
Identification of a Fragment-Based Scaffold that Inhibits the Glycosyltransferase WaaG from Escherichia coli †

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction

2. Results and Discussion
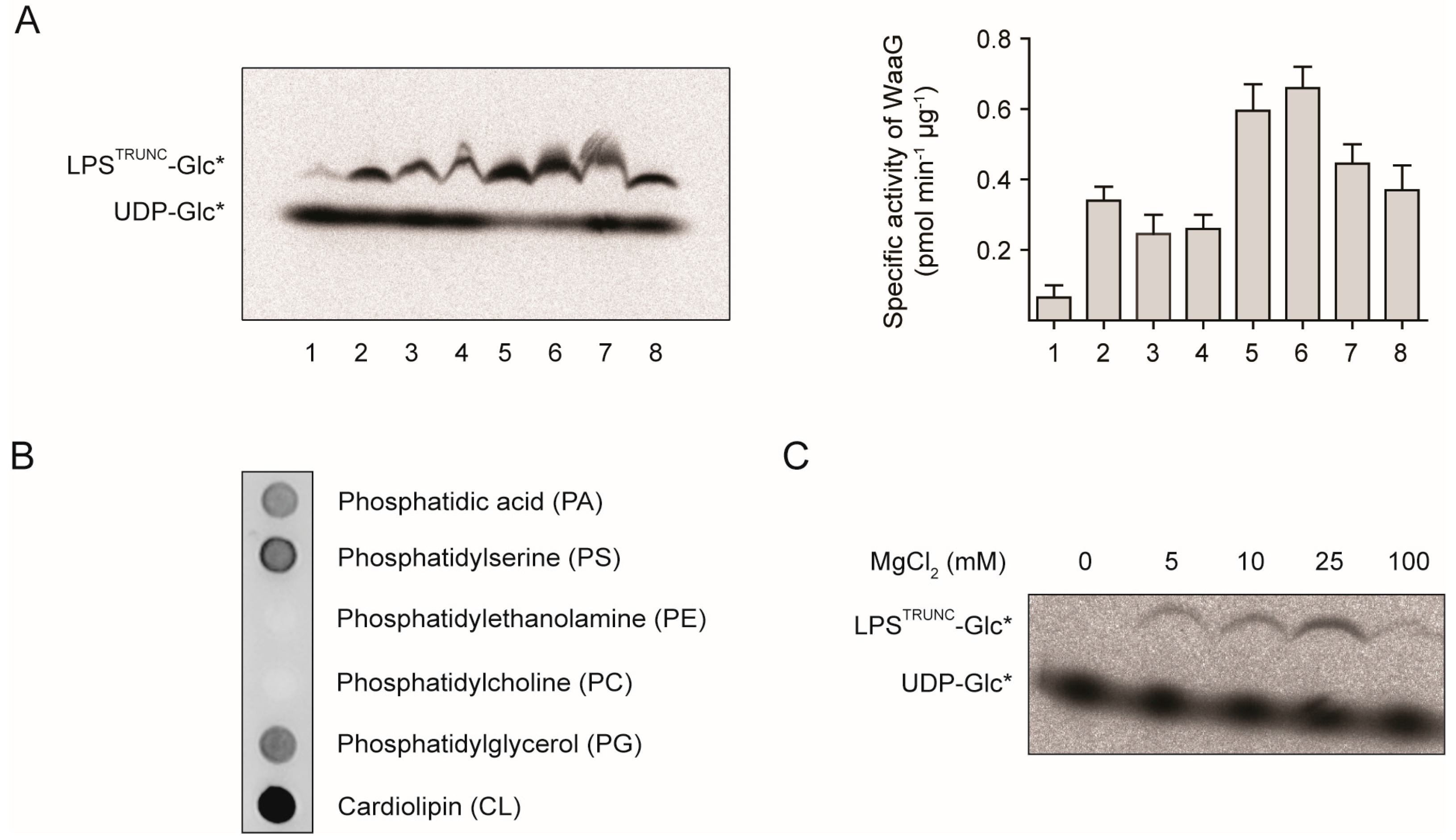
2.1. An in Vitro Assay for WaaG

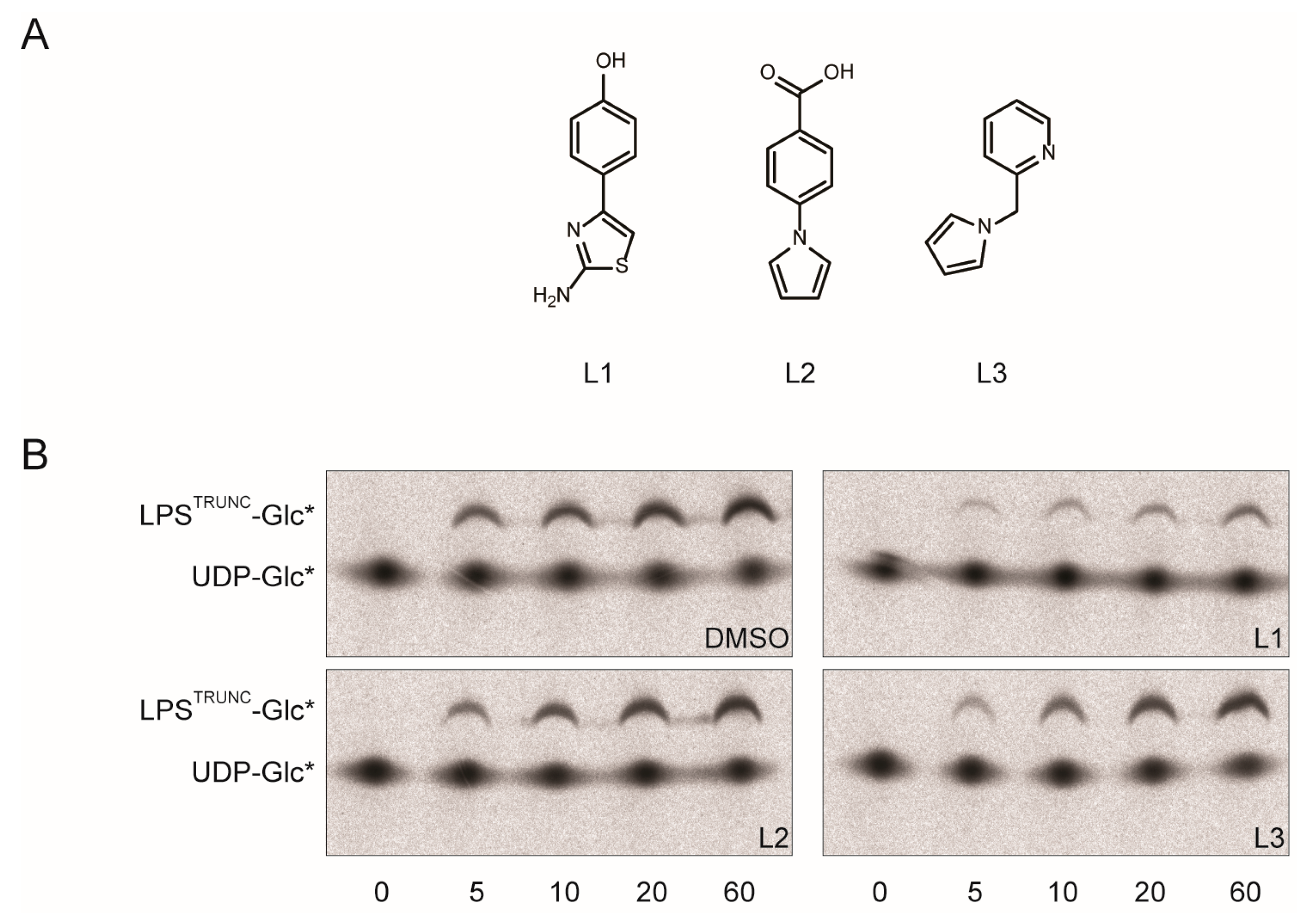
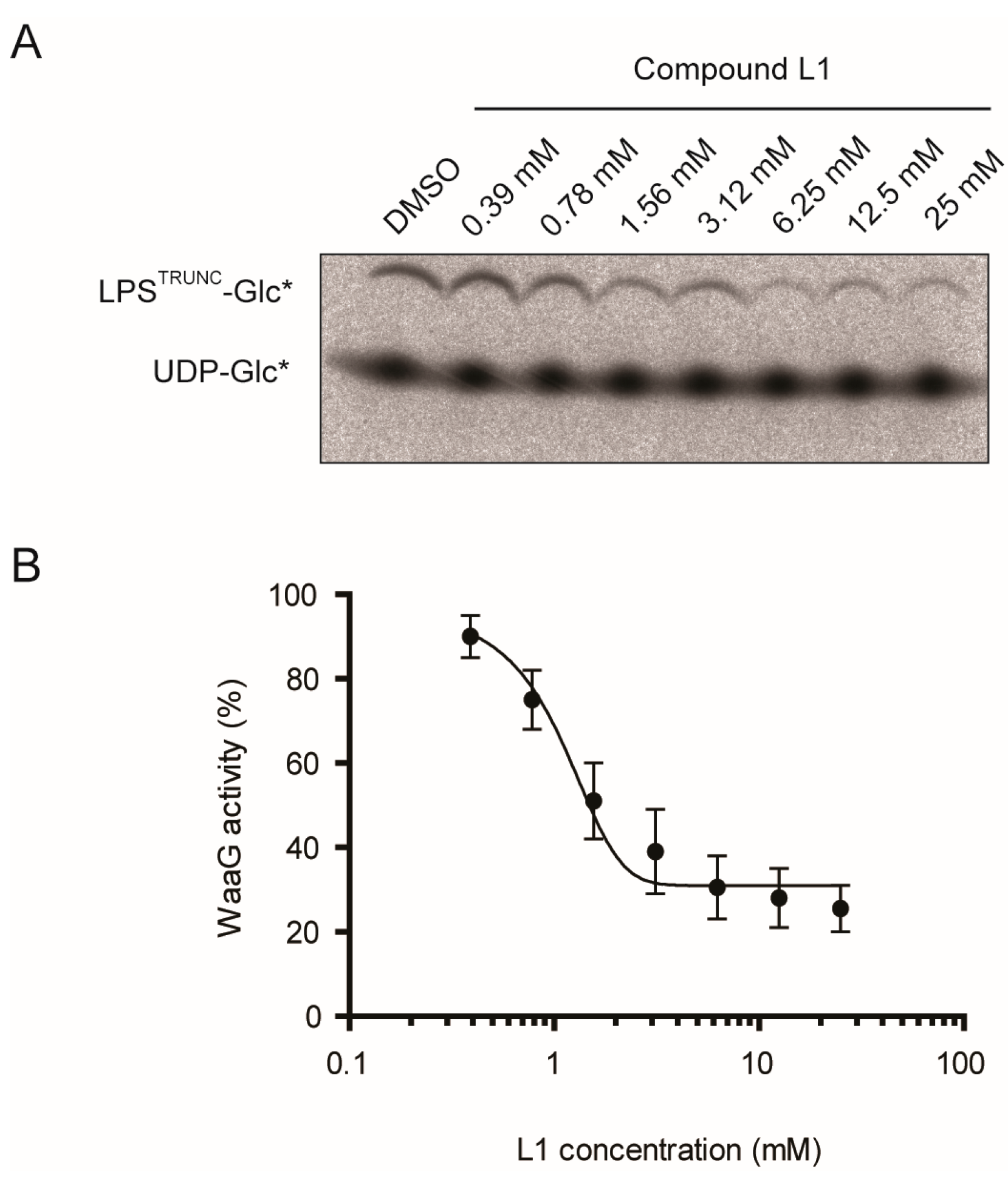
2.2. L1 Can Inhibit WaaG in Vitro



3. Experimental Section
3.1. Bacterial Strains, Plasmids and Chemicals
3.2. Protein Expression and Purification of E. coli His-WaaG
3.3. LPS Extraction
3.4. O-Deacylation of LPS in Aqueous NH4OH
3.5. In Vitro Lipid Binding Assay
3.6. WaaG Activity Assay
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| The following abbreviations are used in this manuscript: |
| CHAPS: | 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate |
| CL: | cardiolipin |
| DHPC: | 1,2-dihexanoyl-sn-glycero-3-phosphocholine |
| Hep: | l-glycero-d-manno-heptose |
| HepII: | l-glycero-d-manno-heptose-II |
| Kdo: | 3-deoxy-d-manno-oct-2-ulosonic acid |
| LPS: | lipopolysaccharide |
| PA: | phosphatidic acid |
| PC: | phosphatidylcholine |
| PE: | phosphatidylethanolamine |
| PG: | phosphatidylglycerol |
| PS: | phosphatidylserine |
References
- Amor, K.; Heinrichs, D.E.; Frirdich, E.; Ziebell, K.; Johnson, R.P.; Whitfield, C. Distribution of core oligosaccharide types in lipopolysaccharides from Escherichia coli. Infect. Immun. 2000, 68, 1116–1124. [Google Scholar] [CrossRef] [PubMed]
- Stenutz, R.; Weintraub, A.; Widmalm, G. The structures of Escherichia coli O-polysaccharide antigens. FEMS Microbiol. Rev. 2006, 30, 382–403. [Google Scholar] [CrossRef] [PubMed]
- Nikaido, H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 2003, 67, 593–656. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Tran, L.; Becket, E.; Lee, K.; Chinn, L.; Park, E.; Tran, K.; Miller, J.H. Antibiotic sensitivity profiles determined with an Escherichia coli gene knockout collection: Generating an antibiotic bar code. Antimicrob. Agents Chemother. 2010, 54, 1393–1403. [Google Scholar] [CrossRef] [PubMed]
- Laxminarayan, R.; Duse, A.; Wattal, C.; Zaidi, A.K.M.; Wertheim, H.F.L.; Sumpradit, N.; Vlieghe, E.; Hara, G.L.; Gould, I.M.; Goossens, H.; et al. Antibiotic resistance—The need for global solutions. Lancet Infect. Dis. 2013, 13, 1057–1098. [Google Scholar] [CrossRef]
- Onishi, H.R.; Pelak, B.A.; Gerckens, L.S.; Silver, L.L.; Kahan, F.M.; Chen, M.H.; Patchett, A.A.; Galloway, S.M.; Hyland, S.A.; Anderson, M.S.; et al. Antibacterial agents that inhibit lipid A biosynthesis. Science 1996, 274, 980–982. [Google Scholar] [CrossRef] [PubMed]
- Sherman, D.J.; Okuda, S.; Denny, W.A.; Kahne, D. Validation of inhibitors of an ABC transporter required to transport lipopolysaccharide to the cell surface in Escherichia coli. Bioorg. Med. Chem. 2013, 21, 4846–4851. [Google Scholar] [CrossRef] [PubMed]
- De Leon, G.P.; Elowe, N.H.; Koteva, K.P.; Valvano, M.A.; Wright, G.D. An in vitro screen of bacterial lipopolysaccharide biosynthetic enzymes identifies an inhibitor of ADP-heptose biosynthesis. Chem. Biol. 2006, 13, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, N.; Jetter, P.; Ueberbacher, B.J.; Werneburg, M.; Zerbe, K.; Steinmann, J.; van der Meijden, B.; Bernardini, F.; Lederer, A.; Dias, R.L.A.; et al. Peptidomimetic antibiotics target outer-membrane biogenesis in Pseudomonas aeruginosa. Science 2010, 327, 1010–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.R.; Lee, J.H.; Jeong, B.C.; Lee, S.H. Lipid A biosynthesis of multidrug-resistant pathogens—A novel drug target. Curr. Pharm. Des. 2013, 19, 6534–6550. [Google Scholar] [CrossRef] [PubMed]
- Yethon, J.A.; Vinogradov, E.; Perry, M.B.; Whitfield, C. Mutation of the lipopolysaccharide core glycosyltransferase encoded by waaG destabilizes the outer membrane of Escherichia coli by interfering with core phosphorylation. J. Bacteriol. 2000, 182, 5620–5623. [Google Scholar] [CrossRef] [PubMed]
- Clifton, L.A.; Skoda, M.W.; le Brun, A.P.; Ciesielski, F.; Kuzmenko, I.; Holt, S.A.; Lakey, J.H. Effect of divalent cation removal on the structure of gram-negative bacterial outer membrane models. Langmuir 2015, 31, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Hoare, A.; Bittner, M.; Carter, J.; Alvarez, S.; Zaldivar, M.; Bravo, D.; Valvano, M.A.; Contreras, I. The outer core lipopolysaccharide of Salmonella enterica serovar typhi is required for bacterial entry into epithelial cells. Infect. Immun. 2006, 74, 1555–1564. [Google Scholar] [CrossRef] [PubMed]
- Hajduk, P.J.; Greer, J. A decade of fragment-based drug design: Strategic advances and lessons learned. Nat. Rev. Drug Discov. 2007, 6, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Rees, D.C.; Congreve, M.; Murray, C.W.; Carr, R. Fragment-based lead discovery. Nat. Rev. Drug Discov. 2004, 3, 660–672. [Google Scholar] [CrossRef] [PubMed]
- De Kloe, G.E.; Bailey, D.; Leurs, R.; de Esch, I.J. Transforming fragments into candidates: Small becomes big in medicinal chemistry. Drug Discov. Today 2009, 14, 630–646. [Google Scholar] [CrossRef] [PubMed]
- Landström, J.; Persson, K.; Rademacher, C.; Lundborg, M.; Wakarchuk, W.; Peters, T.; Widmalm, G. Small molecules containing hetero-bicyclic ring systems compete with UDP-Glc for binding to WaaG glycosyltransferase. Glycoconj. J. 2012, 29, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Congreve, M.; Carr, R.; Murray, C.; Jhoti, H. A “rule of three” for fragment-based lead discovery? Drug Discov. Today 2003, 8, 876–877. [Google Scholar] [CrossRef]
- Albesa-Jove, D.; Giganti, D.; Jackson, M.; Alzari, P.M.; Guerin, M.E. Structure-function relationships of membrane-associated GT-B glycosyltransferases. Glycobiology 2014, 24, 108–124. [Google Scholar] [CrossRef] [PubMed]
- Liebau, J.; Pettersson, P.; Szpryngiel, S.; Maler, L. Membrane interaction of the glycosyltransferase WaaG. Biophys. J. 2015, 109, 552–563. [Google Scholar] [CrossRef] [PubMed]
- Dowhan, W. Molecular genetic approaches to defining lipid function. J. Lipid Res. 2009, 50, S305–S310. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Fleites, C.; Proctor, M.; Roberts, S.; Bolam, D.N.; Gilbert, H.J.; Davies, G.J. Insights into the synthesis of lipopolysaccharide and antibiotics through the structures of two retaining glycosyltransferases from family GT4. Chem. Biol. 2006, 13, 1143–1152. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Sass, H.J.; Zahringer, U.; Grzesiek, S. Structure and dynamics of 13C,15N-labeled lipopolysaccharides in a membrane mimetic. Angew. Chem. Int. Ed. Engl. 2008, 47, 9870–9874. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.H.; Garrett, T.A.; Raetz, C.R.H. In vitro assembly of the outer core of the lipopolysaccharide from Escherichia coli K-12 and Salmonella typhimurium. Biochemistry 2014, 53, 1250–1262. [Google Scholar] [CrossRef] [PubMed]
- De Castro, C.; Parrilli, M.; Holst, O.; Molinaro, A. Microbe-associated molecular patterns in innate immunity: Extraction and chemical analysis of gram-negative bacterial lipopolysaccharides. Methods Enzymol. 2010, 480, 89–115. [Google Scholar] [PubMed]
- Galanos, C.; Luderitz, O.; Westphal, O. A new method for the extraction of R lipopolysaccharides. Eur. J. Biochem. 1969, 9, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Ali, T.; Weintraub, A.; Widmalm, G. Structural determination of the O-antigenic polysaccharide from the Shiga toxin-producing Escherichia coli O171. Carbohydr. Res. 2006, 341, 1878–1883. [Google Scholar] [CrossRef] [PubMed]
- Tedaldi, L.; Wagner, G.K. Beyond substrate analogues: New inhibitor chemotypes for glycosyltransferases. Med. Chem. Commun. 2014, 5, 1106–1125. [Google Scholar] [CrossRef]
- Wang, S.; Vidal, S. Recent design of glycosyltransferase inhibitors. Carbohydr. Chem. 2013, 39, 78–101. [Google Scholar] [CrossRef] [PubMed]
- Devine, S.M.; Mulcair, M.D.; Debono, C.O.; Leung, E.W.; Nissink, J.W.; Lim, S.S.; Chandrashekaran, I.R.; Vazirani, M.; Mohanty, B.; Simpson, J.S.; et al. Promiscuous 2-Aminothiazoles (PrATs): A frequent hitting scaffold. J. Med. Chem. 2015, 58, 1205–1214. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muheim, C.; Bakali, A.; Engström, O.; Wieslander, Å.; Daley, D.O.; Widmalm, G. Identification of a Fragment-Based Scaffold that Inhibits the Glycosyltransferase WaaG from Escherichia coli . Antibiotics 2016, 5, 10. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics5010010
Muheim C, Bakali A, Engström O, Wieslander Å, Daley DO, Widmalm G. Identification of a Fragment-Based Scaffold that Inhibits the Glycosyltransferase WaaG from Escherichia coli . Antibiotics. 2016; 5(1):10. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics5010010
Chicago/Turabian StyleMuheim, Claudio, Amin Bakali, Olof Engström, Åke Wieslander, Daniel O. Daley, and Göran Widmalm. 2016. "Identification of a Fragment-Based Scaffold that Inhibits the Glycosyltransferase WaaG from Escherichia coli " Antibiotics 5, no. 1: 10. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics5010010