
Human-Associated Methicillin-Resistant Staphylococcus aureus Clonal Complex 80 Isolated from Cattle and Aquatic Environments

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of Solid Samples
2.2. Isolation and Identification of MRSA
2.3. DNA Extraction
2.4. Detection of ARGs from Isolated MRSA Strains
2.5. Enterotoxins Detection in MRSA Isolates
2.6. Multilocus Typing of MRSA Isolates
2.7. Bioinformatic Analysis
2.8. Statistical Analysis
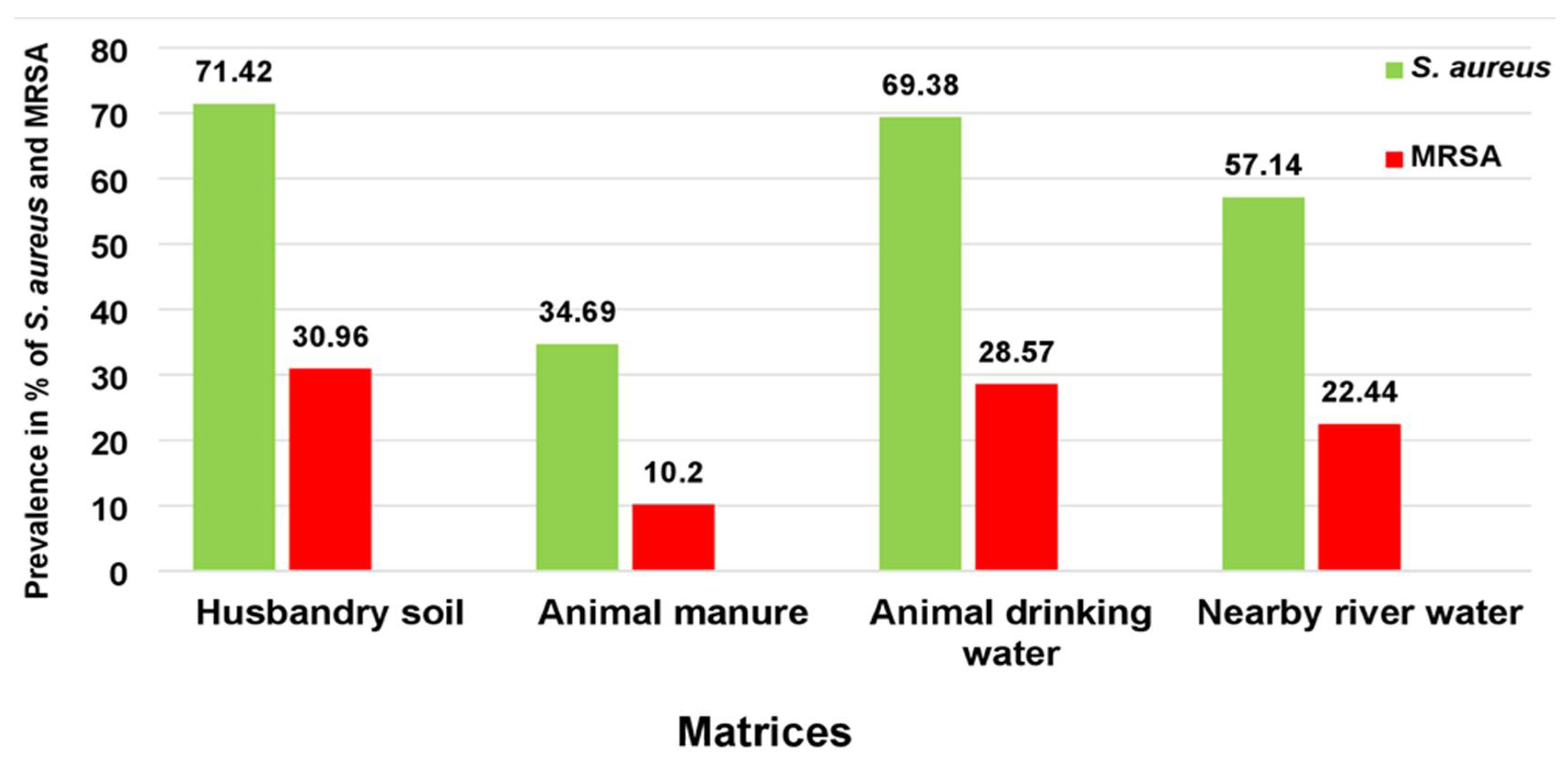
3. Results
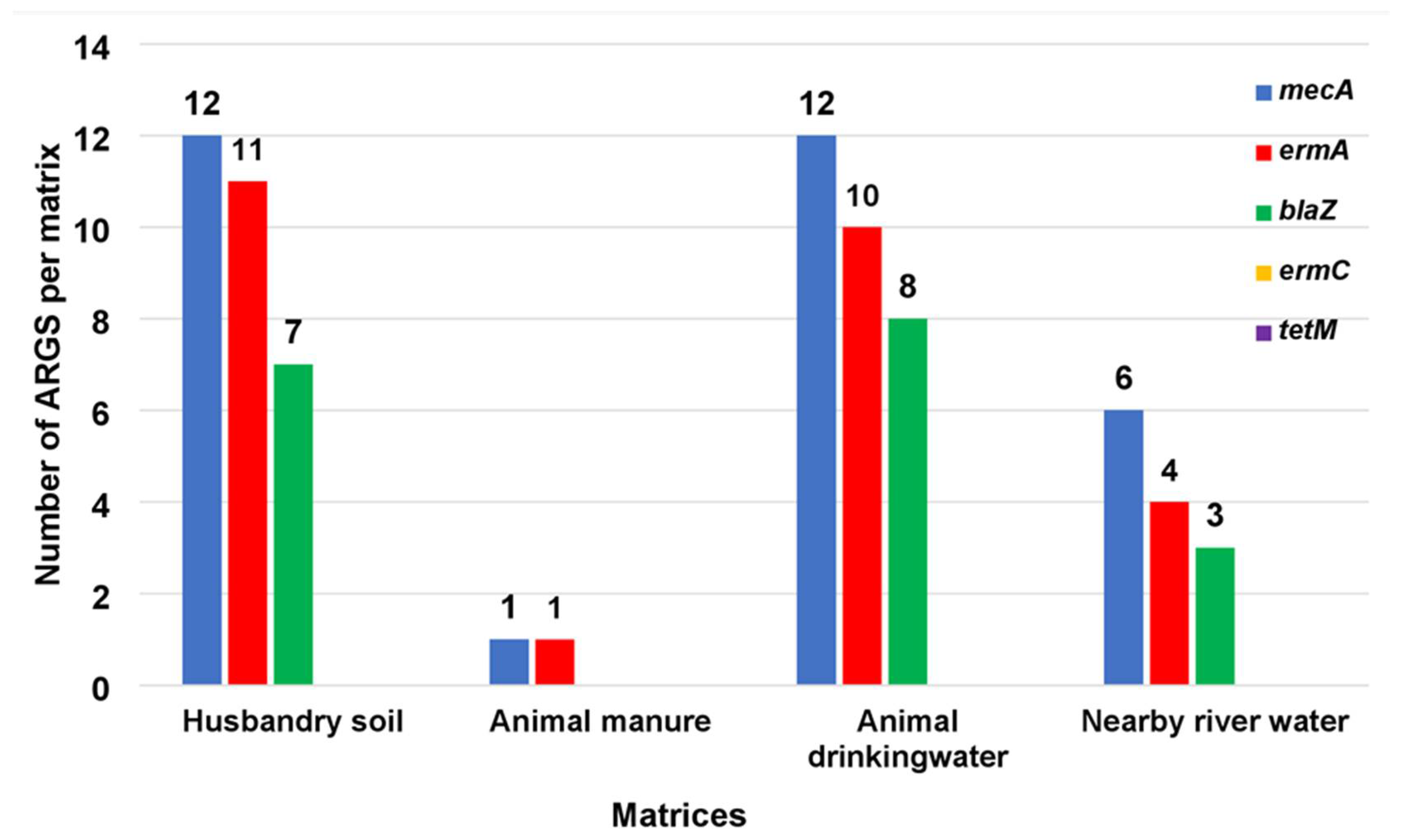
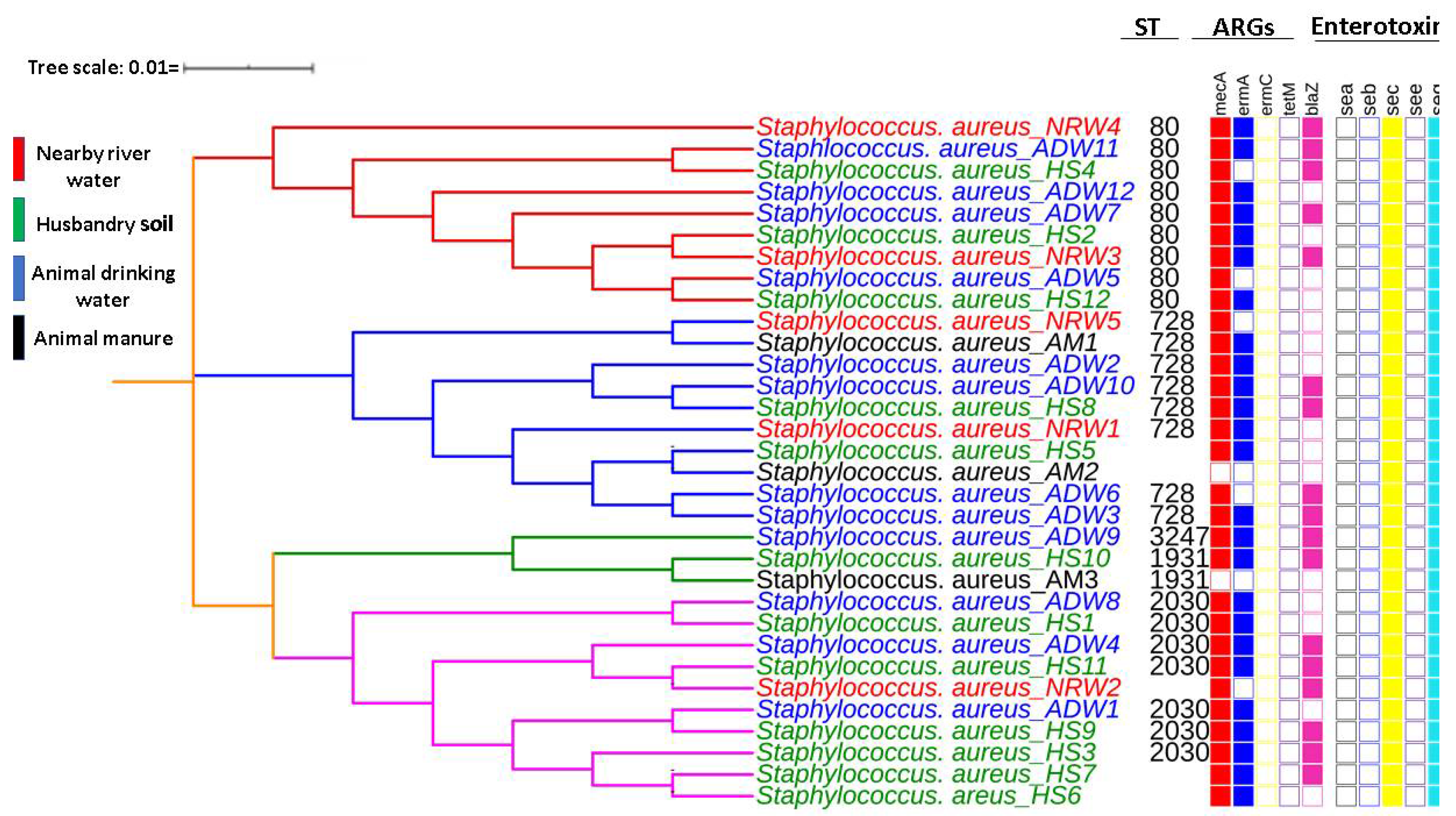
3.1. ARGs Typed in MRSA Isolates
3.2. Enterotoxins Detected in Isolated MRSA
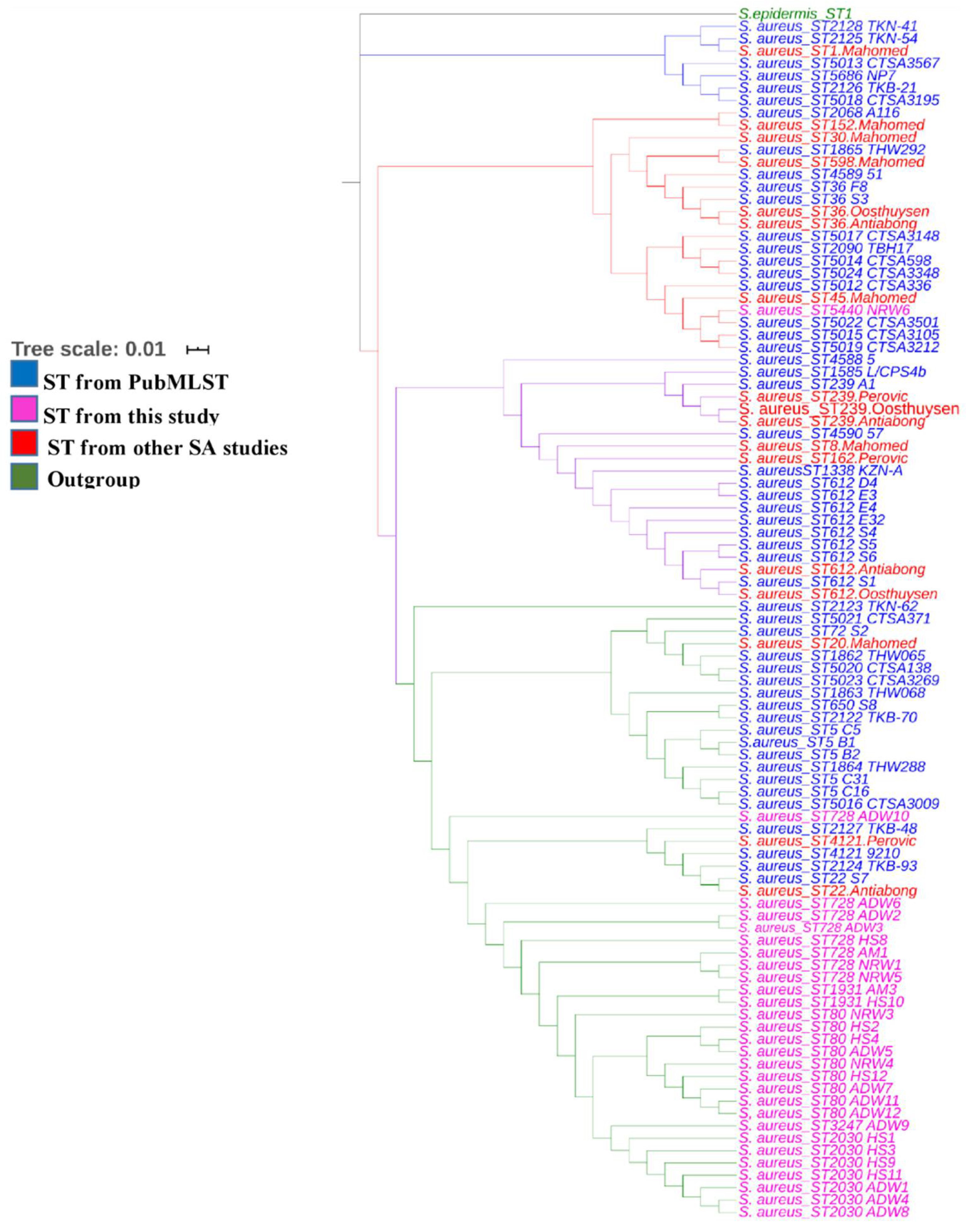
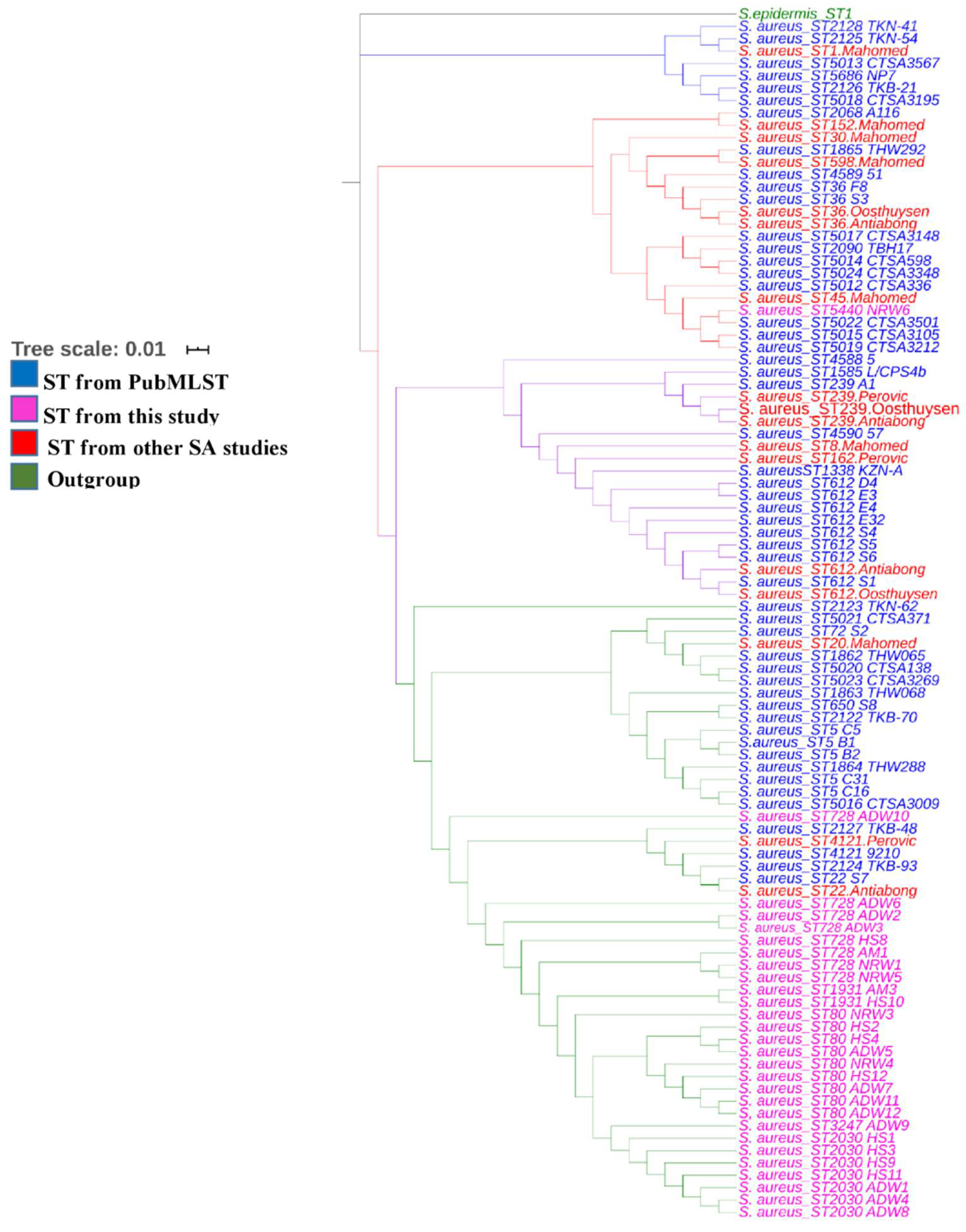
3.3. Sequence Types and MLST-Based Dendrogram
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cabral, J.P.S. Water microbiology. Bacterial pathogens and water. Int. J. Environ. Res. Public Health 2010, 7, 3657–3703. [Google Scholar] [CrossRef] [PubMed]
- Amarasiri, M.; Sano, D.; Suzuki, S. Understanding human health risks caused by antibiotic resistant bacteria (ARB) and antibiotic resistance genes (ARG) in water environments: Current knowledge and questions to be answered. Crit. Rev. Environ. Sci. Technol. 2019, 1–44. [Google Scholar] [CrossRef]
- Edokpayi, J.N.; Rogawski, E.T.; Kahler, D.M.; Hill, C.L.; Reynolds, C.; Nyathi, E.; Smith, J.A.; Odiyo, J.O.; Samie, A.; Bessong, P.; et al. Challenges to sustainable safe drinking water: A case study of water quality and use across seasons in rural communities in Limpopo Province, South Africa. Water 2018, 10, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verlicchi, P.; Grillini, V. Surface Water and Groundwater Quality in South Africa and Mozambique—Analysis of the Most Critical Pollutants for Drinking Purposes and Challenges in Water Treatment Selection. Water 2020, 12, 305. [Google Scholar] [CrossRef] [Green Version]
- Van den Honert, M.S.; Gouws, P.A.; Hoffman, L.C. Importance and implications of antibiotic resistance development in livestock and wildlife farming in South Africa: A Review. South Afr. J. Anim. Sci. 2018, 48, 401–412. [Google Scholar] [CrossRef] [Green Version]
- Ekwanzala, M.D.; Dewar, J.B.; Kamika, I.; Momba, M.N.B. Systematic review in South Africa reveals antibiotic resistance genes shared between clinical and environmental settings. Infect. Drug Resist. 2018, 11, 1907–1920. [Google Scholar] [CrossRef] [Green Version]
- Gajdács, M. The continuing threat of methicillin-resistant Staphylococcus aureus. Antibiotics 2019, 8, 52. [Google Scholar] [CrossRef] [Green Version]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef]
- Perovic, O.; Iyaloo, S.; Kularatne, R.; Lowman, W.; Bosman, N.; Wadula, J.; Seetharam, S.; Duse, A.; Mbelle, N.; Bamford, C.; et al. Prevalence and Trends of Staphylococcus aureus Bacteraemia in Hospitalized Patients in South Africa, 2010 to 2012: Laboratory-Based Surveillance Mapping of Antimicrobial Resistance and Molecular Epidemiology. PLoS ONE 2015, 10, e0145429. [Google Scholar] [CrossRef] [PubMed]
- Cuny, C.; Wieler, L.H.; Witte, W. Livestock-Associated MRSA: The Impact on Humans. Antibiotics 2015, 4, 521–543. [Google Scholar] [CrossRef]
- Naidoo, R.; Nuttall, J.; Whitelaw, A.; Eley, B. Epidemiology of Staphylococcus aureus bacteraemia at a tertiary children’s hospital in Cape Town, South Africa. PLoS ONE 2013, 8, e78396. [Google Scholar] [CrossRef] [Green Version]
- Mkize, N.; Zishiri, O.T.; Mukaratirwa, S. Genetic characterisation of antimicrobial resistance and virulence genes in Staphylococcus aureus isolated from commercial broiler chickens in the Durban metropolitan area, South Africa. J. S. Afr. Vet. Assoc. 2017, 88, e1–e7. [Google Scholar] [CrossRef] [PubMed]
- van Rensburg, M.J.; Eliya Madikane, V.; Whitelaw, A.; Chachage, M.; Haffejee, S.; Gay Elisha, B. The dominant methicillin-resistant Staphylococcus aureus clone from hospitals in Cape Town has an unusual genotype: ST612. Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 2011, 17, 785–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antiabong, J.F.; Kock, M.M.; Maphanga, T.G.; Salawu, A.M.; Mbelle, N.M.; Ehlers, M.M. Trends in the Genetic Background of Methicillin-Resistant Staphylococcus Aureus Clinical Isolates in a South African Hospital: An Institutional-Based Observational Study. Open Microbiol. J. 2017, 11, 339–351. [Google Scholar] [CrossRef]
- Henton, M.M.; Eagar, H.A.; Swan, G.E.; van Vuuren, M. Part VI. Antibiotic management and resistance in livestock production. South Afr. Med. J. 2011, 101, 583–586. [Google Scholar] [CrossRef]
- Van Boeckel, T.P.; Gandra, S.; Ashok, A.; Caudron, Q.; Grenfell, B.T.; Levin, S.A.; Laxminarayan, R. Global antibiotic consumption 2000 to 2010: An analysis of national pharmaceutical sales data. Lancet Infect. Dis. 2017, 14, 742–750. [Google Scholar] [CrossRef]
- Kraemer, S.A.; Ramachandran, A.; Perron, G.G. Antibiotic Pollution in the Environment: From Microbial Ecology to Public Policy. Microorganisms 2019, 7, 180. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Yuan, Q.; Mathieu, J.; Stadler, L.; Senehi, N.; Sun, R.; Alvarez, P.J.J. Antibiotic resistance genes from livestock waste: Occurrence, dissemination, and treatment. NPJ Clean Water 2020, 3, 4. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, J. Tackling drug-resistant infections globally. Arch. Pharm. Pract. 2016, 7, 110. [Google Scholar] [CrossRef]
- Anjum, M.F.; Marco-Jimenez, F.; Duncan, D.; Marín, C.; Smith, R.P.; Evans, S.J. Livestock-Associated Methicillin-Resistant Staphylococcus aureus From Animals and Animal Products in the UK. Front. Microbiol. 2019, 10, 2136. [Google Scholar] [CrossRef] [Green Version]
- Van Lochem, S.; Thompson, P.N.; Annandale, C.H. Prevalence of methicillin-resistant Staphylococcus aureus among large commercial pig herds in South Africa. Onderstepoort J. Vet. Res. 2018, 85, e1–e4. [Google Scholar] [CrossRef]
- Checcucci, A.; Trevisi, P.; Luise, D.; Modesto, M.; Blasioli, S.; Braschi, I.; Mattarelli, P. Exploring the Animal Waste Resistome: The Spread of Antimicrobial Resistance Genes Through the Use of Livestock Manure. Front. Microbiol. 2020, 11, 1416. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, S. Understanding the contribution of environmental factors in the spread of antimicrobial resistance. Environ. Health Prev. Med. 2015, 20, 243–252. [Google Scholar] [CrossRef] [Green Version]
- Manaia, C.M. Assessing the Risk of Antibiotic Resistance Transmission from the Environment to Humans: Non-Direct Proportionality between Abundance and Risk. Trends Microbiol. 2017, 25, 173–181. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-J.; Hu, H.-W.; Chen, Q.-L.; Singh, B.K.; Yan, H.; Chen, D.; He, J.-Z. Transfer of antibiotic resistance from manure-amended soils to vegetable microbiomes. Environ. Int. 2019, 130, 104912. [Google Scholar] [CrossRef] [PubMed]
- Han, X.-M.; Hu, H.-W.; Chen, Q.-L.; Yang, L.-Y.; Li, H.-L.; Zhu, Y.-G.; Li, X.-Z.; Ma, Y.-B. Antibiotic resistance genes and associated bacterial communities in agricultural soils amended with different sources of animal manures. Soil Biol. Biochem. 2018, 126, 91–102. [Google Scholar] [CrossRef]
- Manyi-Loh, C.; Mamphweli, S.; Meyer, E.; Okoh, A. Antibiotic Use in Agriculture and Its Consequential Resistance in Environmental Sources: Potential Public Health Implications. Molecules 2018, 23, 795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massé, D.I.; Saady, N.M.C.; Gilbert, Y. Potential of Biological Processes to Eliminate Antibiotics in Livestock Manure: An Overview. Anim. Open Access J. Mdpi 2014, 4, 146–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kane, T.L.; Carothers, K.E.; Lee, S.W. Virulence Factor Targeting of the Bacterial Pathogen Staphylococcus aureus for Vaccine and Therapeutics. Curr. Drug Targets 2018, 19, 111–127. [Google Scholar] [CrossRef]
- Amoako, D.G.; Somboro, A.M.; Abia, A.L.K.; Allam, M.; Ismail, A.; Bester, L.A.; Essack, S.Y. Whole-Genome Shotgun Sequence of Drug-Resistant Staphylococcus aureus Strain SA9, Isolated from a Slaughterhouse Chicken Carcass in South Africa. Microbiol. Resour. Announc. 2019, 8, e00489-19. [Google Scholar] [CrossRef] [Green Version]
- Enright, M.C.; Day, N.P.J.; Davies, C.E.; Peacock, S.J.; Spratt, B.G. Multilocus Sequence Typing for Characterization of Methicillin-Resistant and Methicillin-Susceptible Clones of Staphylococcus aureus. J. Clin. Microbiol. 2000, 38, 1008–1015. [Google Scholar] [CrossRef] [Green Version]
- Ekwanzala, M.D.; Dewar, J.B.; Kamika, I.; Momba, M.N.B. Comparative genomics of vancomycin-resistant Enterococcus spp. revealed common resistome determinants from hospital wastewater to aquatic environments. Sci. Total Environ. 2020, 719, 137275. [Google Scholar] [CrossRef] [PubMed]
- Abia, L.K.A.; Ubomba-Jaswa, E.; Ssemakalu, C.C.; Momba, M.N.B. Development of a rapid approach for the enumeration of Escherichia coli in riverbed sediment: Case study, the Apies River, South Africa. J. Soils Sediments 2015, 15, 2425–2432. [Google Scholar] [CrossRef]
- Strommenger, B.; Kettlitz, C.; Werner, G.; Witte, W. Multiplex PCR assay for simultaneous detection of nine clinically relevant antibiotic resistance genes in Staphylococcus aureus. J. Clin. Microbiol. 2003, 41, 4089–4094. [Google Scholar] [CrossRef] [Green Version]
- Martineau, F.; Picard, F.J.; Lansac, N.; Ménard, C.; Roy, P.H.; Ouellette, M.; Bergeron, M.G. Correlation between the Resistance Genotype Determined by Multiplex PCR Assays and the Antibiotic Susceptibility Patterns of Staphylococcus aureus andStaphylococcus epidermidis. Antimicrob. Agents Chemother. 2000, 44, 231–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saadati, M.; Barati, B.; Doroudian, M.; Shirzad, H.; Hashemi, M.; Hosseini, S.; Chaleshtari, A.; Bahmani, M.; Hosseinzadeh, S.; Imani, S. Detection of Sea, Seb, Sec, Seq genes in staphylococcus aureus isolated from nasal carriers in Tehran province, Iran; by multiplex PCR. J. Paramed. Sci. 2011, 2. Available online: https://www.sid.ir/FileServer/JE/127220110205.pdf (accessed on 21 July 2021).
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016, 44, W242–W245. [Google Scholar] [CrossRef]
- Oosthuysen, W.F.; Orth, H.; Lombard, C.J.; Sinha, B.; Wasserman, E. Population structure analyses of Staphylococcus aureus at Tygerberg Hospital, South Africa, reveals a diverse population, a high prevalence of Panton-Valentine leukocidin genes, and unique local methicillin-resistant S. aureus clones. Clin. Microbiol. Infect. Off. Publ. Eur. Soc. Clin. Microbiol. Infect. Dis. 2014, 20, 652–659. [Google Scholar] [CrossRef] [Green Version]
- Perovic, O.; Singh-Moodley, A.; Govender, N.P.; Kularatne, R.; Whitelaw, A.; Chibabhai, V.; Naicker, P.; Mbelle, N.; Lekalakala, R.; Quan, V.; et al. A small proportion of community-associated methicillin-resistant Staphylococcus aureus bacteraemia, compared to healthcare-associated cases, in two South African provinces. Eur. J. Clin. Microbiol. Infect. Dis. Off. Publ. Eur. Soc. Clin. Microbiol. 2017, 36, 2519–2532. [Google Scholar] [CrossRef]
- Goolam Mahomed, T.; Kock, M.M.; Masekela, R.; Hoosien, E.; Ehlers, M.M. Genetic relatedness of Staphylococcus aureus isolates obtained from cystic fibrosis patients at a tertiary academic hospital in Pretoria, South Africa. Sci. Rep. 2018, 8, 12222. [Google Scholar] [CrossRef]
- Moodley, A.S.; Perovic, O. Characterisation of Staphylococcus Aureus Bloodstream Isolates From Gauteng and Western Cape Provinces, South Africa, 2016 and 2017. Public Health Surveill. Bull 2017, 16, 99–107. [Google Scholar]
- Abdulgader, S.M.; van Rijswijk, A.; Whitelaw, A.; Newton-Foot, M. The association between pathogen factors and clinical outcomes in patients with Staphylococcus aureus bacteraemia in a tertiary hospital, Cape Town. Int. J. Infect. Dis. 2020, 91, 111–118. [Google Scholar] [CrossRef] [Green Version]
- Dweba, C.C.; Zishiri, O.T.; El Zowalaty, M.E. Isolation and Molecular Identification of Virulence, Antimicrobial and Heavy Metal Resistance Genes in Livestock-Associated Methicillin-Resistant Staphylococcus aureus. Pathogens 2019, 8, 79. [Google Scholar] [CrossRef] [Green Version]
- Friese, A.; Schulz, J.; Zimmermann, K.; Tenhagen, B.-A.; Fetsch, A.; Hartung, J.; Rösler, U. Occurrence of livestock-associated methicillin-resistant Staphylococcus aureus in Turkey and broiler barns and contamination of air and soil surfaces in their vicinity. Appl. Environ. Microbiol. 2013, 79, 2759–2766. [Google Scholar] [CrossRef] [Green Version]
- Thanner, S.; Drissner, D.; Walsh, F. Antimicrobial Resistance in Agriculture. mBio 2016, 7, e02227-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manyi-Loh, C.E.; Mamphweli, S.N.; Meyer, E.L.; Makaka, G.; Simon, M.; Okoh, A.I. An Overview of the Control of Bacterial Pathogens in Cattle Manure. Int. J. Environ. Res. Public Health 2016, 13, 843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramessar, K.; Olaniran, A.O. Antibiogram and molecular characterization of methicillin-resistant Staphylococcus aureus recovered from treated wastewater effluent and receiving surface water in Durban, South Africa. World J. Microbiol. Biotechnol. 2019, 35, 142. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.-G.; Zhao, Y.; Zhu, D.; Gillings, M.; Penuelas, J.; Ok, Y.S.; Capon, A.; Banwart, S. Soil biota, antimicrobial resistance and planetary health. Environ. Int. 2019, 131, 105059. [Google Scholar] [CrossRef]
- Cycoń, M.; Mrozik, A.; Piotrowska-Seget, Z. Antibiotics in the Soil Environment-Degradation and Their Impact on Microbial Activity and Diversity. Front. Microbiol. 2019, 10, 338. [Google Scholar] [CrossRef] [PubMed]
- Pekana, A.; Green, E. Antimicrobial Resistance Profiles of Staphylococcus aureus Isolated from Meat Carcasses and Bovine Milk in Abattoirs and Dairy Farms of the Eastern Cape, South Africa. Int. J. Environ. Res. Public Health 2018, 15, 2223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akanbi, O.E.; Njom, H.A.; Fri, J.; Otigbu, A.C.; Clarke, A.M. Antimicrobial Susceptibility of Staphylococcus aureus Isolated from Recreational Waters and Beach Sand in Eastern Cape Province of South Africa. Int. J. Environ. Res. Public Health 2017, 14, 1001. [Google Scholar] [CrossRef] [Green Version]
- Hoseini Alfatemi, S.M.; Motamedifar, M.; Hadi, N.; Sedigh Ebrahim Saraie, H. Analysis of Virulence Genes Among Methicillin Resistant Staphylococcus aureus (MRSA) Strains. Jundishapur J. Microbiol. 2014, 7, e10741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mairi, A.; Touati, A.; Lavigne, J.-P. Methicillin-Resistant Staphylococcus aureus ST80 Clone: A Systematic Review. Toxins 2020, 12, 119. [Google Scholar] [CrossRef] [Green Version]
- Harastani, H.H.; Tokajian, S.T. Community-associated methicillin-resistant Staphylococcus aureus clonal complex 80 type IV (CC80-MRSA-IV) isolated from the Middle East: A heterogeneous expanding clonal lineage. PLoS ONE 2014, 9, e103715. [Google Scholar] [CrossRef] [Green Version]
- Butaye, P.; Argudín, M.A.; Smith, T.C. Livestock-Associated MRSA and Its Current Evolution. Curr. Clin. Microbiol. Rep. 2016, 3, 19–31. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Abbreviation | Primer Sequence (F: Forward, R: Reverse) 5′ to 3′ | Product Size (bp) | Annealing Temp (°C) | References |
|---|---|---|---|---|
| Antibiotics resistance genes | ||||
| mecA | F-AAAATCGATGGTAAAGGTTGGC R-AGTTCTGCAGTACCGGATTTGC | 532 | 55 | [34] |
| ermA | F-AAGCGGTAAACCCCTCTGA R-TTCGCAAATCCCTTCTCAAC | 190 | 55 | |
| ermC | F-AATCGTCAATTCCTGCATGT R-TAATCGTGGAATACGGGTTTG | 299 | 55 | |
| tetM | F-AGTGGAGCGATTACAGAA RCATATGTCCTGGCGTGTCTA | 158 | 55 | |
| blaZ | F-ACTTCAACACCTGCTGCTTTC R-TGACCACTTTTATCAGCAACC | 173 | 55 | [35] |
| Enterotoxins | ||||
| sea | F-TTGCGAAAAAAGTCTGAA TTGC R-ATTAACCGAAGGTTCTGTAGAAGTA | 552 | 55 | [36] |
| seb | F-TCGCATCAAACTGACAAACG R-AGGTACTCTATAAGTGCCTGCCT | 477 | 55 | |
| sec | F-CTCAAGAACTAGACATAAAAGCTAGG RTTATATCAAAATCGGATTAACATTATC | 271 | 55 | |
| see | F-AGGTTTTTTCACAGGTCATCC R-CTTTTTTTTCTTCGGTCAATC | 178 | 55 | |
| seq | F-AATCTCTGGGTCAATGGTAAGC R-TTGTATTCGTTTTGTAGGTATTTTCG | 122 | 55 | |
| Housekeeping genes | ||||
| arcC | F-TTGATTCACCAGCGCGTATTGTC R-AGGTATCTGCTTCAATCAGCG | 456 | 55 | [31] |
| aroE | F-ATCGGAAATCCTATTTCACATTC R-GGTGTTGTATTAATAACGATATC | 456 | 55 | |
| glpF | F-CTAGGAACTGCAATCTTAATCC R-TGGTAAAATCGCATGTCCAATTC | 465 | 55 | |
| gmk | F-ATCGTTTTATCGGGACCATC R-TCATTAACTACAACGTAATCGTA | 429 | 55 | |
| pta | F-GTTAAAATCGTATTACCTGAAGG R-GACCCTTTTGTTGAAAAGCTTAA | 474 | 55 | |
| tpi | F-TCGTTCATTCTGAACGTCGTGAA R-TTTGCACCTTCTAACAATTGTAC | 402 | 55 | |
| yqiL | F-CAGCATACAGGACACCTATTGGC R-CGTTGAGGAATCGATACTGGAAC | 516 | 55 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramaite, K.; Ekwanzala, M.D.; Dewar, J.B.; Momba, M.N.B. Human-Associated Methicillin-Resistant Staphylococcus aureus Clonal Complex 80 Isolated from Cattle and Aquatic Environments. Antibiotics 2021, 10, 1038. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics10091038
Ramaite K, Ekwanzala MD, Dewar JB, Momba MNB. Human-Associated Methicillin-Resistant Staphylococcus aureus Clonal Complex 80 Isolated from Cattle and Aquatic Environments. Antibiotics. 2021; 10(9):1038. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics10091038
Chicago/Turabian StyleRamaite, Khuliso, Mutshiene Deogratias Ekwanzala, John Barr Dewar, and Maggy Ndombo Benteke Momba. 2021. "Human-Associated Methicillin-Resistant Staphylococcus aureus Clonal Complex 80 Isolated from Cattle and Aquatic Environments" Antibiotics 10, no. 9: 1038. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics10091038