 1024 | 512 | 1024 | 4 |
| Substrate | NOV | ERY | TPP | R6G | NA |
|---|---|---|---|---|---|
| BW25113∆acrA containing | |||||
| / | 4–8 | 2 | 4 | 4 | 1 |
| WT | 64 | 32 | 256 | 128 | 4 |
| L132D | 8 | 2 | 4 | 4 | ND |
| R128D | 4 | 2 | 4 | 4 | ND |
| S139D | 4 | 2 | 8–16 | 4 | ND |
| A113D | 64 | 4 | 32 | 16 | ND |
| A155D | 8 | 4 | 8 | 4 | ND |
| A113D, A155D | 8 | 2 | 8 | 4 | ND |
| L50C | 32 | 8 | 32 | 32 | ND |
| I52C | 32 | 4 | 16 | 32 | ND |
| E229C | 32 | 4 | 32 | 16 | ND |
| R225C | 16 | 8 | 64 | 16 | ND |
| L50C, R225C | 4 | 8 | 8 | 4 | ND |
| I52C, R225C | 4 | 4 | 8 | 8 | ND |
| I52C, E229C | 4 | 2 | 4 | 4 | ND |
| BW25113 containing | |||||
| / | 512 | 64 | 1024 | 512 | 4 |
| R128D | 512 | 64 | 1024 | 512 | 4 |
| L132D | 512 | 64 | 1024 | 512 | 4 |
| S139D | 256 | 64 | 1024 | 512 | 4 |
| A113D | 512 | 64 | 1024 | 512 | 4 |
| A155D | 512 | 64 | 1024 | 512 | 4 |
| A113D, A155D | 512 | 64 | 1024 | 512 | 4 |
| L50C | 512 | 64 | 1024 | 512 | 4 |
| I52C | 256 | 64 | 1024 | 512 | 4 |
| E229C | 512 | 64 | 1024 | 256 | 4 |
| R225C | 512 | 64 | 1024 | 512 | 4 |
| L50C, R225C | 512 | 64 | 1024 | 256 | 4 |
| I52C, R225C | 512 | 128 | 1024 | 512 | 4 |
| I52C, E229C | 512 | 128 | 1024 | 512 | 4 |
| Substrate | NOV | ERY | TPP | R6G |
|---|---|---|---|---|
| / | 512 | 128 | 2048 | 1024 |
| R128D | 512 | 128 | 2048 | 1024 |
| L132D | 512 | 128 | 2048 | 1024 |
| S139D | 512 | 128 | 2048 | 512 |
| A113D | 512 | 128 | 2048 | 512 |
| A155D | 512 | 128 | 2048 | 512 |
| A113D, A155D | 512 | 128 | 2048 | 512 |
| L50C, R225C | 512 | 128 | 2048 | 512 |
| I52C, R225C | 512 | 128 | 2048 | 1024 |
| I52C, E229C | 512 | 128 | 2048 | 1024 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rajapaksha, P.; Ojo, I.; Yang, L.; Pandeya, A.; Abeywansha, T.; Wei, Y. Insight into the AcrAB-TolC Complex Assembly Process Learned from Competition Studies. Antibiotics 2021, 10, 830. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics10070830
Rajapaksha P, Ojo I, Yang L, Pandeya A, Abeywansha T, Wei Y. Insight into the AcrAB-TolC Complex Assembly Process Learned from Competition Studies. Antibiotics. 2021; 10(7):830. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics10070830
Chicago/Turabian StyleRajapaksha, Prasangi, Isoiza Ojo, Ling Yang, Ankit Pandeya, Thilini Abeywansha, and Yinan Wei. 2021. "Insight into the AcrAB-TolC Complex Assembly Process Learned from Competition Studies" Antibiotics 10, no. 7: 830. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics10070830