
Erythromycin Modification That Improves Its Acidic Stability while Optimizing It for Local Drug Delivery


Abstract
:1. Introduction
2. Results and Discussion
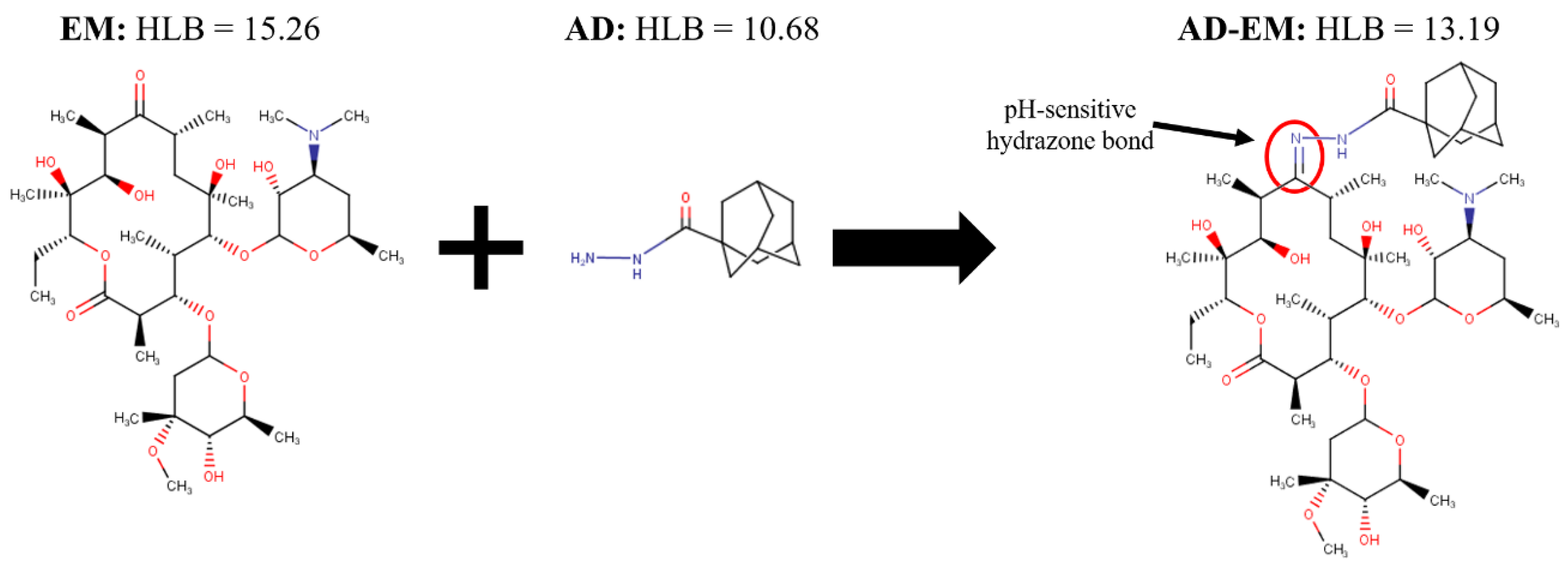
2.1. Synthesis of Adamantane-Modified EM (AD-EM)
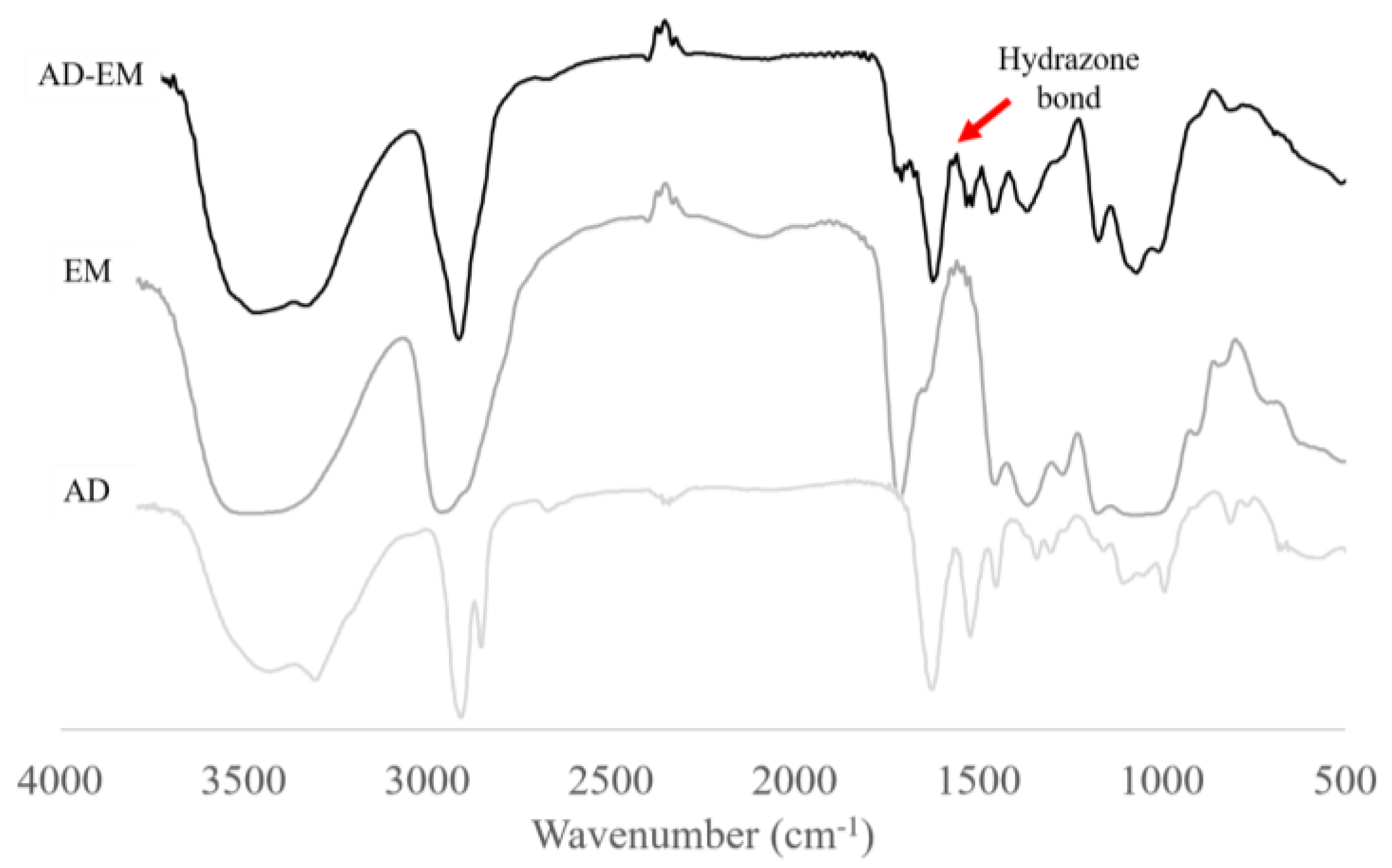
2.2. FTIR Spectrum of AD, EM, and AD-EM
2.3. 1H NMR of EM and AD-EM
2.4. MALDI Mass Spectrometry of EM and AD-EM
2.5. Hydrophilic-Lipophilic Balance Calculations
2.6. Solubility Study
2.7. Acidic Stability Spectral Scan Analysis
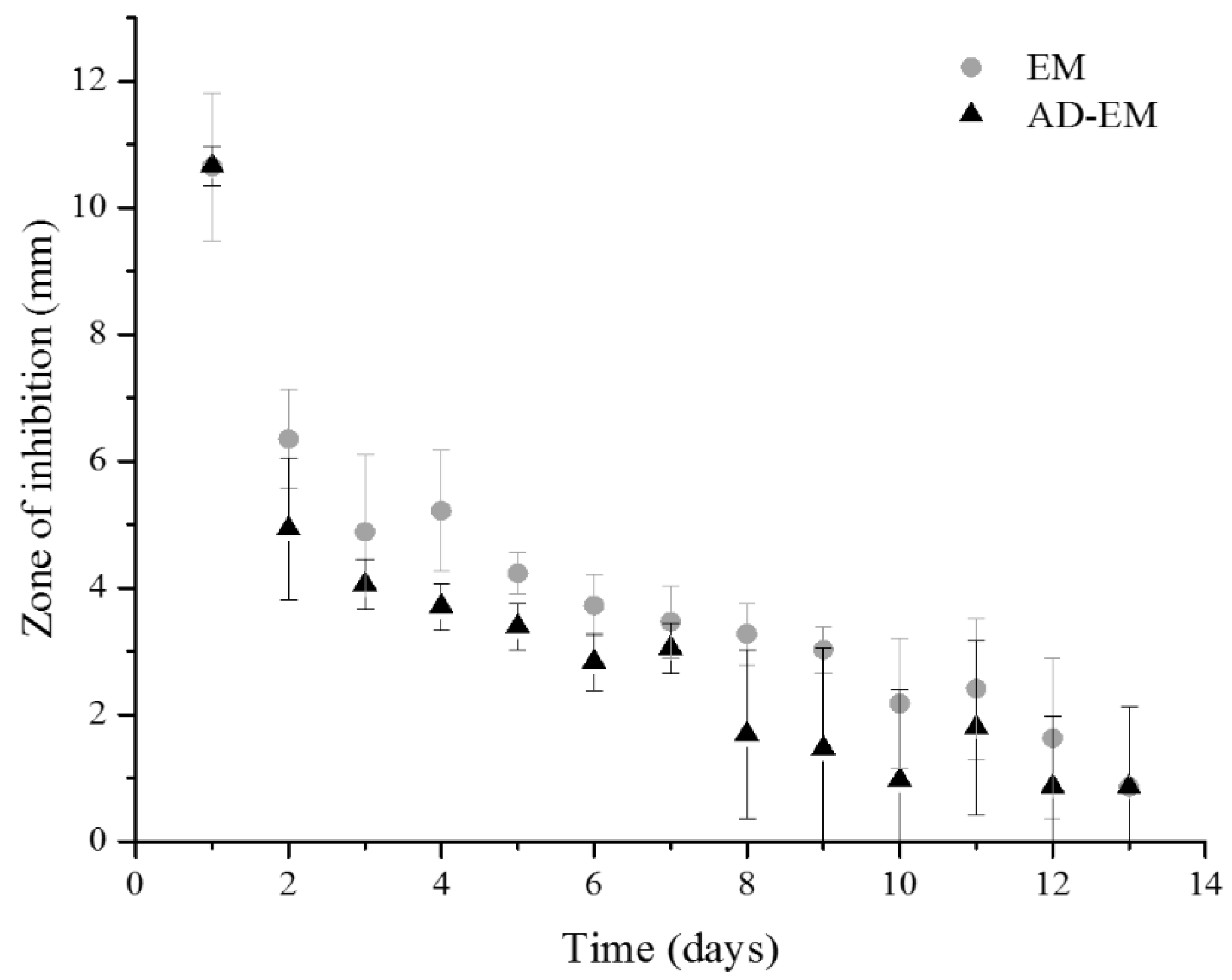
2.8. Zone of Inhibition Antibacterial Study
2.9. Bacterial Biofilm Penetration Studies
2.10. pH-Dependent Drug Release Study
3. Materials and Methods
3.1. Synthesis of Adamantane-Modified EM (AD-EM)
3.2. FTIR Study
3.3. 1H NMR of EM and AD-EM
3.4. MALDI of EM and AD-EM
3.5. Hydrophilic-Lipophilic Balance Calculations
3.6. Solubility Study
3.7. Acidic Conversion Spectral Scans
3.8. Synthesis of Insoluble Cyclodextrin Polymer Disks and Antibiotic Loading
3.9. Zone of Inhibition Antibacterial Study
3.10. Bacterial Biofilm Penetration Study
3.11. Proof-of-Concept Drug Release Study
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Alvarez-Elcoro, S.; Enzler, M.J. The macrolides: Erythromycin, clarithromycin, and azithromycin. Mayo Clin. Proc. 1999, 74, 613–634. [Google Scholar] [CrossRef] [PubMed]
- White, R.J. Why use erythromycin? Thorax 1994, 49, 944–945. [Google Scholar] [CrossRef] [PubMed]
- Braun, P. Hepatotoxicity of erythromycin. J. Infect. Dis. 1969, 119, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Hassanzadeh, A.; Barber, J.; Morris, G.A.; Gorry, P.A. Mechanism for the degradation of erythromycin A and erythromycin A 2′-ethyl succinate in acidic aqueous solution. J. Phys. Chem. A 2007, 111, 10098–10104. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, S.; Takiyama, K.; Fujiki, S.; Iwao, Y.; Miura, K.; Itai, S. Polymorphic transformation of antibiotic clarithromycin under acidic condition. J. Pharm. Sci. 2014, 103, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Zheng, T.; Nolan, E.M. Evaluation of (acyloxy)alkyl ester linkers for antibiotic release from siderophore-antibiotic conjugates. Bioorganic Med. Chem. Lett. 2015, 25, 4987–4991. [Google Scholar] [CrossRef] [PubMed]
- Mishra, N.M.; Briers, Y.; Lamberigts, C.; Steenackers, H.; Robijns, S.; Landuyt, B.; Vanderleyden, J.; Schoofs, L.; Lavigne, R.; Luyten, W.; et al. Evaluation of the antibacterial and antibiofilm activities of novel CRAMP-vancomycin conjugates with diverse linkers. Org. Biomol. Chem. 2015, 13, 7477–7486. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Xu, Y.; Liu, Y.; Chen, X.; Zhao, Z.; Lei, P. Synthesis of 4″-O-desosaminyl clarithromycin derivatives and their anti-bacterial activities. Bioorganic Med. Chem. Lett. 2013, 23, 6274–6279. [Google Scholar] [CrossRef] [PubMed]
- Lewandowski, E.M.; Skiba, J.; Torelli, N.J.; Rajnisz, A.; Solecka, J.; Kowalski, K.; Chen, Y. Antibacterial properties and atomic resolution X-ray complex crystal structure of a ruthenocene conjugated β-lactam antibiotic. Chem. Commun. 2015, 51, 6186–6189. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.A.; Panda, S.S.; Birs, A.S.; Serrano, J.C.; Gonzalez, C.F.; Alamry, K.A.; Katritzky, A.R. Synthesis and antibacterial evaluation of amino acid-antibiotic conjugates. Bioorganic Med. Chem. Lett. 2014, 24, 1856–1861. [Google Scholar] [CrossRef] [PubMed]
- Wanka, L.; Iqbal, K.; Schreiner, P.R. The lipophilic bullet hits the targets: Medicinal chemistry of adamantane derivatives. Chem. Rev. 2013, 113, 3516–3604. [Google Scholar] [CrossRef] [PubMed]
- Zefirova, O.N.; Nurieva, E.V.; Shishov, D.V.; Baskin, I.I.; Fuchs, F.; Lemcke, H.; Schrӧder, F.; Weiss, D.G.; Zefirov, N.S.; Kuznetsov, S.A. Synthesis and SAR requirements of adamantane-colchicine conjugates with both microtubule depolymerizing and tubulin clustering activities. Bioorganic Med. Chem. 2011, 19, 5529–5538. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.F.; Xu, X.D.; Zhang, J.; Yang, J.; Gong, Y.H.; Lei, Q.; Jia, H.Z.; Li, C.; Zhuo, R.X.; Zhang, X.Z. Encapsulation of an adamantane-doxorubicin prodrug in pH-responsive polysaccharide capsules for controlled release. ACS Appl. Mater. Interfaces 2012, 4, 5317–5324. [Google Scholar] [CrossRef] [PubMed]
- Cyphert, E.L.; Fu, A.S.; von Recum, H.A. Chemotherapeutic delivery using pH-responsive, affinity-based release. Exp. Biol. Med. 2017, 242, 692–699. [Google Scholar] [CrossRef] [PubMed]
- Kouatly, O.; Geronikaki, A.; Kamoutsis, C.; Hadjipavlou-Litina, D.; Eleftheriou, P. Adamantane derivatives of thiazolyl-N-substituted amide, as possible non-steroidal anti-inflammatory agents. Eur. J. Med. Chem. 2009, 44, 1198–1204. [Google Scholar] [CrossRef] [PubMed]
- Pagire, S.H.; Pagire, H.S.; Lee, G.B.; Han, S.J.; Kwak, H.J.; Kim, J.Y.; Kim, K.Y.; Rhee, S.D.; Ryu, J.I.; Song, J.S.; et al. Discovery and optimization of adamantane carboxylic acid derivatives as potent diacylglycerol acyltransferase 1 inhibitors for the potential treatment of obesity and diabetes. Eur. J. Med. Chem. 2015, 101, 716–735. [Google Scholar] [CrossRef] [PubMed]
- Creel, C.J.; Lovich, M.A.; Edelman, E.R. Arterial paclitaxel distribution and deposition. Circ. Res. 2000, 86, 879–884. [Google Scholar] [CrossRef] [PubMed]
- Copeland, R.A. The drug-target residence time model: A 10-year retrospective. Nat. Rev. Drug Discov. 2016, 15, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Fiese, E.F.; Steffen, S.H. Comparison of the acid stability of azithromycin and erythromycin A. J. Antimicrob. Chemother. 1990, 25, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Hill, D.R. 9-Hydrazone and 9-Azine Erythromycin Derivatives and a Process for Making the Same. U.S. Patent RE39,531 E, 27 March 2007. [Google Scholar]
- Whitman, M.S.; Tunkel, A.R. Azithromycin and clarithromycin: Overview and comparison with erythromycin. Infect. Control Hosp. Epidemiol. 1992, 13, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Pereira, J.M.; Mejia-Ariza, R.; Ilevbare, G.A.; McGettigan, H.E.; Sriranganathan, N.; Taylor, L.S.; Davis, R.M.; Edgar, K.J. Interplay of degradation, dissolution and stabilization of clarithromycin and its amorphous solid dispersions. Mol. Pharm. 2013, 10, 4640–4653. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-B.; Park, Y.-J.; Kang, C.-Y.; Lee, B.-J. Modulation of microenvironmental pH and utilization of alkalizers in crystalline solid dispersions for enhanced solubility and stability of clarithromycin. Arch. Pharm. Res. 2015, 38, 839–848. [Google Scholar] [CrossRef] [PubMed]
- Ogwal, S.T.U.X. Bioavailability and stability of erythromycin delayed release tablets. Afr. Health Sci. 2001, 1, 90–96. [Google Scholar] [PubMed]
- Zhang, H.; Wu, H.; Fan, L.; Li, F.; Gu, C.; Jia, M. Preparation and characteristics of pH-sensitive derivated dextran hydrogel nanoparticles. Polym. Compos. 2009, 30, 1243–1250. [Google Scholar] [CrossRef]
- Bronner, S.; Monteil, H.; Prévost, G. Regulation of virulence determinants in Staphylococcus aureus: Complexity and applications. FEMS Microbiol. Rev. 2004, 28, 183–200. [Google Scholar] [CrossRef] [PubMed]
- Granadero, D.; Bordello, J.; Perez-Alvite, M.J.; Novo, M.; Al-Soufi, W. Host-guest complexation studied fluorescence correlation spectroscopy: Adamantane-cyclodextrin inclusion. Int. J. Mol. Sci. 2010, 11, 173–188. [Google Scholar] [CrossRef] [PubMed]
- Belskaya, N.P.; Dehaen, W.; Bakulev, V.A. Synthesis and properties of hydrazones bearing amide, thioamide, and amidine functions. Online J. Org. Chem. 2010, 1, 275–332. [Google Scholar]
- Everett, J.R.; Tyler, J.W. An analysis of the 1H and 13C N.M.R. spectra of erythromycin a using two-dimensional methods. J. Chem. Soc. Perkin Trans. 1985, 1, 2599–2603. [Google Scholar] [CrossRef]
- Crisalli, P.; Hernández, A.R.; Kool, E.T. Fluorescence quenchers for hydrazone and oxime orthoganol bioconjugation. Bioconjug. Chem. 2012, 23, 1969–1980. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, C.A.; Kenar, J.A.; Price, N. Preparation of saturated and unsaturated fatty acid hydrazides and long chain C-glycoside ketohydrazones. Green Chem. 2010, 12, 2012–2018. [Google Scholar] [CrossRef]
- Kalia, J; Raines, R.T. Hydrolytic stability of hydrazones and oximes. Agnew. Chem. Int. Ed. Engl. 2008, 47, 7523–7526. [Google Scholar] [CrossRef] [PubMed]
- Szisz, D. HLB Predictor. ChemAxon Docs. 2015. Available online: https://docs.chemaxon.com/display/docs/HLB+Predictor (accessed on 11 November 2016).
- Van den Bossche, L.; Lodi, A.; Schaar, J.; Shaakov, S.; Zorzan, M.; Tranquillini, M.E.; Overballe-Petersen, C.; Hoogmartens, J.; Adams, E. An interlaboratory study on the suitability of a gradient LC-UV method as a compendial method for the determination of erythromycin and its related substances. J. Pharm. Biomed. Anal. 2010, 53, 109–112. [Google Scholar] [CrossRef] [PubMed]
- Ford, J.H.; Prescott, G.C.; Hinman, J.W.; Caron, E.L. Colorimetric determination of erythromycin. Anal. Chem. 1953, 25, 1195–1197. [Google Scholar] [CrossRef]
- Danielson, N.D.; Holeman, J.A.; Bristol, D.C.; Kirzner, D.H. Simple methods for the qualitative identification and quantitative determination of macrolide antibiotics. J. Pharm. Biomed. Anal. 1993, 11, 121–130. [Google Scholar] [CrossRef]
- Gallagher, P.A.; Danielson, N.D. Colorimetric determination of macrolide antibiotics using ferric ion. Talanta 1995, 42, 1425–1432. [Google Scholar] [CrossRef]
- Rivera-Delgado, E.; Ward, E.; von Recum, H.A. Providing sustained transgene induction through affinity-based drug delivery. J. Biomed. Mater. Res. A 2016, 104, 1135–1142. [Google Scholar] [CrossRef] [PubMed]
- Keller, B.O.; Li, L. Three-layer matrix/sample preparation method for MALDI MS analysis of low nanomolar protein samples. J. Am. Soc. Mass Spectrom. 2006, 17, 780–785. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.T. A quantitative kinetic theory of emulsion type, I. Physical chemistry of the emulsifying agent. In Proceedings of the Second International Congress of Surface Activity; Butterworths: London, UK, 1957; pp. 426–438. [Google Scholar]
- Guo, X.; Rong, Z.; Ying, X. Calculation of hydrophile-lipophile balance for polyethoxylated surfactants by group contribution method. J. Colloid Interface Sci. 2006, 298, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Griffin, W.C. Calculation of HLB values of non-ionic surfactants. J. Soc. Cosmet. Chem. 1954, 5, 249–256. [Google Scholar]
- Kabri, T.; Arab-Tehrany, E.; Belhaj, N.; Linder, M. Physico-chemical characterization of nano-emulsions in cosmetic matrix enriched on omega-3. J. Nanobiotechnol. 2011, 9. [Google Scholar] [CrossRef] [PubMed]
- Manna, P.K.; Kumaran, V.; Mohanta, G.P.; Manvalan, R. Preparation and evaluation of a new erythromycin derivative-erythromycin taurate. Acta Pharm. 2004, 54, 231–242. [Google Scholar] [PubMed]
- Varanda, F.; Pratas de Melo, M.J.; Caço, A.I.; Dohrn, R.; Makrydaki, F.A.; Voutsas, E.; Tassios, D.; Marrucho, I.M. Solubility of antibiotics in different solvents. 1. Hydrochloride forms of tetracycline, moxifloxacin, and ciprofloxacin. Ind. Eng. Chem. Res. 2006, 45, 6368–6374. [Google Scholar] [CrossRef]
- Thatiparti, T.R.; von Recum, H.A. Cyclodextrin complexation for affinity-based antibiotic delivery. Macromol. Biosci. 2010, 10, 82–90. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Solvent | EM (mg/mL) | AD-EM (mg/mL) |
|---|---|---|
| Water | 1.5 ± 0.2 | 0.33 ± 0.03 |
| Phosphate Buffered Saline (PBS), pH 7.4 | 1.6 ± 0.4 | 0.34 ± 0.01 |
| Acetate buffer, pH 5.0 | 15 | 1.39 ± 0.04 |
| Methanol | >40 | 7.3 ± 0.2 |
| Ethyl Acetate | >40 | >40 |
| Acetone | >40 | 37.5 |
| Drug Incubation Time (Hours) | AD-EM (% Control Colonies* Remaining) | EM (% Control Colonies* Remaining) |
|---|---|---|
| 1 | 62.7% ± 22.1% | 71.6% ± 40.2% |
| 7 | 14.8% ± 13.7% | 10.5% ± 6.8% |
| 24 | 11.7% ± 7.9% | 8.3% ± 6.6% |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cyphert, E.L.; Wallat, J.D.; Pokorski, J.K.; Von Recum, H.A. Erythromycin Modification That Improves Its Acidic Stability while Optimizing It for Local Drug Delivery. Antibiotics 2017, 6, 11. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics6020011
Cyphert EL, Wallat JD, Pokorski JK, Von Recum HA. Erythromycin Modification That Improves Its Acidic Stability while Optimizing It for Local Drug Delivery. Antibiotics. 2017; 6(2):11. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics6020011
Chicago/Turabian StyleCyphert, Erika L., Jaqueline D. Wallat, Jonathan K. Pokorski, and Horst A. Von Recum. 2017. "Erythromycin Modification That Improves Its Acidic Stability while Optimizing It for Local Drug Delivery" Antibiotics 6, no. 2: 11. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics6020011