
Docking into Mycobacterium tuberculosis Thioredoxin Reductase Protein Yields Pyrazolone Lead Molecules for Methicillin-Resistant Staphylococcus aureus

 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
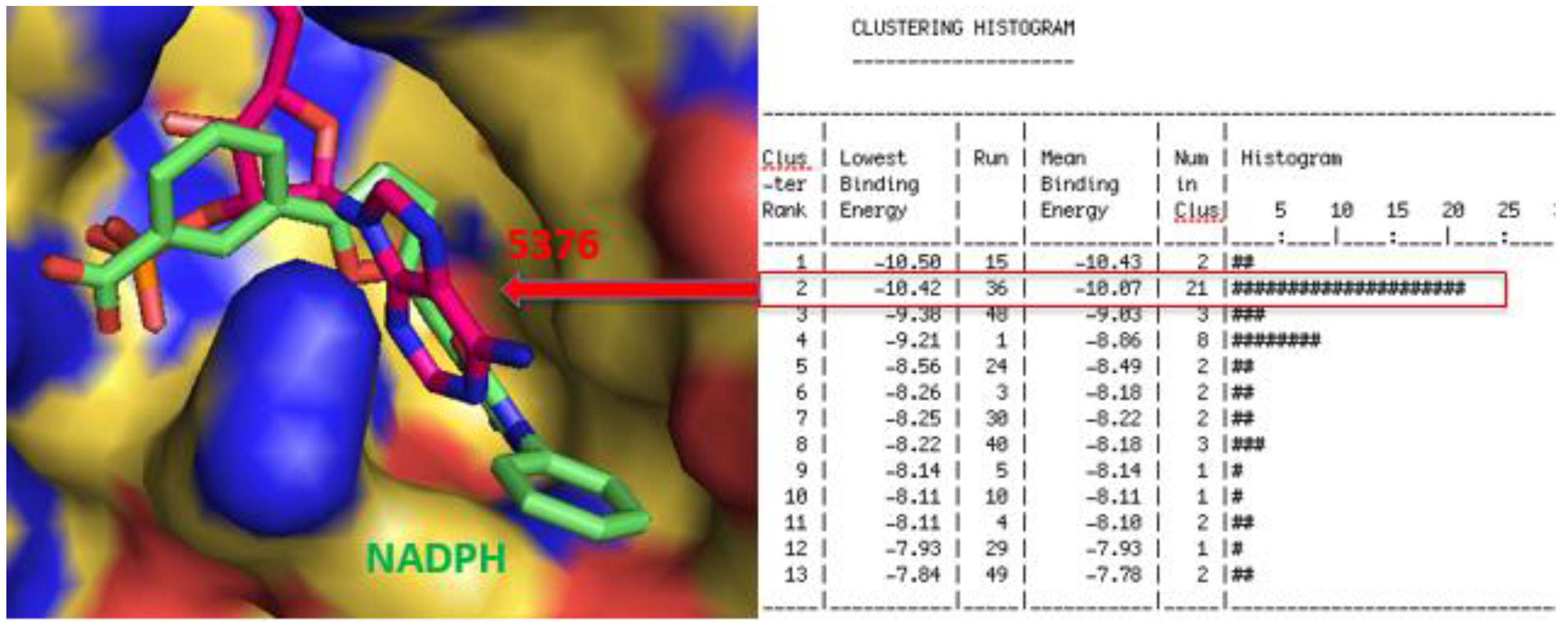
2.1. Initial Virtual Screening and Bacterial Assays
2.2. Ability of Analogues of 5376 to Kill Certain Strains of S. aureus
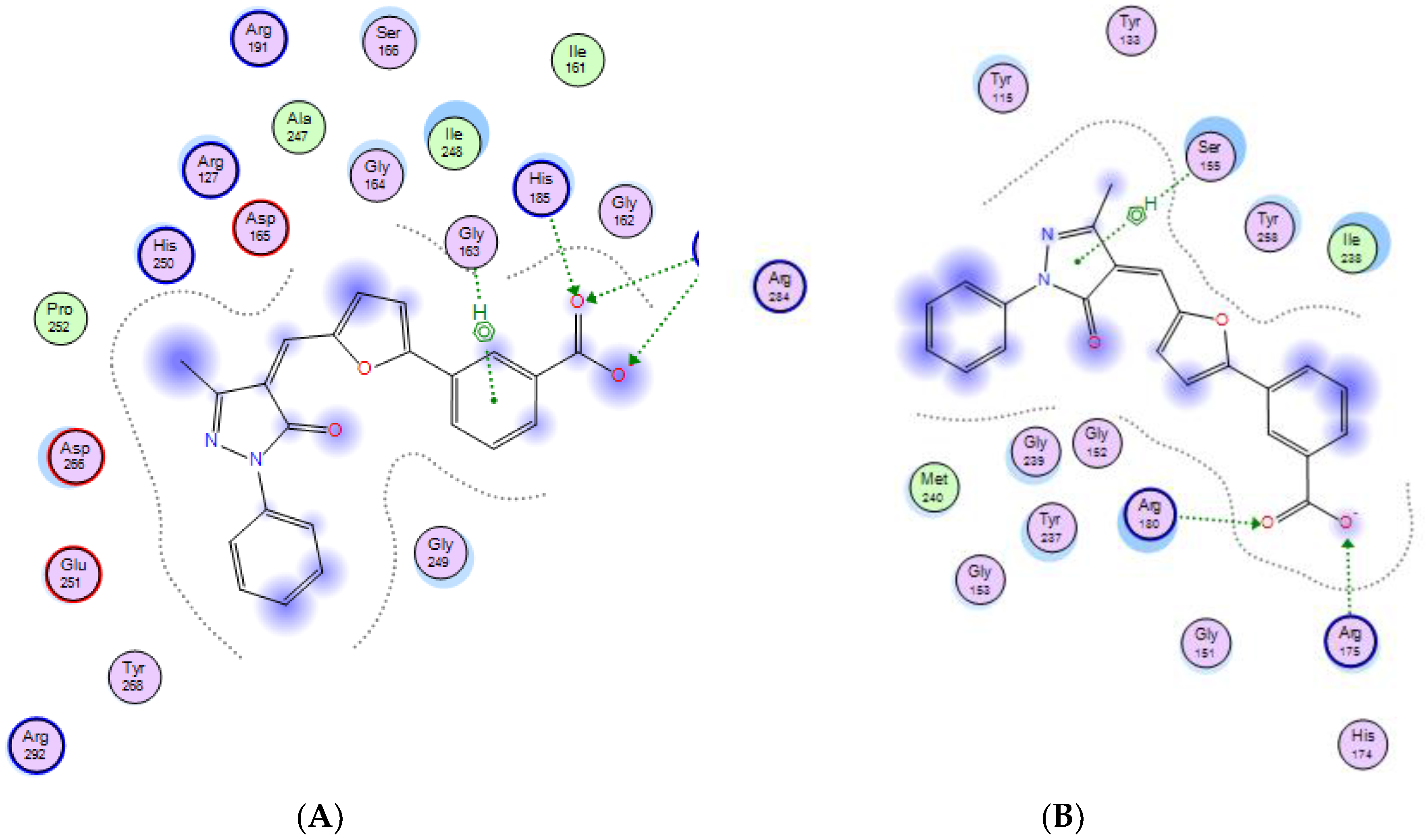
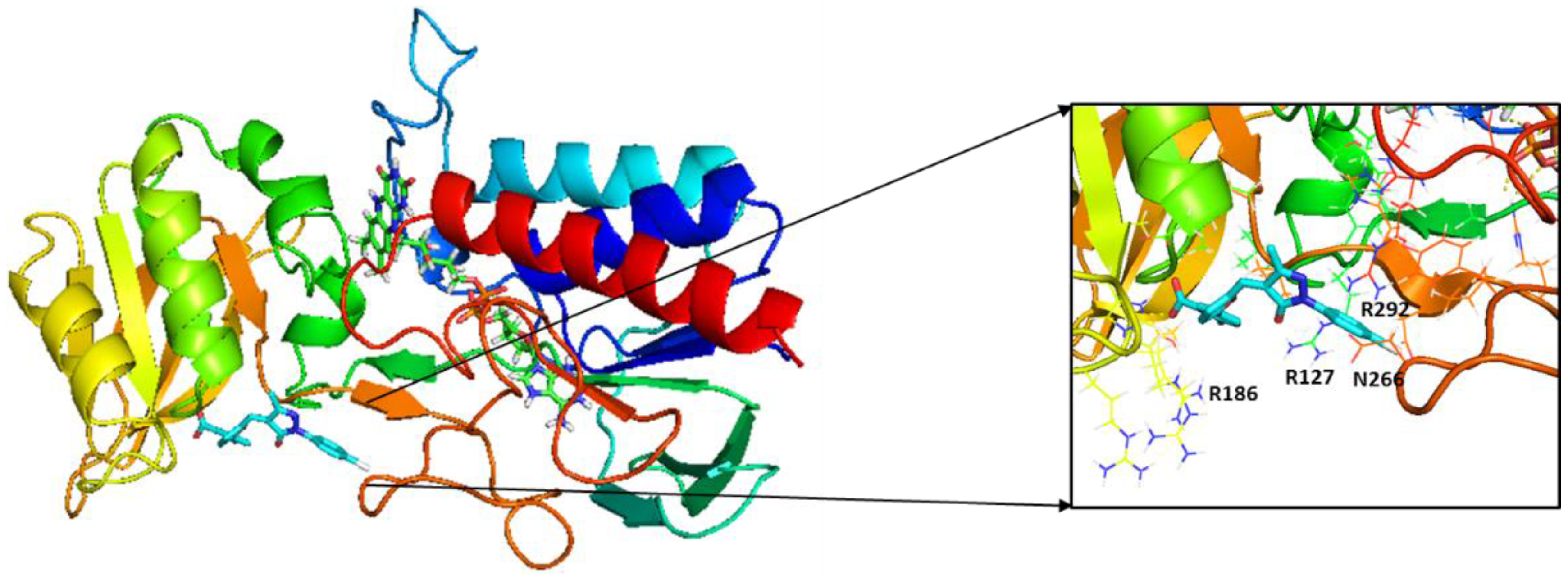
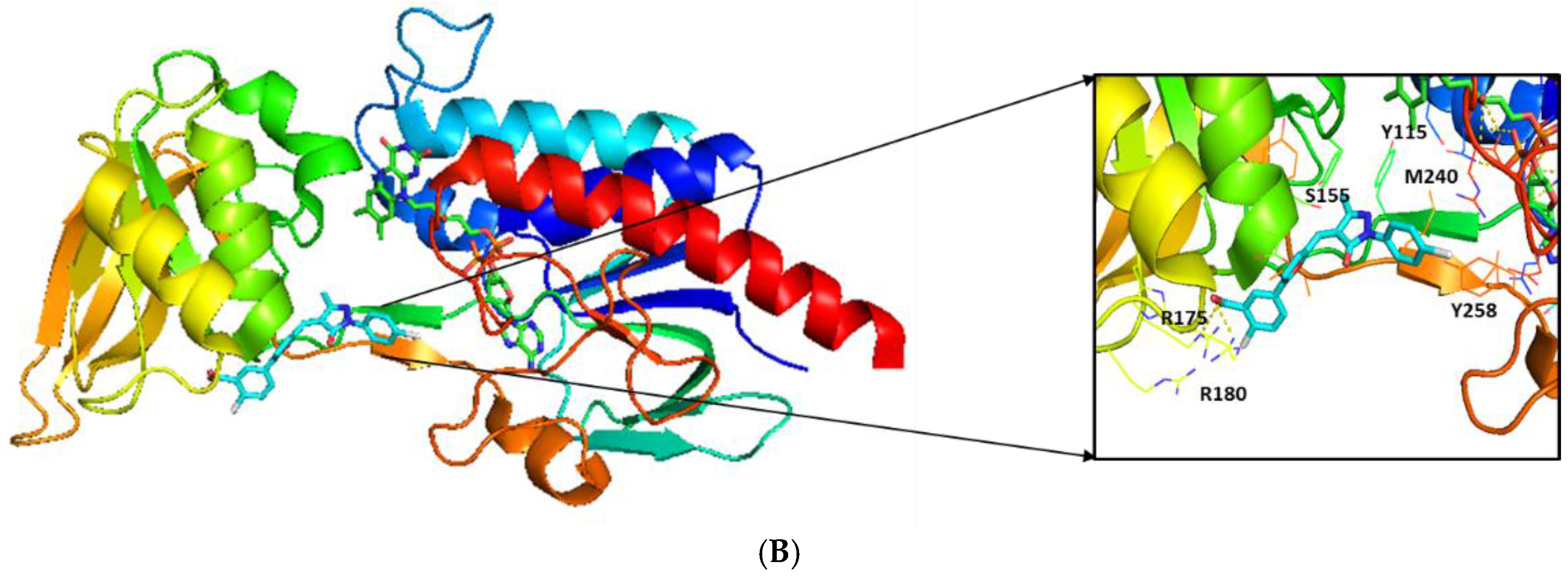
2.3. Molecular Modeling
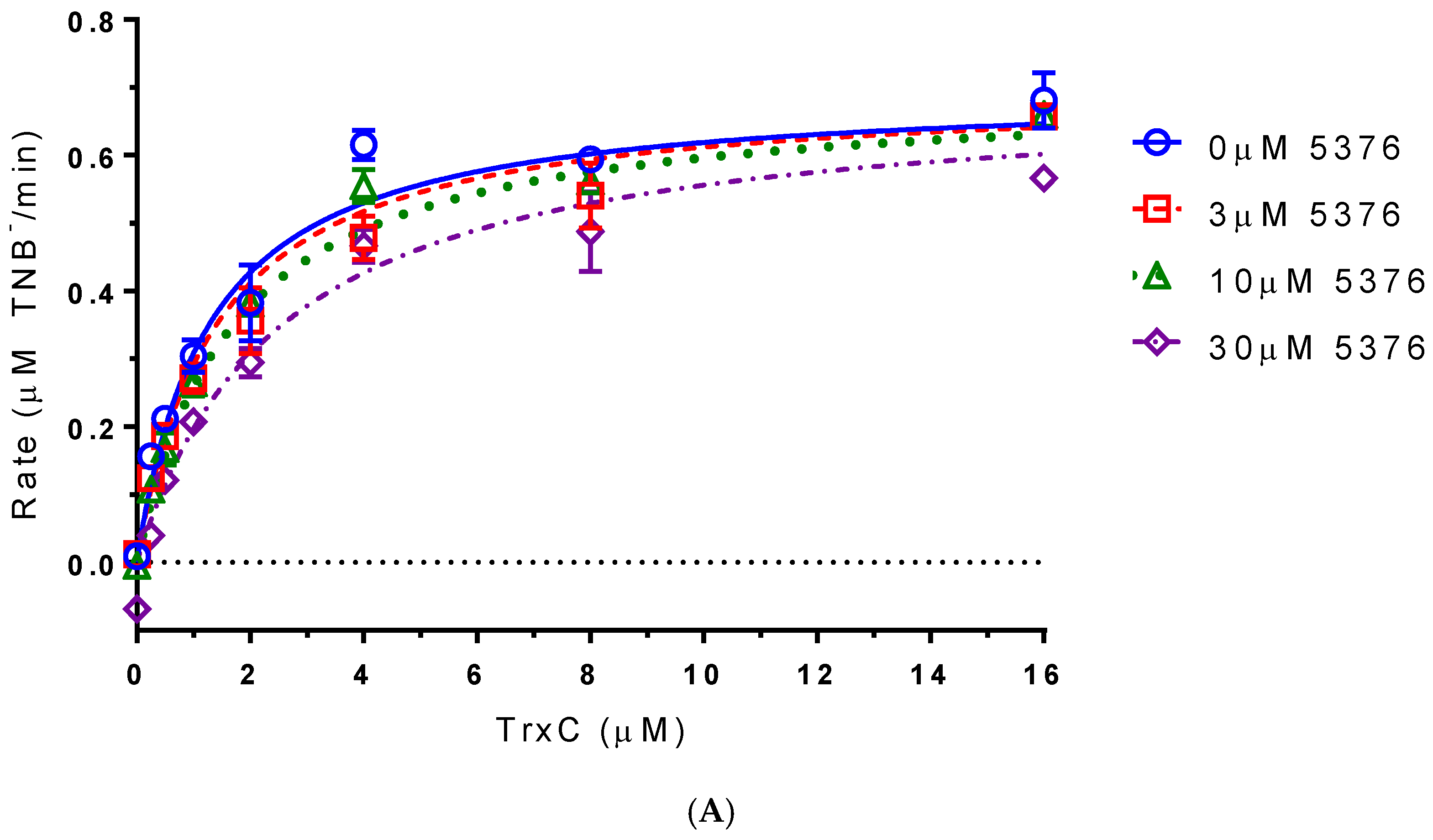
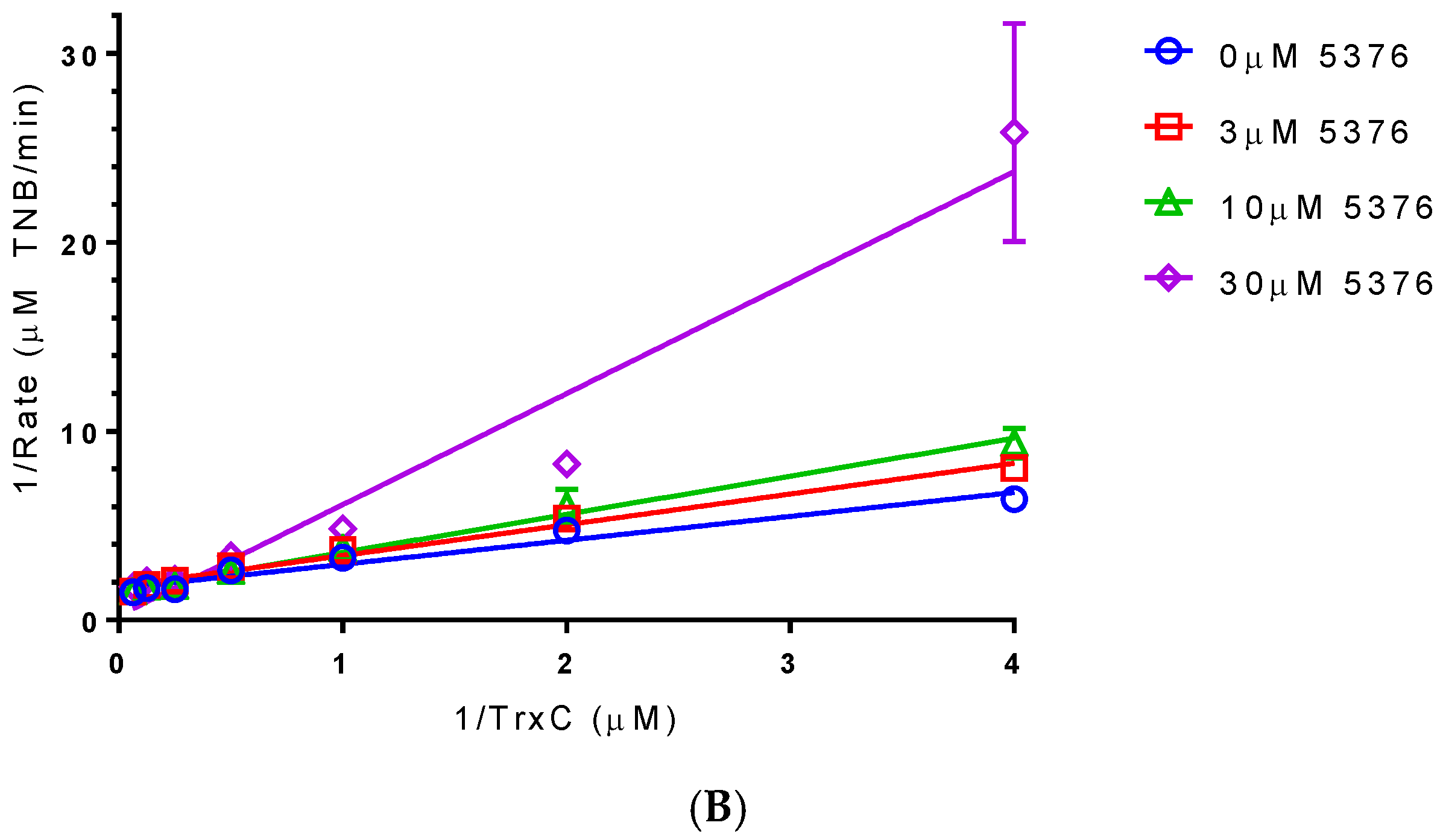
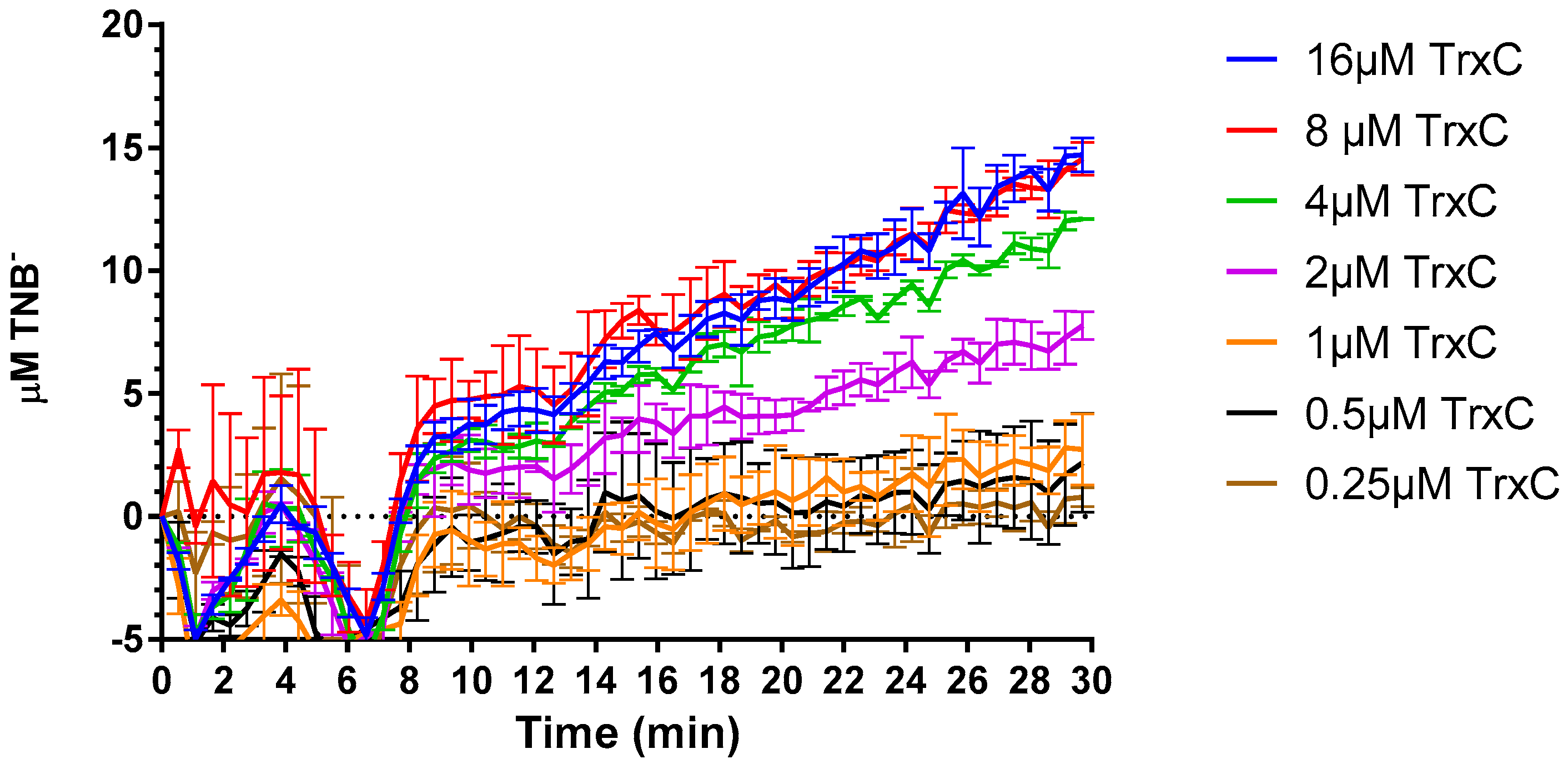
2.4. Compound 5376 Inhibits M. tuberculosis TrxR Activity and Binding of M. tuberculosis Thioredoxin C (TrxC) to M. tuberculosis TrxR
3. Experimental Section
3.1. Bacterial Species
3.2. Minimum Inhibitory Concentration (MIC)/Minimum Bactericial Concentration (MBC)
3.3. Thioredoxin Reductase-DTNB Assay
3.4. Docking
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| DTNB | Ellman′s Reagent (5,5-dithio-bis-(2-nitrobenzoic acid)) |
| DTT | 1,4-Dithiothreitol |
| TrxR | Thioredoxin Reductase |
| Trx | Thioredoxin |
| PDB | Protein Data Bank |
Appendix A

References
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.H., Jr. Mechanism and structure of thioredoxin reductase from Escherichia coli. FASEB J. 1995, 9, 1267–1276. [Google Scholar] [PubMed]
- Holmgren, A. Thioredoxin. Annu. Rev. Biochem. 1985, 54, 237–271. [Google Scholar] [CrossRef] [PubMed]
- Saccoccia, F.A.F.; Boumis, G.; Carotti, D.; Desiato, G.; Miele, A.E.; Bellelli, A. Thioredoxin Reductase and Its Inhibitors. Curr. Protein Pept. Sci. 2014, 15, 621–646. [Google Scholar] [CrossRef] [PubMed]
- Zumla, A.; Nahid, P.; Cole, S.T. Advances in the development of new tuberculosis drugs and treatment regimens. Nat. Rev. Drug Discov. 2013, 12, 388–404. [Google Scholar] [CrossRef] [PubMed]
- Kruh, N.A.; Troudt, J.; Izzo, A.; Prenni, J.; Dobos, K.M. Portrait of a pathogen: The Mycobacterium tuberculosis proteome in vivo. PLoS ONE 2010, 5, e13938. [Google Scholar] [CrossRef] [PubMed]
- Akif, M.; Suhre, K.; Verma, C.; Mande, S.C. Conformational flexibility of Mycobacterium tuberculosis thioredoxin reductase: Crystal structure and normal-mode analysis. Acta Crystallogr. 2005, 61, 1603–1611. [Google Scholar]
- Suaya, J.A.; Mera, R.M.; Cassidy, A.; O’Hara, P.; Amrine-Madsen, H.; Burstin, S.; Miller, L.G. Incidence and cost of hospitalizations associated with Staphylococcus aureus skin and soft tissue infections in the United States from 2001 through 2009. BMC Infect. Dis. 2014. [Google Scholar] [CrossRef] [PubMed]
- Uziel, O.; Borovok, I.; Schreiber, R.; Cohen, G.; Aharonowitz, Y. Transcriptional regulation of the Staphylococcus aureus thioredoxin and thioredoxin reductase genes in response to oxygen and disulfide stress. J. Bacteriol. 2003, 186, 326–334. [Google Scholar] [CrossRef]
- Liang, S.Y.; Lin, S.Y.; Chiang, I.C.; Shih, Y.L. Quantitative inner membrane proteome datasets of the wild-type and the Δmin mutant of Escherichia coli. Data Brief 2016, 8, 304–307. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, T.N.; Osman, H.; Werngren, J.; Hoffner, S.; Engman, L.; Holmgren, A. Ebselen and analogs as inhibitors of Bacillus anthracis thioredoxin reductase and bactericidal antibacterials targeting Bacillus species, Staphylococcus aureus and Mycobacterium tuberculosis. Biochim. Biophys. Acta 2016, 1860, 1265–1271. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Weiner, W.S.; Schroeder, C.E.; Simpson, D.S.; Hanson, A.M.; Sweeney, N.L.; Marvin, R.K.; Ndjomou, J.; Kolli, R.; Isailovic, D.; et al. Ebselen inhibits hepatitis C virus NS3 helicase binding to nucleic acid and prevents viral replication. ACS Chem. Biol. 2014, 9, 2393–2403. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Vlamis-Gardikas, A.; Kandasamy, K.; Zhao, R.; Gustafsson, T.N.; Engstrand, L.; Hoffner, S.; Engman, L.; Holmgren, A. Inhibition of bacterial thioredoxin reductase: An antibiotic mechanism targeting bacteria lacking glutathione. FASEB J. 2013, 27, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Owings, J.P.; McNair, N.N.; Mui, Y.F.; Gustafsson, T.N.; Holmgren, A.; Contel, M.; Goldberg, J.B.; Mead, J.R. Auranofin and N-heterocyclic carbene gold-analogs are potent inhibitors of the bacteria Helicobacter pylori. FEMS Microbiol. Lett. 2016. [Google Scholar] [CrossRef] [PubMed]
- Harbut, M.B.; Vilcheze, C.; Luo, X.; Hensler, M.E.; Guo, H.; Yang, B.; Chatterjee, A.K.; Nizet, V.; Jacobs, W.R., Jr.; Schultz, P.G.; et al. Auranofin exerts broad-spectrum bactericidal activities by targeting thiol-redox homeostasis. Proc. Natl. Acad. Sci. USA 2015, 112, 4453–4458. [Google Scholar] [CrossRef] [PubMed]
- Hikisz, P.; Szczupak, L.; Koceva-Chyla, A.; Gu Spiel, A.; Oehninger, L.; Ott, I.; Therrien, B.; Solecka, J.; Kowalski, K. Anticancer and Antibacterial Activity Studies of Gold(I)-Alkynyl Chromones. Molecules 2015, 20, 19699–19718. [Google Scholar] [CrossRef] [PubMed]
- Walters, W.P.; Stahl, M.T.; Murcko, M.A. Virtual screening—An overview. Drug Discov. Today 1998, 3, 160–178. [Google Scholar] [CrossRef]
- Meng, X.; Zhang, H.; Mezei, M.; Cui, M. Molecular Docking: A powerful approach for structure-based drug discovery. Curr. Comput. Aided Drug Discov. 2011, 7, 146–157. [Google Scholar] [CrossRef]
- Cosconati, S.; Forli, S.; Perryman, A.L.; Harris, R.; Goodsell, D.S.; Olson, A.J. Virtual Screening with AutoDock: Theory and Practice. Expert Opin. Drug Discov. 2010, 5, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Patel J, C.I.F.; Bradford, P.; Eliopoulos, G.; Hindler, J. Performance Standards for Antimicrobial Susceptibility Testing: Seventeenth Informational Supplement; Clinical and Laboratory Standards Institute: Wayne, HJ, USA, 2007; pp. 13–231. [Google Scholar]
- Shahjahan Kabir, M.; Engelbrecht, K.; Polanowski, R.; Krueger, S.M.; Ignasiak, R.; Rott, M.; Schwan, W.R.; Stemper, M.E.; Reed, K.D.; et al. New classes of Gram-positive selective antibacterials: Inhibitors of MRSA and surrogates of the causative agents of anthrax and tuberculosis. Bioorg. Med. Chem. Lett. 2008, 18, 5745–5749. [Google Scholar] [CrossRef] [PubMed]
- Akif, M.; Khare, G.; Tyagi, A.K.; Mande, S.C.; Sardesai, A.A. Functional studies of multiple thioredoxins from Mycobacterium tuberculosis. J. Bacteriol. 2008, 190, 7087–7095. [Google Scholar] [CrossRef] [PubMed]
- Olson, A.L.; Neumann, T.S.; Cai, S.; Sem, D.S. Solution structures of Mycobacterium tuberculosis thioredoxin C and models of intact thioredoxin system suggest new approaches to inhibitor and drug design. Proteins 2013, 81, 675–689. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.; Henrick, K.; Nakamura, H. Announcing the worldwide Protein Data Bank. Nat. Struct. Mol. Biol. 2003. [Google Scholar] [CrossRef] [PubMed]
- Lang, P.T.; Brozell, S.R.; Mukherjee, S.; Pettersen, E.F.; Meng, E.C.; Thomas, V.; Rizzo, R.C.; Case, D.A.; James, T.L.; Kuntz, I.D. DOCK 6: Combining techniques to modelRNA-small molecule complexes. RNA 2009, 15, 1219–1230. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MIC (μg/mL) | ||||||
|---|---|---|---|---|---|---|
| Compound | S. aureus | E. faecalis | E. coli | P. aeruginosa | M. smegmatis | M. marinum |
| 8973 | 64 (64) a | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) |
| 6702 | 64 (64) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) |
| 1108 | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) |
| 2628 | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) |
| 9010 | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | 128 (128) | >128 (>128) |
| 1632 | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) |
| 5376 | 8 (4-8) | 128 (128) | >128 (>128) | >128 (>128) | 128 (128) | >128 (>128) |
| 1882 | 32 (32) | 32 (32) | >128 (>128) | >128 (>128) | 128 (128) | >128 (>128) |
| 3719 | 64 (64) | 128 (128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) |
| Tetracycline | 0.5 (0.25–0.5) | 32 (32) | 2 (1–2) | 16 (16) | ND | ND |
| Isoniazid | ND b | ND | ND | ND | 4 | 8 |
| MIC (μg/mL) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | ATCC a | MW2 | JE2 | Newman | N315 | MC7606 b | MC7769 | MC7827 | MC7846 |
| 8973 | >128 c (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) |
| 5376 | 8 (4-8) | 4 (4) | 8 (8) | 4 (4) | 4 (2–4) | 2 (2) | 4 (4–8) | 8 (8) | 4 (4) |
| 5741518 | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) |
| 5870804 | 16 (16) | 32 (32) | 32 (32) | 64 (64) | 32 (32) | 4 (4) | 64 (64) | 32 (32–64) | 16 (16) |
| 2082-0182 | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) |
| 2083-1665 | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) |
| 2083-1773 | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) | >128 (>128) |
| Tetracycline | 0.25 (0.25–0.5) | 0.25 (0.25) | 0.25 (0.25) | 0.25 (0.25) | 0.25 (0.25) | 32 (16–32) | 0.25 (0.25) | 32 (32) | 0.125 (0.125) |
| Oxacillin | 0.25 (0.25) | 8 (8–16) | 8 (8–6) | 0.5 (0.5) | 16 (16) | 4 (4) | 4 (4) | 4 (4) | 4 (4–8) |
| Vancomycin | 0.5 (0.5) | 1 (1) | 1 (1) | 1 (1) | 1 (1) | 1 (1) | 1 (1) | 1 (1) | 4 (4) |
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | R | X | Y | MIC (µg/mL) MC7606 | MIC (µg/mL) MC7769 | MIC (µg/mL) MC7827 | MIC (µg/mL) MC7846 | Docking Energy (kcal/mol) |
| 5376 | O | 4-Br | 4-Cl | 2 | 4 | 8 | 4 | −9.3 |
| 5741518 | O | 4-CH3 | 4-Cl | >128 | >128 | >128 | >128 | −8.7 |
| 5870804 | O | 4-Br | H | 4 | 64 | 32 | 16 | −10.9 |
| 2082-0182 | O | H | 4-Cl | >128 | >128 | >128 | >128 | −9.0 |
| 2083-1665 | OCH3 | 4-Br | 4-Cl | >128 | >128 | >128 | >128 | −8.6 |
| 2083-1773 | O | 3-Br | 4-Cl | >128 | >128 | >128 | >128 | −9.8 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sweeney, N.L.; Lipker, L.; Hanson, A.M.; Bohl, C.J.; Engel, K.E.; Kalous, K.S.; Stemper, M.E.; Sem, D.S.; Schwan, W.R. Docking into Mycobacterium tuberculosis Thioredoxin Reductase Protein Yields Pyrazolone Lead Molecules for Methicillin-Resistant Staphylococcus aureus. Antibiotics 2017, 6, 4. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics6010004
Sweeney NL, Lipker L, Hanson AM, Bohl CJ, Engel KE, Kalous KS, Stemper ME, Sem DS, Schwan WR. Docking into Mycobacterium tuberculosis Thioredoxin Reductase Protein Yields Pyrazolone Lead Molecules for Methicillin-Resistant Staphylococcus aureus. Antibiotics. 2017; 6(1):4. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics6010004
Chicago/Turabian StyleSweeney, Noreena L., Lauren Lipker, Alicia M. Hanson, Chris J. Bohl, Katie E. Engel, Kelsey S. Kalous, Mary E. Stemper, Daniel S. Sem, and William R. Schwan. 2017. "Docking into Mycobacterium tuberculosis Thioredoxin Reductase Protein Yields Pyrazolone Lead Molecules for Methicillin-Resistant Staphylococcus aureus" Antibiotics 6, no. 1: 4. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics6010004