
Adapting Clofazimine for Treatment of Cutaneous Tuberculosis by Using Self-Double-Emulsifying Drug Delivery Systems


Abstract
:1. Introduction
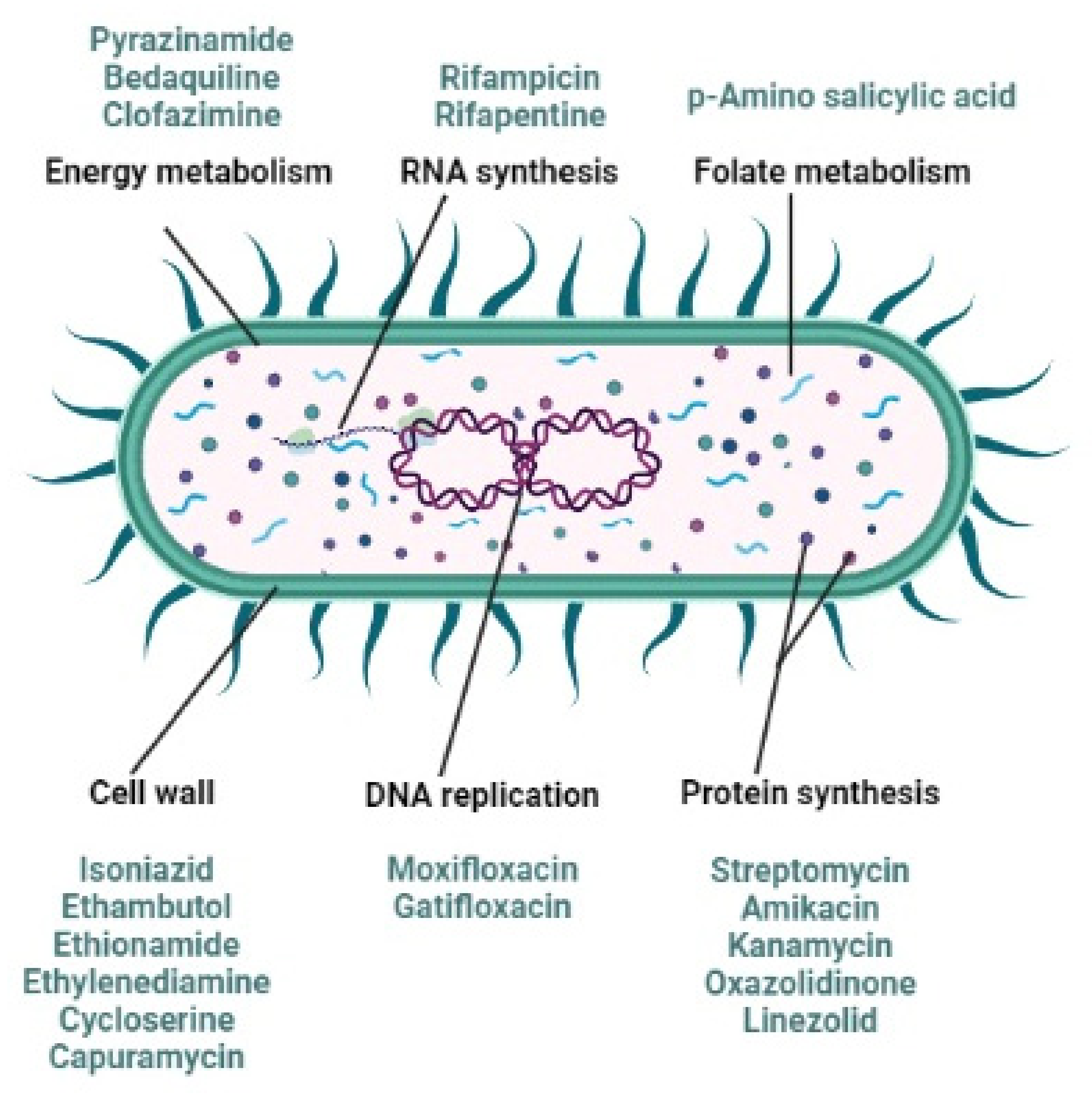
2. Anti-Tubercular Drug Resistance
- patient noncompliance (failure to complete the correct full course of treatment),
- reinfection with Mtb post treatment,
- the inappropriate utilization of medicines such as when health-care providers prescribe the incorrect treatment regime (incorrect employment of antibiotics),
- prescribing an incorrect dosage, or length of time for treatment (incorrect treatment regime),
- an inconsistent treatment (drug availability),
- restricted access to treatment, or
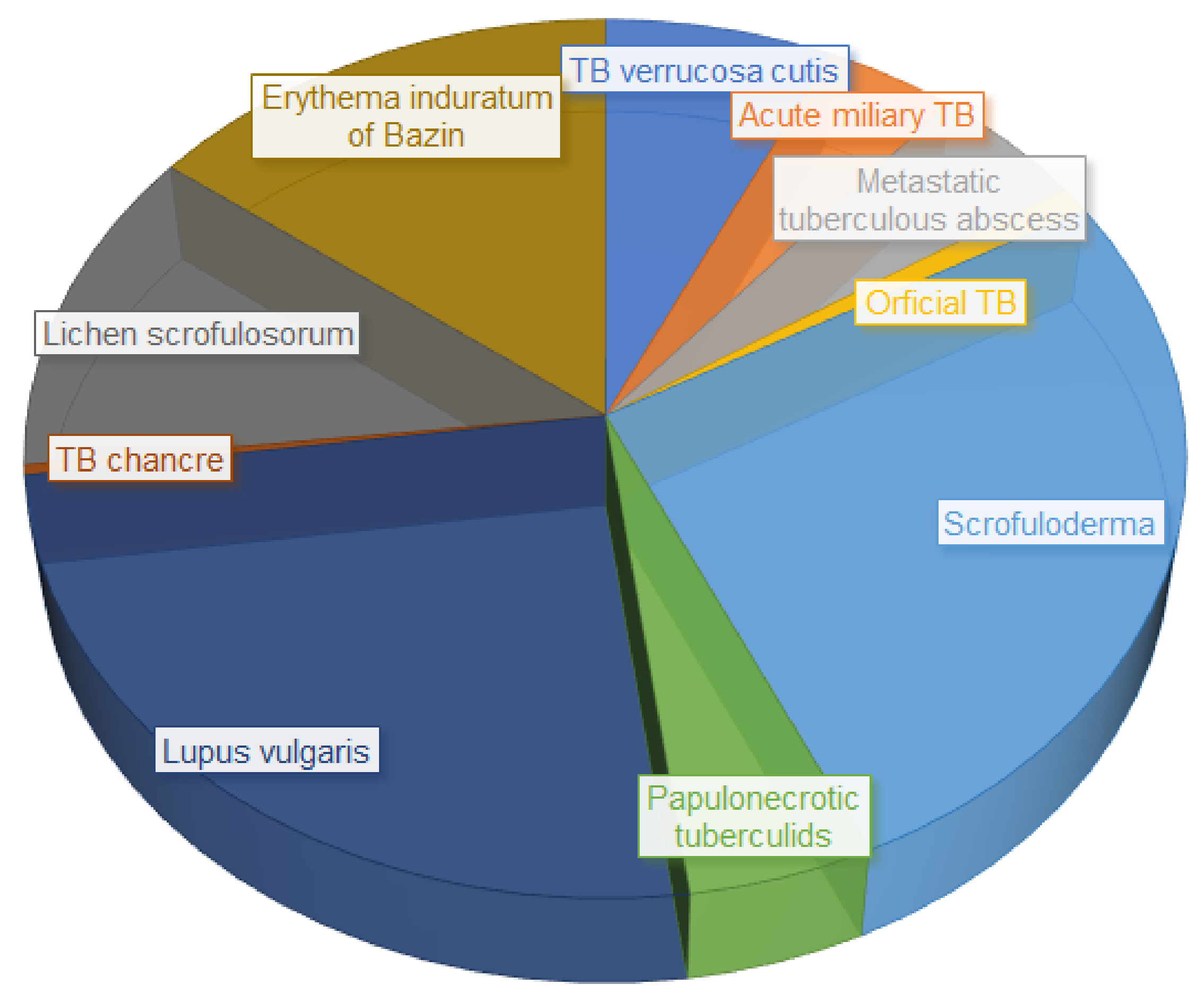
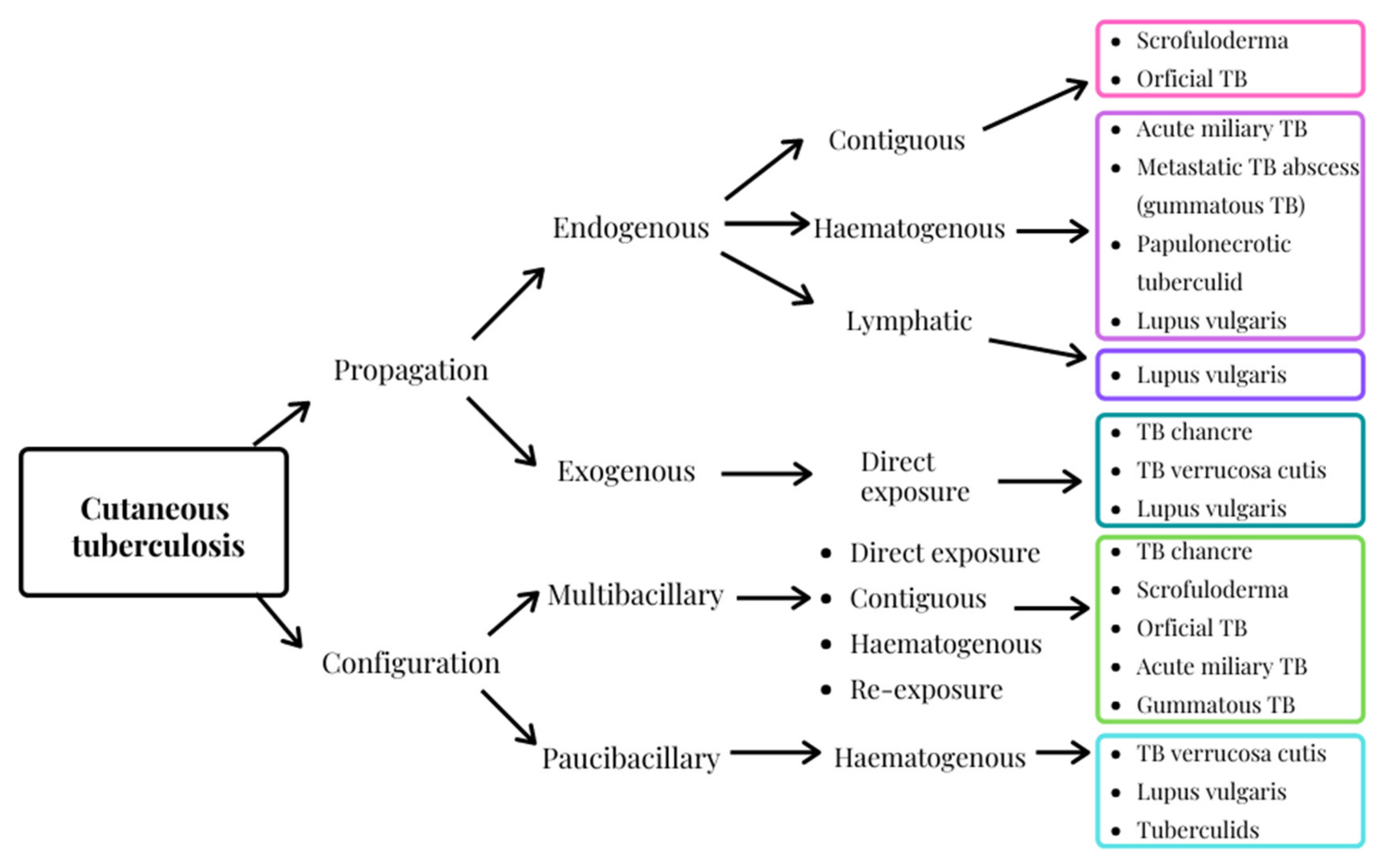
3. Cutaneous Tuberculosis, an Extra-Pulmonary Infection
4. Clofazimine, an Orphan Drug
5. An Alternative Strategy to Deliver Clofazimine More Effectively
5.1. Contemplating Topical Delivery
5.2. Clofazimine Characteristics Challenging the Topical Route
5.3. Auxiliary Natural Excipients to Enhance Topical Clofazimine Delivery
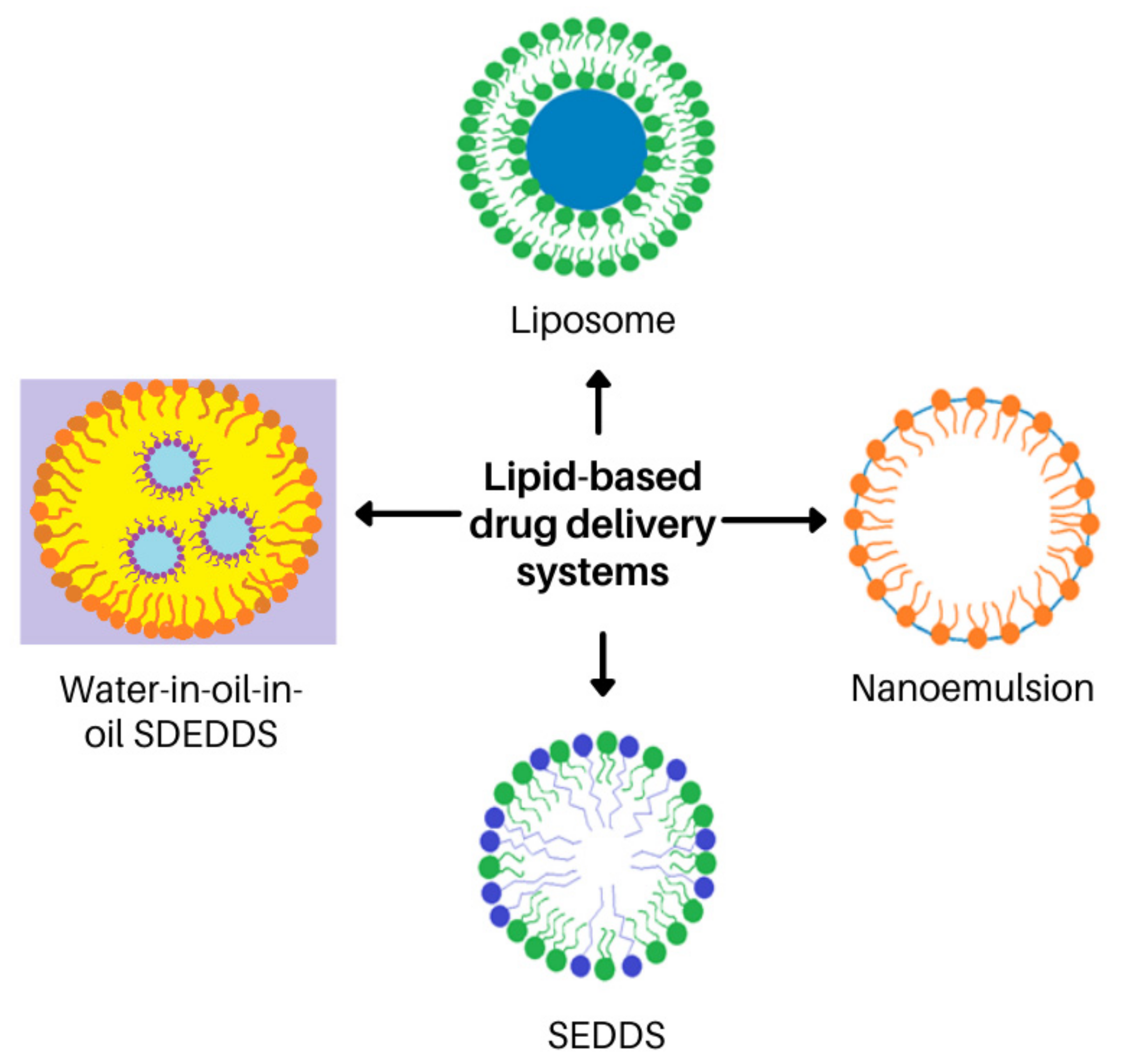
6. The Prospect of Self-Double-Emulsifying Drug Delivery Systems for Enhancing Topical Delivery of Clofazimine
6.1. Lipid-Based Carrier Systems Ideal for Lipophilic Drugs
6.2. Why SDEDDSs Are Considered More Appropriate Than Other Lipid-Based Carrier Systems
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Disclaimer
References
- Ankrah, A.O.; Glaudemans, A.W.J.M.; Maes, A.; van der Wiele, C.; Dierckx, R.A.J.O.; Vorster, M.; Sathekge, M.M. Tuberculosis. Semin. Nucl. Med. 2018, 48, 108–130. [Google Scholar] [CrossRef] [PubMed]
- Sotgiu, G.; Tiberi, S.; Centis, R.; D’Ambrosio, L.; Fuentes, Z.; Zumla, A.; Migliori, G.B. Applicability of the shorter ‘Bangladesh regimen’ in high multidrug-resistant tuberculosis settings. Int. J. Infect. Dis. 2017, 56, 190–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grace, A.G.; Mittal, A.; Jain, S.; Tripathy, J.P.; Satyanarayana, S.; Tharyan, P.; Kirubakaran, R. Shortened treatment regimens versus the standard regimen for drug-sensitive pulmonary tuberculosis. Cochrane Database Syst. Rev. 2018, 1, CD012918. [Google Scholar] [CrossRef] [Green Version]
- Nellums, L.B.; Thompson, H.; Holmes, A.; Castro-Sánchez, E.; Otter, J.A.; Norredam, M.; Friedland, J.S.; Hargreaves, S. Antimicrobial resistance among migrants in Europe: A systematic review and meta-analysis. Lancet Infect. Dis. 2018, 18, 796–811. [Google Scholar] [CrossRef] [Green Version]
- Walker, T.M.; Merker, M.; Knoblauch, A.M.; Helbling, P.; Schoch, O.D.; van der Werf, M.J.; Kranzer, K.; Fiebig, L.; Kröger, S.; Haas, W.; et al. A cluster of multidrug-resistant Mycobacterium tuberculosis among patients arriving in Europe from the Horn of Africa: A molecular epidemiological study. Lancet Infect. Dis. 2018, 18, 431–440. [Google Scholar] [CrossRef] [Green Version]
- Dicko, A.; Faye, O.; Fofana, Y.; Soumoutera, M.; Berthé, S.; Touré, S.; Traoré, B.; Guindo, B.; Tall, K.; Keita, A.; et al. Cutaneous tuberculosis in Bamako, Mali. Pan. Afr. Med. J. 2017, 27, 102. [Google Scholar] [CrossRef]
- Boudville, D.A.; Joshi, R.; Rijkers, G.T. Migration and tuberculosis in Europe. J. Clin. Tuberc. Other Mycobact. Dis. 2020, 18, 100143. [Google Scholar] [CrossRef]
- Olaru, I.D.; Albert, H.; Zallet, J.; Werner, U.E.; Ahmed, N.; Rieder, H.L.; Salfinger, M.; Kranzer, K. Impact of quality improvement in tuberculosis laboratories in low-and lower-middle-income countries: A systematic review. Int. J. Tuberc. Lung Dis. 2018, 22, 309–320. [Google Scholar] [CrossRef]
- Monedero-Recuero, I. Drug-Resistant Tuberculosis in Europe. What Are We Waiting For? Am. J. Respir. Crit. Care Med. 2018, 198, 302–304. [Google Scholar] [CrossRef]
- Bainomugisa, A.; Pande, S.; Donnan, E.; Simpson, G.; Foster, J.B.; Lavu, E.; Hiasihri, S.; McBryde, E.S.; Moke, R.; Vincent, S.; et al. Cross-border movement of highly drug-resistant mycobacterium tuberculosis from Papua New Guinea to Australia through Torres Strait Protected Zone, 2010–2015. Emerg. Infect. Dis. 2019, 25, 406. [Google Scholar] [CrossRef] [Green Version]
- Toms, C.; Stapledon, R.; Waring, J.; Douglas, P. Tuberculosis notifications in Australia, 2012 and 2013. Commun. Dis. Intell. Q. Rep. 2015, 39, E217–E235. [Google Scholar]
- Lönnroth, K.; Mor, Z.; Erkens, C.; Bruchfeld, J.; Nathavitharana, R.R.; van der Werf, M.J.; Lange, C. Tuberculosis in migrants in low-incidence countries: Epidemiology and intervention entry points. Int. J. Tuberc. Lung Dis. 2017, 21, 624–636. [Google Scholar] [CrossRef]
- Smith, S.E.; Pratt, R.; Trieu, L.; Barry, P.M.; Thai, D.T.; Ahuja, S.D.; Shah, S. Epidemiology of pediatric multidrug-resistant tuberculosis in the United States, 1993–2014. Clin. Infect. Dis 2017, 65, 437–1443. [Google Scholar] [CrossRef]
- Marais, B.J. Improving access to tuberculosis preventive therapy and treatment for children. Int. J. Infect. Dis. 2017, 56, 122–125. [Google Scholar] [CrossRef] [Green Version]
- Dodd, P.J.; Yuen, C.M.; Sismanidis, C.; Seddon, J.A.; Jenkins, H.E. The global burden of tuberculosis mortality in children: A mathematical modelling study. Lancet Glob. Health 2017, 5, e898–e906. [Google Scholar] [CrossRef] [Green Version]
- Graham, S.M.; Sismanidis, C.; Menzies, H.J.; Marais, B.J.; Detjen, A.K. Black RE. Importance of tuberculosis control to address child survival. Lancet 2014, 383, 1605–1607. [Google Scholar] [CrossRef] [Green Version]
- Tacconelli, E.; Pezzani, M.D. Public health burden of antimicrobial resistance in Europe. Lancet Infect. Dis. 2019, 19, 4–6. [Google Scholar] [CrossRef] [Green Version]
- Baker, S.J.; Payne, D.J.; Rappuoli, R.; De Gregorio, E. Technologies to address antimicrobial resistance. Proc. Natl. Acad. Sci. USA 2018, 115, 12887–12895. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization (WHO). The End TB Strategy. 2016. Available online: http://www.who.int/tb/post2015_TBstrategy.pdf?ua=1 (accessed on 18 March 2022).
- World Health Organization (WHO). UN General Assembly High-Level Meeting on the Fight Against Tuberculosis. 2018. Available online: https://www.who.int/news-room/events/un-general-assembly-high-level-meeting-on-ending-tb (accessed on 10 April 2021).
- Stop TB Partnership. 2019. Available online: http://www.stoptb.org/global/advocacy/unhlm_whatis.asp (accessed on 20 May 2021).
- General Assembly of the United Nations. United Nations High-Level Meeting on the Fight to End Tuberculosis 26 September 2018, UNHQ, New York. 2018. Available online: https://www.un.org/pga/73/event/fight-to-end-tuberculosis/ (accessed on 20 March 2022).
- United Nations: Meetings Coverage and Press Releases. World Leaders Reaffirm Commitment to End Tuberculosis by 2030, as General Assembly Adopts Declaration Outlining Actions for Increased Financing, Treatment Access. 2018. Available online: https://www.un.org/press/en/2018/ga12067.doc.htm (accessed on 20 May 2021).
- World Health Organization (WHO). Global Tuberculosis Report 2021; World Health Organization (WHO): Geneva, Switzerland, 2021; ISBN 9789240037021. (Electronic Version). [Google Scholar]
- Lange, C.; Chesov, D.; Heyckendorf, J. Clofazimine for the treatment of multidrug-resistant tuberculosis. Clin. Microbiol. Infect. 2019, 25, 128–130. [Google Scholar] [CrossRef] [Green Version]
- van Zyl, L.; Viljoen, J.M.; Haynes, R.K.; Aucamp, M.; Ngwane, A.H.; du Plessis, J. Topical delivery of artemisone, clofazimine and decoquinate encapsulated in vesicles and their in vitro efficacy against Mycobacterium tuberculosis. AAPS Pharm. Sci. Tech. 2019, 20, 33–44. [Google Scholar] [CrossRef]
- Osuchukwu, O.; Nuῆez, M.; Packard, S.; Ehiri, J.; Rosales, C.; Hawkins, E.; Gerardo Avilés, J.G.; Gonzalez-Salazar, F.; Oren, E. Latent Tuberculosis Infection Screening Acceptability among Migrant Farmworkers. Int. Migr. 2017, 55, 62–74. [Google Scholar] [CrossRef]
- Cardona, P.J. Pathogenesis of tuberculosis and other mycobacteriosis. Enferm. Infecc. Microbiol. Clin. 2018, 36, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Centis, R.; D’Ambrosio, L.; Zumla, A.; Migliori, G.B. Shifting from tuberculosis control to elimination: Where are we? What are the variables and limitations? Is it achievable? Int. J. Infect. Dis. 2017, 56, 30–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- dos Santos, J.B.D.; Figueiredo, A.R.; Ferraz, C.E.; Oliveira, M.H.D.; Silva, P.G.D.; Medeiros, V.L.S.D. Cutaneous tuberculosis: Epidemiologic, etiopathogenic and clinical aspects-part I. An. Bras. Dermatol. 2014, 89, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Churchyard, G. This Kills More South Africans Than Any Other Disease. There’s a New Way to Stop it. BHEKISISA Centre for Health Journalism. 2019. Available online: https://bhekisisa.org/article/2019-03-28-00-3hp-latent-tb-treatment-short-course-guidelines-regimen-isoniazid-rifapentine (accessed on 19 February 2022).
- van Zyl, L.; du Plessis, J.; Viljoen, J. Cutaneous tuberculosis overview and current treatment regimens. Tuberculosis 2015, 95, 629–638. [Google Scholar] [CrossRef]
- Chen, S.T.; Cahalane, A.M.; Ryan, E.T.; Foreman, R.K. Case 2-2019: A 36-year-old man with rash, abdominal pain, and lymphadenopathy. N. Engl. J. Med. 2019, 380, 275–283. [Google Scholar] [CrossRef] [Green Version]
- Mohanty, N.; Nayak, B.B. Cutaneous tuberculosis, tuberculosis verrucosa cutis. Med. J. Dr. D. Y. Patil. Univ. 2014, 7, 53–55. [Google Scholar] [CrossRef]
- World Health Organization (WHO). Global Tuberculosis Report 2018; World Health Organization (WHO): Geneva, Switzerland, 2018; ISBN 9789241565646. [Google Scholar]
- Yao, L.; LiangLiang, C.; JinYue, L.; WanMei, S.; Lili, S.; YiFan, L.; HuaiChen, L. Ambient air pollution exposures and risk of drug-resistant tuberculosis. Environ. Int. 2019, 124, 161–169. [Google Scholar] [CrossRef]
- Venkatesh, U.; Srivastava, D.K.; Srivastava, A.K.; Tiwari, H.C. Epidemiological profile of multidrug-resistant tuberculosis patients in Gorakhpur Division, Uttar Pradesh, India. J. Fam. Med. Prim. Care 2018, 7, 589–595. [Google Scholar] [CrossRef]
- Velayati, A.A.; Farnia, P.; Hoffner, S. Drug-resistant Mycobacterium tuberculosis: Epidemiology and role of morphological alterations. J. Glob. Antimicrob. Resist. 2018, 12, 192–196. [Google Scholar] [CrossRef]
- McBryde, E.S.; Meehan, M.T.; Doan, T.N.; Ragonnet, R.; Marais, B.J.; Guernier, V.; Trauer, J.M. The risk of global epidemic replacement with drug-resistant Mycobacterium tuberculosis strains. Int. J. Infect. Dis. 2017, 56, 14–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aaron, L.; Saadoun, D.; Calatroni, I.; Launay, O.; Memain, N.; Vincent, V.; Marchal, G.; Dupont, B.; Bouchaud, O.; Valeyre, D.; et al. Tuberculosis in HIV-infected patients: A comprehensive review. Clin. Microbiol. Infect. 2004, 10, 388–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pourakbari, B.; Sadeghi, R.H.; Mahmoudi, S.; Parvaneh, N.; Valian, S.K.; Mamishi, S. Evaluation of interleukin-12 receptor β1 and interferon gamma receptor 1 deficiency in patients with disseminated BCG infection. Allergol. Immunopathol. 2019, 47, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Caminero, J.A.; Cayla, J.A.; García-García, J.M.; García-Pérez, F.J.; Palacios, J.J.; Ruiz-Manzano, J. Diagnosis and treatment of drug-resistant tuberculosis. Arch. Bronconeumol. 2017, 53, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Tuberculosis (TB). Division of Tuberculosis Elimination. Centers for Disease Control and Prevention. CDC Twenty Four Seven. Saving Lives, Protecting People. 2016. Available online: https://www.cdc.gov/tb/publications/factsheets/drtb/mdrtb.htm (accessed on 15 February 2022).
- Alexander, P.E.; De, P. The emergence of extensively drug-resistant tuberculosis (TB): TB/HIV coinfection, multidrug-resistant TB and the resulting public health threat from extensively drug-resistant TB, globally and in Canada. Can. J. Infect. Dis. Med. Microbiol. 2007, 18, 289–291. [Google Scholar] [CrossRef]
- Sharma, S.K.; Turaga, K.K.; Balamurugan, A.; Saha, P.K.; Pandey, R.M.; Jain, N.K.; Katoch, V.M.; Mehrab, N.K. Clinical and genetic risk factors for the development of multi-drug resistant tuberculosis in non-HIV infected patients at a tertiary care center in India: A case-control study. Infect. Genet. Evol. 2003, 3, 183–188. [Google Scholar] [CrossRef]
- Munsiff, S.S.; Joseph, S.; Ebrahimzadeh, A.; Frieden, T.R. Rifampin monoresistant tuberculosis in New York City, 1993–1994. Clin. Infect. Dis. 1997, 25, 1465–1467. [Google Scholar] [CrossRef] [Green Version]
- Bhat, Z.S.; Rather, M.A.; Maqbool, M.; Ahmad, Z. Drug targets exploited in Mycobacterium tuberculosis: Pitfalls and promis-es on the horizon. Biomed Pharm. 2018, 103, 1733–1747. [Google Scholar] [CrossRef]
- Tiberi, S.; Muñoz-Torrico, M.; Duarte, R.; Dalcolmo, M.; D’Ambrosio, L.; Migliori, G.B. New drugs and perspectives for new anti-tuberculosis regimens. Pulmonology 2018, 24, 86–98. [Google Scholar] [CrossRef]
- Peña, M.J.M.; García, B.S.; Baquero-Artigao, F.; Pérez, D.M.; Pérez, R.P.; Echevarría, A.M.; Amador, J.T.R.; Durán, D.G.P.; Julian, A.N. Tuberculosis treatment for children: An update. An. de Pediatría 2018, 88, 52.e1–52.e12. [Google Scholar] [CrossRef]
- Maitre, T.; Petitjean, G.; Chauffour, A.; Bernard, C.; El Helali, N.; Jarlier, V.; Reibel, F.; Chavanet, P.; Aubry, A.; Veziris, N. Are moxifloxacin and levofloxacin equally effective to treat XDR tuberculosis? J. Antimicrob. Chemother. 2017, 72, 2326–2333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiberi, S.; Utjesanovic, N.; Galvin, J.; Centis, R.; D’Ambrosio, L.; van den Boom, M.; Zumla, A.; Migliori, G.B. Drug resistant TB–latest developments in epidemiology, diagnostics and management. IJID 2022, (in press). [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Jin, Y.; Vissa, V.; Zhang, L.; Liu, W.; Qin, L.; Wan, K.; Wu, X.; Wang, H.; Liu, W.; et al. Molecular characteristics of mycobacterium tuberculosis strains isolated from cutaneous tuberculosis patients in China. Acta Derm.-Venereol. 2017, 97, 472–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai, U.; Joshi, J.M. Extrapulmonary drug-resistant tu.uberculosis at a drug-resistant tuberculosis center, Mumbai: Our experience–Hope in the midst of despair! Lung India 2019, 36, 3–7. [Google Scholar] [CrossRef]
- Haitz, K.; Ly, A.; Smith, G. Idiopathic granulomatous mastitis. Cutis 2019, 103, 38–42. [Google Scholar]
- Mimesh, S.A.; Memish, Z.A. Cutaneous Tuberculosis. In Extrapulmonary Tuberculosis, 1st ed.; Sener, A., Erdem, H., Eds.; Springer: Cham, Switzerland, 2019; pp. 175–180. [Google Scholar]
- de los Santos Moreno, A.; Gómez, A.S.; Blasco, E.R.; Cuevas, M.C.; Saborido, D.G. Infecciones bacterianas crónicas (I). Tuberculosis. Med.-Programa de Form. Médica Contin. Acreditado 2018, 12, 3115–3123. [Google Scholar] [CrossRef]
- Peirse, M.; Houston, A. Extrapulmonary tuberculosis. Medicine 2017, 45, 747–752. [Google Scholar] [CrossRef]
- Dusthackeer, A.; Sekar, G.; Chidambaram, S.; Kumar, V.; Mehta, P.; Swaminathan, S. Drug resistance among extrapulmonary TB patients: Six years’ experience from a supranational reference laboratory. Indian J. Med. Res. 2015, 142, 568–574. [Google Scholar] [CrossRef]
- Lee, J.Y. Diagnosis and treatment of extrapulmonary tuberculosis. Tuberc. Respir. Dis. 2015, 78, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Mehta, P.K.; Raj, A.; Singh, N.; Khuller, G.K. Diagnosis of extrapulmonary tuberculosis by PCR. FEMS Immunol. Med. Microbiol. 2012, 66, 20–36. [Google Scholar] [CrossRef] [Green Version]
- Joshi, J.M. Tuberculosis chemotherapy in the 21st century: Back to the basics. Lung India 2011, 28, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Mohan, A.; Sharma, S.K. Medical schools and tuberculosis control: Bridging the discordance between what is preached and what is practiced. Indian J. Chest Dis. Allied Sci. 2004, 46, 5–7. [Google Scholar] [PubMed]
- Frankel, A.; Penrose, C.; Emer, J. Cutaneous tuberculosis: A practical case report and review for the dermatologist. J. Clin. Aesthet. Dermatol. 2009, 2, 19–27. [Google Scholar] [PubMed]
- de Mello, R.B.; do Vale, E.C.S.; Baeta, I.G.R. Scrofuloderma: A diagnostic challenge. An. Bras. Dermatol. 2019, 94, 102–104. [Google Scholar] [CrossRef] [Green Version]
- Rullán, J.; Seijo-Montes, R.E.; Vaillant, A.; Sánchez, N.P. Cutaneous manifestations of pulmonary disease. In Atlas of Dermatology in Internal Medicine, 14th ed.; Sánchez, N.P., Ed.; Springer: New York, NY, USA, 2012; pp. 17–30. [Google Scholar]
- Ghosh, S.; Aggarwal, K.; Jain, V.K.; Chaudhuri, S.; Ghosh, E. Tuberculosis verrucosa cutis presenting as diffuse plantar keratoderma: An unusual sight. Indian J. Dermatol. 2014, 59, 80–81. [Google Scholar] [CrossRef]
- Walter, N.; Daley, C.L. Tuberculosis and nontuberculous mycobacterial infections. In Clinical Respiratory Medicine, 4th ed.; Spiro, S.G., Silvestri, G.A., Agustí, A., Eds.; Elsevier Ltd.: Philadelphia, PA, USA, 2012; pp. 383–405. [Google Scholar]
- Jones-Lopez, E.C.; Ellner, J.J. Tuberculosis and atypical mycobacterial infections. In Tropical Infectious Diseases, 3rd ed.; Guerrant, R.L., Walker, D.H., Weller, P.F., Eds.; Elsevier Inc.: London, UK, 2011; pp. 228–247. [Google Scholar]
- Yates, V.M. Mycobacterial infections. In Rook’s Textbook of Dermatology, 8th ed.; Burns, T., Breathnach, S., Cox, N., Griffiths, C., Eds.; Blackwell Publishing Ltd.: West Sussex, UK, 2010; Volume 2, pp. 31.1–31.41. [Google Scholar]
- Gharabaghi, M.A. Cutaneous tuberculosis caused by isoniazid-resistant Mycobacterium tuberculosis. BMJ Case Rep. 2012, 2012, bcr2012006253. [Google Scholar] [CrossRef] [Green Version]
- Hernández Solis, A.; Herrera González, N.E.; Cazarez, F.; Mercadillo Pérez, P.; Olivera Diaz, H.O.; Escobar-Gutierrez, A.; Cortés Ortíz, I.; González González, H.; Reding-Bernal, A.; Sabido, R.C. Skin biopsy: A pillar in the identification of cutaneous Mycobacterium tuberculosis infection. J. Infect. Dev. Ctries. 2012, 6, 626–631. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Fan, Y.K.; Wang, P.; Chen, Q.Q.; Wang, G.; Xu, A.E.; Chen, L.Q.; Hu, R.; Chen, W.; Song, Z.Q.; et al. Cutaneous tuberculosis in China—A multicentre retrospective study of cases diagnosed between 1957 and 2013. J. Eur. Acad. Dermatol. Venereol. 2018, 32, 632–638. [Google Scholar] [CrossRef]
- Ho, C.K.; Ho, M.H.; Chong, L.Y. Cutaneous tuberculosis in Hong Kong: An update. Hong Kong Med. J. 2006, 12, 272–277. [Google Scholar]
- Gopinathan, R.; Pandit, D.; Joshi, J.; Jerajani, H.; Mathur, M. Clinical and morphological variants of cutaneous tuberculosis and its relation to Mycobacterium species. Indian J. Med. Microbl. 2001, 19, 193–196. [Google Scholar]
- Kaul, S.; Kaur, I.; Mehta, S.; Singal, A. Cutaneous tuberculosis. Part I: Pathogenesis, classification, and clinical features. JAAD 2022, (in press). [Google Scholar] [CrossRef] [PubMed]
- De Maio, F.; Trecarichi, E.M.; Visconti, E.; Sanguinetti, M.; Delogu, G.; Sali, M. Understanding cutaneous tuberculosis: Two clinical cases. JMM Case Rep. 2016, 3, e005070–e005076. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, H.C.; Abramo, C.; Munk, M.E. Immunological diagnosis of tuberculosis: Problems and strategies for success. J. Bras. Pneumol. 2007, 33, 323–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sloan, D.J.; Lewis, J.M. Management of multidrug-resistant TB: Novel treatments and their expansion to low resource settings. Trans. R Soc. Trop Med. Hyg. 2016, 110, 163–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Q.; Chen, W.; Hao, F. Cutaneous Tuberculosis: A Great Imitator. Clin. Dermatol. 2019, 37, 192–199. [Google Scholar] [CrossRef]
- Karas, L.; Lu, C.Y.; Agrawal, P.B.; Asgari, M.M. The impact of the Orphan Drug Act on Food and Drug Administration-approved therapies for rare skin diseases and skin-related cancers. J. Am. Acad. Dermatol. 2019, 81, 867–877. [Google Scholar] [CrossRef]
- van Deun, A.; Salim, H.; Kumar Das, A.P.; Bastian, I.; Portaels, F. Results of a standardised regimen for multidrug-resistant tuberculosis in Bangladesh. Int. J. Tuberc. Lung Dis. 2010, 8, 560–567. [Google Scholar]
- Günther, G.; Van Leth, F.; Alexandru, S.; Altet, N.; Avsar, K.; Bang, D.; Barbuta, R.; Bothamley, G.; Ciobanu, A.; Crudu, V.; et al. Clinical management of multidrug-resistant tuberculosis in 16 European countries. Am. J. Respir. Crit. Care Med. 2018, 198, 379–386. [Google Scholar] [CrossRef]
- Shah, M.A.; Shah, I. Increasing Prevalence of Pediatric Drug-resistant Tuberculosis in Mumbai, India, and Its Outcome. Pediatr. Infect. Dis. J. 2018, 37, 1261–1263. [Google Scholar] [CrossRef]
- Cox, H.; Hughes, J.; Black, J.; Nicol, M.P. Precision medicine for drug-resistant tuberculosis in high-burden countries: Is individualised treatment desirable and feasible? Lancet Infect. Dis. 2018, 18, e282–e287. [Google Scholar] [CrossRef]
- Barry, V.C.; Belton, J.G.; Conalty, M.L.; Den-Steny, J.M.; Edward, D.W.; O’Sullivan, J.F.; Twomey, D.; Winder, F. A new series of phenazines (rimino-compounds) with high antituberculosis activity. Nature 1957, 179, 1013–1015. [Google Scholar] [CrossRef] [PubMed]
- Drugbank. Clofazimine. 2017. Available online: https://www.drugbank.ca/drugs/DB00845 (accessed on 3 June 2021).
- Ammerman, N.C.; Swanson, R.V.; Bautista, E.M.; Almeida, D.V.; Saini, V.; Omansen, T.F.; Guo, H.; Chang, Y.S.; Li, S.-Y.; Tapley, A.; et al. Impact of clofazimine dosing on treatment-shortening of the first-line regimen in a mouse model of tuberculosis. Antimicrob. Agents Chemother. 2018, 62, e00636-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGuffin, S.A.; Pottinger, P.S.; Harnisch, J.P. Clofazimine in nontuberculous mycobacterial infections: A growing niche. Open Forum Infect. Dis. 2017, 4, ofx147. [Google Scholar] [CrossRef] [PubMed]
- Swanson, R.V.; Adamson, J.; Moodley, C.; Ngcobo, B.; Ammerman, N.C.; Dorasamy, A.; Moodley, S.; Mgaga, Z.; Tapley, A.; Bester, L.A.; et al. Pharmacokinetics and pharmacodynamics of clofazimine in a mouse model of tuberculosis. Antimicrob. Agents Chemother. 2015, 59, 3042–3051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cholo, M.C.; Steel, H.C.; Fourie, P.B.; Germishuizen, W.A.; Anderson, R. Clofazimine: Current status and future prospects. J. Antimicrob. Chemother. 2012, 67, 290–298. [Google Scholar] [CrossRef]
- Zhu, H.; Fu, L.; Wang, B.; Chen, X.; Zhao, J.; Huang, H.; Lu, Y. Activity of Clofazimine and TBI-166 against Mycobacterium tuberculosis in Different Administration Intervals in Mouse Tuberculosis Models. Antimicrob. Agents Chemother. 2021, 11, e02164-20. [Google Scholar] [CrossRef]
- Browne, S.G.; Hogerzeil, L.M. “B 663” in the treatment of leprosy. Preliminary report of a pilot trial. Lepr. Rev. 1962, 33, 6–10. [Google Scholar] [CrossRef]
- Attwood, M.M.; Rask-Andersen, M.; Schiöth, H.B. Orphan drugs and their impact on pharmaceutical development. Trends Pharmacol. Sci. 2018, 39, 525–535. [Google Scholar] [CrossRef]
- Brennan, P.J.; Young, D.B. Tuberculosis. In Handbook of Anti-Tuberculosis Agents; Elsevier: Amsterdam, The Netherlands, 2008; Volume 88, pp. 85–170. [Google Scholar]
- Cholo, M.C.; Mothiba, M.T.; Fourie, B.; Anderson, R. Mechanisms of action and therapeutic efficacies of the lipophilic antimycobacterial agents clofazimine and bedaquiline. J. Antimicrob. Chemother. 2017, 72, 338–353. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Li, T.; Qu, G.; Pang, Y.; Zhao, Y. In vitro synergistic activity of clofazimine and other antituberculous drugs against multidrug-resistant Mycobacterium tuberculosis isolates. Int. J. Antimicrob. Agents 2015, 45, 71–75. [Google Scholar] [CrossRef]
- Dooley, K.E.; Obuku, E.A.; Durakovic, N.; Belitsky, V.; Mitnick, C.; Nuermberger, E.L. Efficacy Subgroup, RESIST-TB. World Health Organization group 5 drugs for the treatment of drug-resistant tuberculosis: Unclear efficacy or untapped potential? J. Infect. Dis. 2013, 207, 1352–1358. [Google Scholar] [CrossRef] [PubMed]
- Mirnejad, R.; Asadi, A.; Khoshnood, S.; Mirzaei, H.; Heidary, M.; Fattorini, L.; Ghodousi, A.; Darban-Sarokhalil, D. Clofazimine: A useful antibiotic for drug-resistant tuberculosis. Biomed. Pharmacother. 2018, 105, 1353–1359. [Google Scholar] [CrossRef] [PubMed]
- Kuaban, C.; Noeske, J.; Rieder, H.L.; Ait-Khaled, N.; Abena Foe, J.L.; Trébucq, A. High effectiveness of a 12-month regimen for MDR-TB patients in Cameroon. Int. J. Tuberc. Lung Dis. 2015, 19, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Aung, K.J.M.; Van Deun, A.; Declercq, E.; Sarker, M.R.; Das, P.K.; Hossain, M.A.; Rieder, H.L. Successful ‘9-month Bangladesh regimen’ for multidrug-resistant tuberculosis among over 500 consecutive patients. Int. J. Tuberc. Lung Dis. 2014, 18, 1180–1187. [Google Scholar] [CrossRef]
- Piubello, A.; Harouna, S.H.; Souleymane, M.B.; Boukary, I.; Morou, S.; Daouda, M.; Hanki, Y.; Van Deun, A. High cure rate with standardized short-course multidrug-resistant tuberculosis treatment in Niger: No relapses. Int. J. Tuberc. Lung Dis. 2014, 18, 1188–1194. [Google Scholar] [CrossRef]
- Tang, S.; Yao, L.; Hao, X.; Liu, Y.; Zeng, L.; Liu, G.; Li, M.; Li, F.; Wu, M.; Zhu, Y.; et al. Clofazimine for the treatment of multidrug-resistant tuberculosis: Prospective, multicenter, randomized controlled study in China. Clin. Infect. Dis. 2015, 60, 1361–1367. [Google Scholar] [CrossRef] [Green Version]
- Tyagi, S.; Ammerman, N.C.; Li, S.Y.; Adamson, J.; Converse, P.J.; Swanson, R.V.; Almeida, D.V.; Grosset, J.H. Clofazimine shortens the duration of the first-line treatment regimen for experimental chemotherapy of tuberculosis. Proc. Natl. Acad. Sci. USA 2015, 112, 869–874. [Google Scholar] [CrossRef] [Green Version]
- Grosset, J.H.; Tyagi, S.; Almeida, D.V.; Converse, P.J.; Li, S.Y.; Ammerman, N.C.; Bishai, W.R.; Enarson, D.; Trébucq, A. Assessment of clofazimine activity in a second-line regimen for tuberculosis in mice. Am. J. Respir. Crit. Care Med. 2013, 188, 608–612. [Google Scholar] [CrossRef] [Green Version]
- Seung, K.J.; Hewison, C. Now is the time for shorter all-oral regimens for multidrug-resistant tuberculosis. Lancet Glob Health 2019, 7, e706. [Google Scholar] [CrossRef] [Green Version]
- Durusu, İ.Z.; Hüsnügil, H.H.; Ataş, H.; Biber, A.; Gerekçi, S.; Güleç, E.A.; Özen, C. Anti-cancer effect of clofazimine as a single agent and in combination with cisplatin on U266 multiple myeloma cell line. Leuk Res. 2017, 55, 33–40. [Google Scholar] [CrossRef]
- Food and Drug Administration. LAMPRENE® (clofazimine) Capsules, Full Prescribing Information; U.S. Food and Drug Administration; Reference ID: 3956651; Silver Spring, MD, USA, 2016. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/019500s013lbl.pdf (accessed on 5 January 2022).
- Novartis. Lamprene® Prescribing Information; NDA 19-500/S-010; Novartis: East Hanover, NJ, USA, 2002. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2003/19500slr010_lamprene_lbl.pdf (accessed on 5 January 2022).
- Chen, Y.; Yuan, Z.; Shen, X.; Wu, J.; Wu, Z.; Xu, B. Resistance to second-line antituberculosis drugs and delay in drug susceptibility testing among multidrug-resistant tuberculosis patients in Shanghai. BioMed. Res. Int. 2016, 2016, 2628913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalcolmo, M.; Gayoso, R.; Sotgiu, G.; D’Ambrosio, L.; Rocha, J.L.; Borga, L.; Fandinho, F.; Braga, J.U.; Galesi, V.M.N.; Barreira, D.; et al. Effectiveness and safety of clofazimine in multidrug-resistant tuberculosis: A nationwide report from Brazil. Eur. Respir. J. 2017, 49, 1602445. [Google Scholar] [CrossRef] [PubMed]
- Szeto, W.; Garcia-Buitrago, M.T.; Abbo, L.; Rosenblatt, J.D.; Moshiree, B.; Morris, M.I. Clofazimine enteropathy: A rare and under recognized complication of mycobacterial therapy. Open Forum. Infect. Dis. 2016, 2, ofw004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, G.S.; Keswani, R.K.; Sud, S.; Rzeczycki, P.M.; Murashov, M.D.; Koehn, T.A.; Standiford, T.J.; Stringer, K.A.; Rosania, G.R. Clofazimine biocrystal accumulation in macrophages upregulates interleukin 1 receptor antagonist production to induce a systemic anti-inflammatory state. Antimicrobl. Agents Chemothe. 2016, 60, 3470–3479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopal, M.; Padayatchi, N.; Metcalfe, J.Z.; O’Donnell, M.R. Systematic review of clofazimine for the treatment of drug-resistant tuberculosis. Int. J. Tuberc. Lung Dis. 2013, 17, 1001–1007. [Google Scholar] [CrossRef] [Green Version]
- Barot, R.K.; Viswanath, V.; Pattiwar, M.S.; Torsekar, R.G. Crystalline deposition in the cornea and conjunctiva secondary to long-term clofazimine therapy in a leprosy patient. Indian J. Ophthalmol. 2011, 59, 328–329. [Google Scholar] [CrossRef]
- Kaur, I.; Ram, J.; Kumar, B.; Kaur, S.; Sharma, V.K. Effect of clofazimine on eye in multibacillary leprosy. Indian J. Lepr. 1990, 62, 87–90. [Google Scholar]
- Zaccagnino, A.; Managò, A.; Leanza, L.; Gontarewitz, A.; Linder, B.; Azzolini, M.; Biasutto, L.; Zoratti, M.; Peruzzo, R.; Legler, K.; et al. Tumor-reducing effect of the clinically used drug clofazimine in a SCID mouse model of pancreatic ductal adenocarcinoma. Oncotarget 2017, 8, 38276–38293. [Google Scholar] [CrossRef]
- Dixit, V.B.; Chaudhary, S.D.; Jain, V.K. Clofazimine induced nail changes. Indian J. Lepr. 1989, 61, 476–478. [Google Scholar]
- World Health Organization. WHO Model Prescribing Information: Drugs Used in Leprosy; World Health Organization: Geneva, Switzerland, 1998; Available online: http://apps.who.int/medicinedocs/pdf/h2988e/h2988e.pdf (accessed on 5 January 2022).
- National Hansen’s Disease (Leprosy) Program. Recommended Treatment Regimens; US Department of Health and Human Services: Washington, DC, USA, 2018. Available online: https://www.hrsa.gov/hansensdisease/diagnosis/recommendedtreatment.html (accessed on 5 January 2022).
- Hwang, T.J.; Dotsenko, S.; Jafarov, A.; Weyer, K.; Falzon, D.; Lunte, K.; Nunn, P.; Jaramillo, E.; Keshavjee, S.; Wares, D.F. Safety and availability of clofazimine in the treatment of multidrug and extensively drug-resistant tuberculosis: Analysis of published guidance and meta-analysis of cohort studies. BMJ Open 2014, 4, e004143. [Google Scholar] [CrossRef]
- Ozturk, Z.; Tatliparmak, A. Leprosy treatment during pregnancy and breastfeeding: A case report and brief review of literature. Dermatol. Ther. 2017, 30, e12414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drobac, P.C.; del Castillo, H.; Sweetland, A.; Anca, G.; Joseph, J.K.; Furin, J.; Shin, S. Treatment of multidrug-resistant tuberculosis during pregnancy: Long-term follow-up of 6 children with intrauterine exposure to second-line agents. Clin. Infect. Dis. 2005, 40, 1689–1692. [Google Scholar] [CrossRef] [PubMed]
- Levis, W.; Rendini, T. Clofazimine Mechanisms of Action in Mycobacteria, HIV, and Cancer. J. Infect. Dis. 2017, 215, 1488. [Google Scholar] [CrossRef] [PubMed]
- Machado, T.C.; Gelain, A.B.; Rosa, J.; Cardoso, S.G.; Caon, T. Cocrystallization as a novel approach to enhance the transdermal administration of meloxicam. Eur. J. Pharm. Sci. 2018, 123, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Singh Malik, D.; Mital, N.; Kaur, G. Topical drug delivery systems: A patent review. Expert. Opin. Ther. Pat. 2016, 26, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Bhowmik, D.; Gopinath, H.; Kumar, B.P.; Duraivel, S.; Kumar, K.S. Recent advances in novel topical drug delivery system. Pharm. Innov. 2012, 1, 12–31. [Google Scholar]
- Gratieri, T.; Alberti, I.; Lapteva, M.; Kalia, Y.N. Next generation intra-and transdermal therapeutic systems: Using non-and minimally-invasive technologies to increase drug delivery into and across the skin. Eur. J. Pharm. Sci. 2013, 50, 609–622. [Google Scholar] [CrossRef]
- Prausnitz, M.R.; Mitragotri, S.; Langer, R. Current status and future potential of transdermal drug delivery. Nat. Rev. Drug Discov. 2004, 3, 115–124. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, N.; Song, H.; Li, H.; Wen, J.; Tan, Y.; Zheng, W. Design, characterization and comparison of transdermal delivery of colchicine via borneol-chemically-modified and borneol-physically-modified ethosome. Drug Deliv. 2019, 26, 70–77. [Google Scholar] [CrossRef] [Green Version]
- Nan, L.; Liu, C.; Li, Q.; Wan, X.; Guo, J.; Quan, P.; Fang, L. Investigation of the enhancement effect of the natural transdermal permeation enhancers from Ledum palustre L. var. angustum N. Busch: Mechanistic insight based on interaction among drug, enhancers and skin. Eur. J. Pharm Sci. 2018, 124, 105–113. [Google Scholar] [CrossRef]
- Ita, K. Transdermal delivery of drugs with microneedles—potential and challenges. Pharmaceutics 2015, 7, 90–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, M.S.; Mohammed, Y.; Pastore, M.N.; Namjoshi, S.; Yousef, S.; Alinaghi, A.; Haridass, I.N.; Abd, E.; Leite-Silva, V.R.; Benson, H.A.E.; et al. Topical and cutaneous delivery using nanosystems. J. Control. Release 2017, 247, 86–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slominski, A.T.; Manna, P.R.; Tuckey, R.C. On the role of skin in the regulation of local and systemic steroidogenic activities. Steroids 2015, 103, 72–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellefroid, C.; Lechanteur, A.; Evrard, B.; Piel, G. Lipid gene nanocarriers for the treatment of skin diseases: Current state-of-the-art. Eur. J. Pharm. Biopharm. 2019, 137, 95–111. [Google Scholar] [CrossRef] [PubMed]
- Marwah, H.; Garg, T.; Goyal, A.K.; Rath, G. Permeation enhancer strategies in transdermal drug delivery. Drug Deliv. 2016, 23, 564–578. [Google Scholar] [CrossRef]
- Burger, C.; Gerber, M.; Du Preez, J.L.; Du Plessis, J. Optimised transdermal delivery of pravastatin. Int. J. Pharm. 2015, 496, 518–525. [Google Scholar] [CrossRef]
- Das, A.; Ahmed, A.B. Natural permeation enhancer for transdermal drug delivery system and permeation evaluation: A review. Asian J. Pharm. Cli. Res. 2017, 10, 5–9. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Xu, W.; Liang, Y.; Wang, H. The application of skin metabolomics in the context of transdermal drug delivery. Pharmacol. Rep. 2017, 69, 252–259. [Google Scholar] [CrossRef]
- Sun, R.; Celli, A.; Crumrine, D.; Hupe, M.; Adame, L.C.; Pennypacker, S.D.; Park, K.; Uchida, Y.; Feingold, K.R.; Elias, P.M.; et al. Lowered humidity produces human epidermal equivalents with enhanced barrier properties. Tissue Eng. Part. C Methods 2014, 21, 15–22. [Google Scholar] [CrossRef] [Green Version]
- Ramani, R.; Pandya, S.; Motka, U.; Lakhani, D.; Ramaney, D.; Sheth, D. Drug penetration enhancement in transdermal drug delivery system by chemical penetration enhancers. Int. J. Preclin. Pharm. Res. 2013, 4, 10–17. [Google Scholar]
- Naik, A.; Kalia, Y.N.; Guy, R.H. Transdermal drug delivery: Overcoming the skin’s barrier function. Pharm. Sci. Technolo. Today 2000, 3, 318–326. [Google Scholar] [CrossRef]
- British Pharmacopoeia (BP). Clofazomine. 2018. Available online: https://www-pharmacopoeia-com.nwulib.nwu.ac.za/bp-2018/monographs/clofazimine.html?date=2018-01-01&page=21 (accessed on 22 March 2022).
- Alkilani, A.; McCrudden, M.T.; Donnelly, R. Transdermal drug delivery: Innovative pharmaceutical developments based on disruption of the barrier properties of the stratum corneum. Pharmaceutic 2015, 7, 438–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, R.; Fan, W.; Yu, Q.; Dong, X.; Qi, J.; Zhu, Q.; Zhao, W.; Wu, W.; Chen, Z.; Li, Y.; et al. Size-dependent penetration of nanoemulsions into epidermis and hair follicles: Implications for transdermal delivery and immunization. Oncotarget 2017, 8, 38214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gidal, B.E.; Clark, A.M.; Anders, B.; Gilliam, F. The application of half-life in clinical decision making: Comparison of the pharmacokinetics of extended-release topiramate (USL255) and immediate-release topiramate. Epilepsy Res. 2017, 129, 26–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kathe, K.; Kathpalia, H. Film forming systems for topical and transdermal drug delivery. Asian J. Pharm. Sci. 2017, 12, 487–497. [Google Scholar] [CrossRef]
- Moser, K.; Kriwet, K.; Froehlich, C.; Kalia, Y.N.; Guy, R.H. Supersaturation: Enhancement of skin penetration and permeation of a lipophilic drug. Pharm. Res. 2001, 18, 1006–1011. [Google Scholar] [CrossRef]
- Mao, F.; Kong, Q.; Ni, W.; Xu, X.; Ling, D.; Lu, Z.; Li, J. Melting point distribution analysis of globally approved and discontinued drugs: A research for improving the chance of success of drug design and discovery. Chem. Open 2016, 5, 357–368. [Google Scholar] [CrossRef] [Green Version]
- Das, M.K.; Bhattacharya, A.; Ghosal, S.K. Effect of different terpene-containing essential oils on percutaneous absorption of trazodone hydrochloride through mouse epidermis. Drug Deliv. 2006, 13, 425–431. [Google Scholar] [CrossRef]
- Sangana, R.; Gu, H.; Chun, D.Y.; Einolf, H.J. Evaluation of clinical drug interaction potential of clofazimine using static and dynamic modelling approaches. Drug Metab. Dispos. 2018, 46, 26–32. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Feng, J.; McManus, S.A.; Lu, H.D.; Ristroph, K.D.; Cho, E.J.; Dobrijevic, E.L.; Chan, H.K.; Prud’homme, R.K. Design and solidification of fast-releasing clofazimine nanoparticles for treatment of cryptosporidiosis. Mol. Pharm. 2017, 14, 3480–3488. [Google Scholar] [CrossRef] [Green Version]
- N’Da, D. Prodrug strategies for enhancing the percutaneous absorption of drugs. Molecules 2014, 19, 20780–20807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid-Wendtner, M.H.; Korting, H.C. The pH of the skin surface and its impact on the barrier function. Skin Pharmacol. Physiol. 2006, 19, 296–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, A.; Jacob, S.; Al-Dhubiab, B.; Attimarad, M.; Harsha, S. Basic considerations in the dermatokinetics of topical formulations. Braz. J. Pharm. Sci. 2013, 49, 423–434. [Google Scholar] [CrossRef] [Green Version]
- Dhote, V.; Bhatnagar, P.; Mishra, P.K.; Mahajan, S.C.; Mishra, D.K. Iontophoresis: A Potential Emergence of a Transdermal Drug Delivery System. Sci. Pharm. 2012, 8, 1–28. [Google Scholar] [CrossRef] [Green Version]
- Chaulagain, B.; Jain, A.; Tiwari, A.; Verma, A.; Jain, S.K. Passive delivery of protein drugs through transdermal route. Artif. Cells Nanomed. Biotechnol. 2018, 46, 472–487. [Google Scholar] [CrossRef] [Green Version]
- Pranitha, A.; Lakshmi, P.K. Effect of pH on weakly acidic and basic model drugs and determination of their ex vivo transdermal permeation routes. Braz. J. Pharm. Sci. 2018, 54, e00070. [Google Scholar] [CrossRef]
- Viljoen, J.M.; Cowley, A.; Du Preez, J.; Gerber, M.; du Plessis, J. Penetration enhancing effects of selected natural oils utilized in topical dosage forms. Drug Dev. Ind. Pharm. 2015, 41, 2045–2054. [Google Scholar] [CrossRef]
- Čižinauskas, V.; Elie, N.; Brunelle, A.; Briedis, V. Skin Penetration Enhancement by Natural Oils for Dihydroquercetin Delivery. Molecules 2017, 22, 1536. [Google Scholar] [CrossRef] [Green Version]
- Zaid, A.N.; Jaradat, N.A.; Eid, A.M.; Al Zabadi, H.; Alkaiyat, A.; Darwish, S.A. Ethnopharmacological survey of home remedies used for treatment of hair and scalp and their methods of preparation in the West Bank-Palestine. BMC Complement. Altern Med. 2017, 17, 355–370. [Google Scholar] [CrossRef] [Green Version]
- Standard for Olive Oils and Olive Pomace Oils CODEX STAN 33–1981. In International Food Standards: World Health Organization Food and Agriculture Organization of the United Nations; WHO: Geneva, Switzerland, 2015; Available online: https://www.fao.org/fao-who-codexalimentarius/sh-proxy/en/?lnk=1&url=https%253A%252F%252Fworkspace.fao.org%252Fsites%252Fcodex%252FStandards%252FCXS%2B33-1981%252FCXS_033e.pdf (accessed on 2 February 2022).
- Lin, T.K.; Zhong, L.; Santiago, J. Anti-inflammatory and skin barrier repair effects of topical application of some plant oils. Int. J. Mol. Sci. 2018, 19, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaughn, A.R.; Clark, A.K.; Sivamani, R.K.; Shi, V.Y. Natural oils for skin-barrier repair: Ancient compounds now backed by modern science. Am. J. Clin. Dermatol. 2018, 19, 103–117. [Google Scholar] [CrossRef]
- Cooke, A.; Cork, M.; Danby, S. A national survey of UK maternity and neonatal units regarding the use of oil for baby skincare. Br. J. Midwifery 2011, 19, 354–362. [Google Scholar] [CrossRef]
- Kulkarni, A.; Kaushik, J.S.; Gupta, P.; Sharma, H.; Agrawal, R.K. Massage and touch therapy in neonates: The current evidence. Indian Pediatr. 2010, 47, 771–776. [Google Scholar] [CrossRef]
- Loden, M. Role of topical emollients and moisturizers in the treatment of dry skin barrier disorders. Am. J. Clin. Dermatol. 2003, 4, 771–788. [Google Scholar] [CrossRef]
- Fox, L.T.; Gerber, M.; Plessis, J.D.; Hamman, J.H. Transdermal drug delivery enhancement by compounds of natural origin. Molecules 2011, 16, 10507–10540. [Google Scholar] [CrossRef]
- Yang, J.Y.; Li, J.; Wang, M.; Zou, X.G.; Peng, B.; Yin, Y.L.; Deng, Z.Y. A novel aqueous extraction for camellia oil by emulsified oil: A frozen/thawed method. Eur. J. Lipid Sci. Tech. 2019, 121, 1800431. [Google Scholar] [CrossRef]
- Sparr, E.; Millecamps, D.; Isoir, M.; Burnier, V.; Larsson, Å.; Cabane, B. Controlling the hydration of the skin though the application of occluding barrier creams. J. Royal Soc. Interface 2013, 10, 20120788. [Google Scholar] [CrossRef]
- Carranco, N.; Farrés-Cebriá, M.; Saurina, J.; Núñez, O. Authentication and quantitation of fraud in extra virgin olive oils based on HPLC-UV fingerprinting and multivariate calibration. Foods 2018, 7, 44. [Google Scholar] [CrossRef] [Green Version]
- Roni, M.A.; Jalil, R.U. Comparative study of ibuprofen solubility in synthetic and natural lipid vehicles. Dhaka Univ. J. Pharml. Sci. 2011, 10, 65–66. [Google Scholar] [CrossRef]
- Elmowafy, M. Skin penetration/permeation success determinants of nanocarriers: Pursuit of a perfect formulation. Colloids Surf. B Biointerfaces 2021, 203, 111748. [Google Scholar] [CrossRef]
- Rahman, M.A.; Hussain, A.; Hussain, M.S.; Mirza, M.A.; Iqbal, Z. Role of excipients in successful development of self-emulsifying/microemulsifying drug delivery system (SEDDS/SMEDDS). Drug Dev. Ind. Pharm. 2013, 39, 1–19. [Google Scholar] [CrossRef]
- Bernkop-Schnürch, A.; Jalil, A. Do drug release studies from SEDDS make any sense? J. Control. Releas 2018, 271, 55–59. [Google Scholar] [CrossRef]
- Morgen, M.; Saxena, A.; Chen, X.-Q.; Miller, W.; Nkansah, R.; Goodwin, A.; Cape, J.; Haskell, R.; Su, C.; Gudmundsson, O.; et al. Lipophilic salts of poorly soluble compounds to enable high-dose lipidic SEDDS formulations in drug discovery. Eur. J. Pharm. Biopharm. 2017, 117, 212–223. [Google Scholar] [CrossRef]
- Balganesh, T.S.; Alzari, P.M.; Cole, S.T. Rising standards for tuberculosis drug development. Trends Pharmacol. Sci. 2008, 29, 576–581. [Google Scholar] [CrossRef]
- Chatterjee, B.; Hamed Almurisi, S.; Ahmed Mahdi Dukhan, A.; Mandal, U.K.; Sengupta, P. Controversies with self-emulsifying drug delivery system from pharmacokinetic point of view. Drug Deliv. 2016, 23, 3639–3652. [Google Scholar] [CrossRef] [Green Version]
- Gonçalves, A.; Nikmaram, N.; Roohinejad, S.; Estevinho, B.N.; Rocha, F.; Greiner, R.; Mc Clements, D.J. Production, properties, and applications of solid self-emulsifying delivery systems (S-SEDS) in the food and pharmaceutical industries. Colloids Surf. A Physicochem. Eng. Asp. 2018, 538, 108–126. [Google Scholar] [CrossRef]
- Watanabe, A.; Murayama, S.; Karasawa, K.; Yamamoto, E.; Morikawa, S.; Takita, R.; Murata, S.; Kato, M. A simple and easy method of monitoring doxorubicin release from a liposomal drug formulation in the serum using fluorescence spectroscopy. Chem. Pharm. Bull. 2019, 67, 367–371. [Google Scholar] [CrossRef] [Green Version]
- Gupta, P.K.; Bhandari, N.; Shah, H.N.; Khanchandani, V.; Keerthana, R.; Nagarajan, V.; Hiremath, L. An update on nanoemulsions using nanosized liquid in liquid colloidal systems. In Nanoemulsions—Properties, Fabrications and Applications; Online First; IntechOpen: London, UK, 2019. [Google Scholar] [CrossRef] [Green Version]
- Chime, S.A.; Kenechukwu, F.C.; Attama, A.A. Nanoemulsions—Advances in formulation, characterization and applications in drug delivery. In Application of Nanotechnology in Drug Delivery; IntechOpen: London, UK, 2014. [Google Scholar]
- Williams, A.C. Topical and transdermal drug delivery. In Aulton’s Pharmaceutics: The Design and Manufacture of Medicines, 5th ed.; Aulton, M.E., Taylor, K.M.G., Eds.; Churchill Livingstone: Edinburgh, Scotland, 2018; pp. 675–697. [Google Scholar]
- Barry, B.W. Novel mechanisms and devices to enable successful transdermal drug delivery. Eur. J. Pharm. Sci. 2001, 14, 101–114. [Google Scholar] [CrossRef]
- El Maghraby, G.M.M.; Williams, A.C.; Barry, B.W. Oestradiol skin delivery from ultradeformable liposomes: Refinement of surfactant concentration. Int. J. Pharm. 2000, 196, 63–74. [Google Scholar] [CrossRef]
- Patil, J.M.; Yadava, S.K.; Mokale, V.J.; Naik, J. Development of surfactant free nanoparticles by a single emulsion high pressure homogenization technique and effect of formulation parameters on the drug entrapment and release. Int. J. Pharm. 2013, 3, 843–852. [Google Scholar]
- Rohrer, J.; Zupančič, O.; Hetenyi, G.; Kurpiers, M.; Bernkop-Schnürch, A. Design and evaluation of SEDDS exhibiting high emulsifying properties. J. Drug Deliv. Sci. Technol. 2018, 44, 366–372. [Google Scholar] [CrossRef]
- Köllner, S.; Nardin, I.; Markt, R.; Griesser, J.; Prüfert, F.; Bernkop-Schnürch, A. Self-emulsifying drug delivery systems: Design of a novel vaginal delivery system for curcumin. Eur. J. Pharm. Biopharm. 2017, 115, 268–275. [Google Scholar] [CrossRef]
- Pattewar, S.V.; Kasture, S.B.; Pande, V.V.; Sharma, S.K. A new self microemulsifying mouth dissolving film. Indian J. Pharm. Educ. Res. 2016, 50, S191–S199. [Google Scholar] [CrossRef] [Green Version]
- ElKasabgy, N.A. Ocular supersaturated self-nanoemulsifying drug delivery systems (S-SNEDDS) to enhance econazole nitrate bioavailability. Int. J. Pharm. 2014, 460, 33–44. [Google Scholar] [CrossRef]
- Xiao, L.; Yi, T.; Liu, Y. A new self-microemulsifying mouth dissolving film to improve the oral bioavailability of poorly watersoluble drugs. Drug Dev. Ind. Pharm. 2013, 39, 1284–1290. [Google Scholar] [CrossRef]
- Khan, M.; Nadhman, A.; Sehgal, S.A.; Siraj, S.; Yasinzai, M.M. Formulation and characterization of a self-emulsifying drug delivery system (sedds) of curcumin for the topical application in cutaneous and mucocutaneous leishmaniasis. Curr. Top. Med. Chem. 2018, 18, 1603–1609. [Google Scholar] [CrossRef]
- Pratiwi, L.; Fudholi, A.; Martien, R.; Pramono, S. Self-nanoemulsifying drug delivery system (Snedds) for topical delivery of mangosteen peels (Garcinia mangostana L.): Formulation Design and In vitro Studies. J. Young Pharm. 2017, 9, 341–346. [Google Scholar] [CrossRef] [Green Version]
- Vasconcelos, T.; Marques, S.; Sarmento, B. Measuring the emulsification dynamics and stability of self-emulsifying drug delivery systems. Eur. J. Pharm. Biopharm. 2018, 123, 1–8. [Google Scholar] [CrossRef]
- Rani, S.; Rana, R.; Saraogi, G.K.; Kumar, V.; Gupta, U. Self-emulsifying oral lipid drug delivery systems: Advances and challenges. AAPS Pharm. Sci. Tech. 2019, 20, 129. [Google Scholar] [CrossRef]
- Nikolakakis, I.; Partheniadis, I. Self-emulsifying granules and pellets: Composition and formation mechanisms for instant or controlled release. Pharmaceutics 2017, 9, 50. [Google Scholar] [CrossRef] [Green Version]
- Gurram, A.K.; Deshpande, P.B.; Kar, S.S.; Nayak, U.Y.; Udupa, N.; Reddy, M.S. Role of components in the formation of self-microemulsifying drug delivery systems. Indian J. Pharm. Sci. 2015, 77, 249–257. [Google Scholar] [CrossRef] [Green Version]
- Smith, K.L. Penetrant characteristics influencing skin absorption. In Methods for Skin Absorption, 1st ed.; Kemppainen, B.W., Reifenrath, W.G., Eds.; CRC Press: Boca Raton, FL, USA, 1990; pp. 23–34. [Google Scholar]
- Rajeshwar, V.; Shrivastava, B. Self-emulsifying drug delivery system (SEDDS): A conventional alternative and approach to improve oral bioavailability. Int. J. Pharm. Sci. Res. 2018, 9, 3114–3127. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, I.; Zhang, M.; Pang, Y.; Li, Z.; Zhao, A.; Feng, J. Self-emulsifying drug delivery system and the applications in herbal drugs. Drug Deliv. 2015, 22, 475–486. [Google Scholar] [CrossRef] [Green Version]
- Anton, N.; Vandamme, T.F. Nano-emulsions and micro-emulsions: Clarifications of the critical differences. Pharm. Res. 2011, 28, 978–985. [Google Scholar] [CrossRef]
- Thakare, P.; Mogal, V.; Borase, P.; Dusane, J.; Kshirsagar, S. A review on self-emulsified drug delivery system. Pharm. Biol. Eval. 2016, 3, 140–153. [Google Scholar]
- Senapati, P.C.; Sahoo, S.K.; Sahu, A.N. Mixed surfactant based (SNEDDS) self-nanoemulsifying drug delivery system presenting efavirenz for enhancement of oral bioavailability. Biomed. Pharmacother. 2016, 80, 42–51. [Google Scholar] [CrossRef]
- Hu, C.; Wang, Q.; Ma, C.; Xia, Q. Non-aqueous self-double-emulsifying drug delivery system: A new approach to enhance resveratrol solubility for effective transdermal delivery. Colloids Surf. A Physicochem. 2016, 489, 360–369. [Google Scholar] [CrossRef]
- van Staden, D.; du Plessis, J.; Viljoen, J. Development of topical/transdermal self-emulsifying drug delivery systems, not as simple as expected. Sci. Pharm. 2020, 88, 17. [Google Scholar] [CrossRef] [Green Version]
- van Staden, D.; du Plessis, J.; Viljoen, J. Development of a self-emulsifying drug delivery system for optimized topical delivery of clofazimine. Pharmaceutics 2020, 12, 523. [Google Scholar] [CrossRef]
- Soares, T.B.; Loureiro, L.; Carvalho, A.; Oliveira, M.E.C.R.; Dias, A.; Sarmento, B.; Lúcio, M. Lipid nanocarriers loaded with natural compounds: Potential new therapies for age related neurodegenerative diseases? Prog. Neurobiol. 2018, 168, 21–41. [Google Scholar] [CrossRef]
- Paiva-Santos, A.C.; Gama, M.; Peixoto, D.; Sousa-Oliveira, I.; Ferreira-Faria, I.; Zeinali, M.; Abbaspour-Ravasjani, S.; Mascarenhas-Melo, F.; Hamishehkar, H.; Veiga, F. Nanocarrier-based dermopharmaceutical formulations for the topical management of atopic dermatitis. Int. J. Pharm. 2022, 618, 121656. [Google Scholar] [CrossRef] [PubMed]
- Akula, S.; Gurram, A.K.; Devireddy, S.R. Self-microemulsifying drug delivery systems: An attractive strategy for enhanced therapeutic profile. Int. Sch. Res. Notices 2014, 2014, 964051. [Google Scholar] [CrossRef] [PubMed]
- Jepps, O.G.; Dancik, Y.; Anissimov, Y.G.; Roberts, M.S. Modelling the human skin barrier—Towards a better understanding of dermal absorption. Adv. Drug Deliv. Rev. 2013, 65, 152–168. [Google Scholar] [CrossRef] [PubMed]
- King, M.J.; Michel, D.; Foldvari, M. Evidence for lymphatic transport of insulin by topically applied biphasic vesicles. J. Pharm. Pharmacol. 2003, 55, 1339–1344. [Google Scholar] [CrossRef] [PubMed]
- Elbahwy, I.A.; Lupo, N.; Ibrahim, H.M.; Ismael, H.R.; Kasem, A.A.; Caliskan, C.; Matuszczak, B.; Bernkop-Schnürch, A. Mucoadhesive self-emulsifying delivery systems for ocular administration of econazole. Int. J. Pharm. 2018, 541, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Efiana, N.A.; Mahmood, A.; Lam, H.T.; Zupančič, O.; Leonaviciute, G.; Bernkop-Schnürch, A. Improved mucoadhesive properties of self-nanoemulsifying drug delivery systems (SNEDDS) by introducing acyl chitosan. Int. J. Pharm. 2018, 519, 206–212. [Google Scholar] [CrossRef]
- Leonaviciute, G.; Adamovic, N.T.; Lam, H.T.; Rohrer, J.; Partenhauser, A.; Bernkop-Schnürch, A. Self-emulsifying drug delivery systems (SEDDS): Proof-of-concept how to make them mucoadhesive. Eur. J. Pharm. Biopharm. 2017, 112, 51–57. [Google Scholar] [CrossRef]
- Salimi, E.; Le-Vinh, B.; Zahir-Jouzdani, F.; Matuszczak, B.; Ghaee, A.; Bernkop-Schnürch, A. Self-emulsifying drug delivery systems changing their zeta potential via a flip-flop mechanism. Int. J. Pharm. 2018, 550, 200–206. [Google Scholar] [CrossRef]
- Menzel, C.; Bernkop- Schnürch, A. Enzyme decorated drug carriers: Targeted swords to cleave and overcome the mucus barrier. Adv. Drug Deliv. Rev. 2018, 124, 164–174. [Google Scholar] [CrossRef]
- Xu, Y.; Wu, J.; Liao, S.; Sun, Z. Treating tuberculosis with high doses of anti-TB drugs: Mechanisms and outcomes. Ann. Clin. Microbiol. Antimicrob. 2017, 16, 67–80. [Google Scholar] [CrossRef]
- Bhandari, S.; Rana, V.; Tiwary, A.K. Antimalarial solid self-emulsifying system for oral use: In vitro investigation. Ther. Deliv. 2017, 8, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Nigade, P.M.; Patil, S.L.; Tiwari, S.S. Self-emulsifying drug delivery system (SEDDS): A review. Int. J. Pharm. Biol. 2012, 2, 42–52. [Google Scholar]
- Gaur, R.K. Antibiotic resistance: Alternative approaches. Indian J. Pharmacol. 2017, 49, 208–210. [Google Scholar] [CrossRef]
- Mashele, S.A.; Steel, H.C.; Matjokotja, M.T.; Rasehlo, S.S.M.; Anderson, R.; Cholo, M.C. Assessment of the efficacy of clofazimine alone and in combination with primary agents against Mycobacterium tuberculosis in vitro. J. Glob. Antimicrob. Resist. 2022, 29, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Tanner, L.; Mashabela, G.T.; Omollo, C.C.; de Wet, T.J.; Parkinson, C.J.; Warner, D.F.; Haynes, R.K.; Wiesner, L. Intracellular Accumulation of Novel and Clinically Used TB Drugs Potentiates Intracellular Synergy. Microbiol. Spectr. 2021, 9, e00434-21. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Priority Drug Groups | TB Drug |
|---|---|
| Group A: Incorporate all three drugs as part of regimen | Levofloxacin OR Moxifloxacin Bedaquiline Linezolid |
| Group B: Add one or both drugs to regimen | Clofazimine Cycloserine OR Terizidone |
| Group C: Add to complete regimen and include when drugs from Group A and B cannot be included as influenced by resistance, toxicity, or tolerability | Ethambutol Delamanid Pyrazinamide Imipenem–cilastatin OR Meropenem AND Amoxicillin/Clavulanate Amikacin OR Streptomycin Ethionamide OR Prothionamide Aminosalicylic acid |
| Physicochemical Characteristics | |
|---|---|
| Chemical formula | C27H22Cl2N4 |
| Molecular weight | 473.40 Da |
| Chemical structure |  |
| Melting point | 210–212 °C |
| Appearance | Reddish-brown, fine powder |
| Log P(Octanol-water partition coefficient) | 7.66 |
| UV Detection wavelength | 254 nm |
| Elimination half life | 70 days |
| pKa value | 8.51 |
| Solubility | Soluble in methylene chloride, very slightly soluble in ethanol, aqueous solubility < 0.001 mg/L |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
van Staden, D.; Haynes, R.K.; Viljoen, J.M. Adapting Clofazimine for Treatment of Cutaneous Tuberculosis by Using Self-Double-Emulsifying Drug Delivery Systems. Antibiotics 2022, 11, 806. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics11060806
van Staden D, Haynes RK, Viljoen JM. Adapting Clofazimine for Treatment of Cutaneous Tuberculosis by Using Self-Double-Emulsifying Drug Delivery Systems. Antibiotics. 2022; 11(6):806. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics11060806
Chicago/Turabian Stylevan Staden, Daniélle, Richard K. Haynes, and Joe M. Viljoen. 2022. "Adapting Clofazimine for Treatment of Cutaneous Tuberculosis by Using Self-Double-Emulsifying Drug Delivery Systems" Antibiotics 11, no. 6: 806. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics11060806