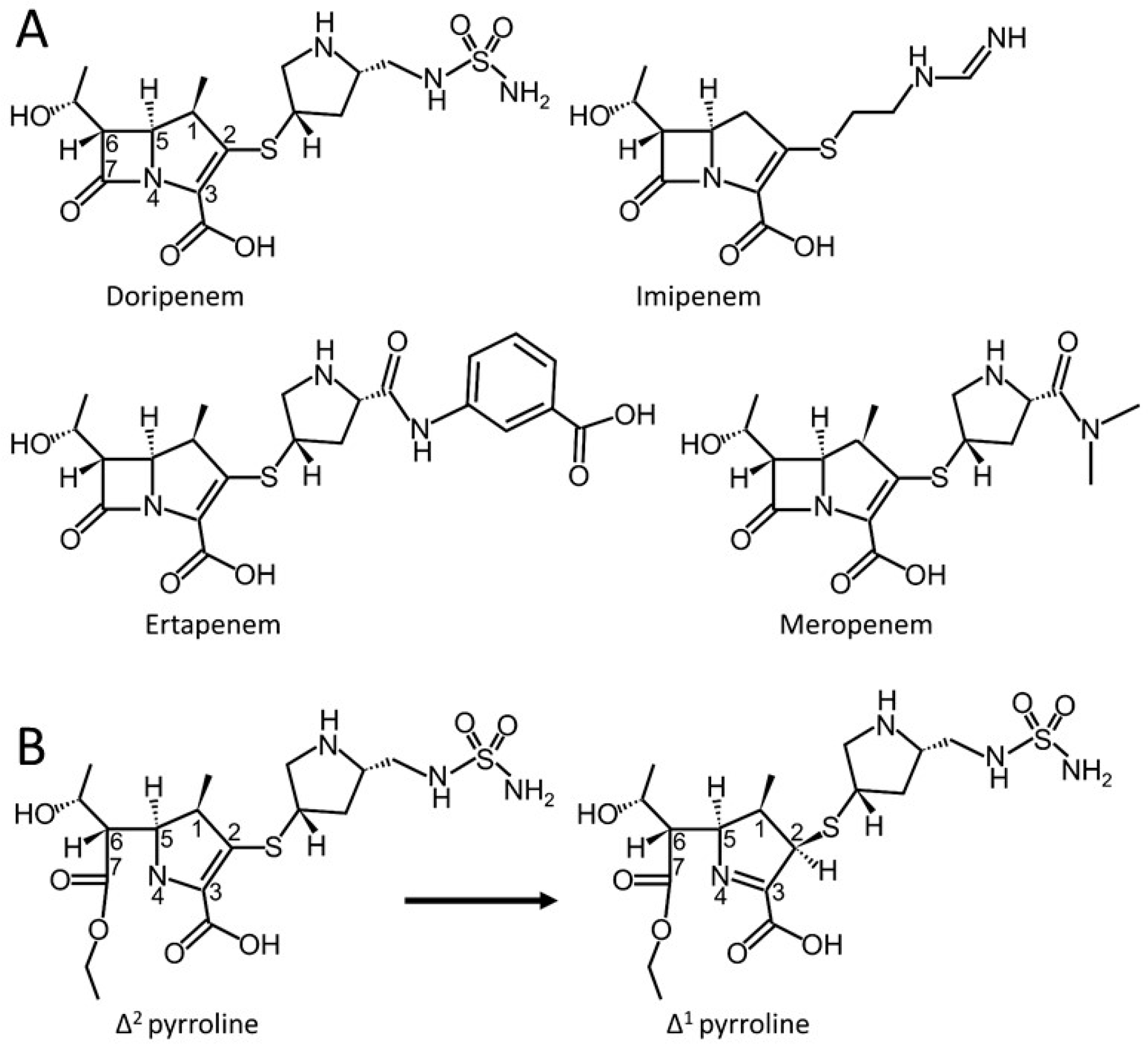
Structural Analysis of The OXA-48 Carbapenemase Bound to A “Poor” Carbapenem Substrate, Doripenem

Abstract
:1. Introduction
2. Materials and Methods
2.1. Cloning of blaOXA-48 K73A
2.2. Expression and Purification of the OXA-48 K73A Variant
2.3. Crystallography
3. Results and Discussion
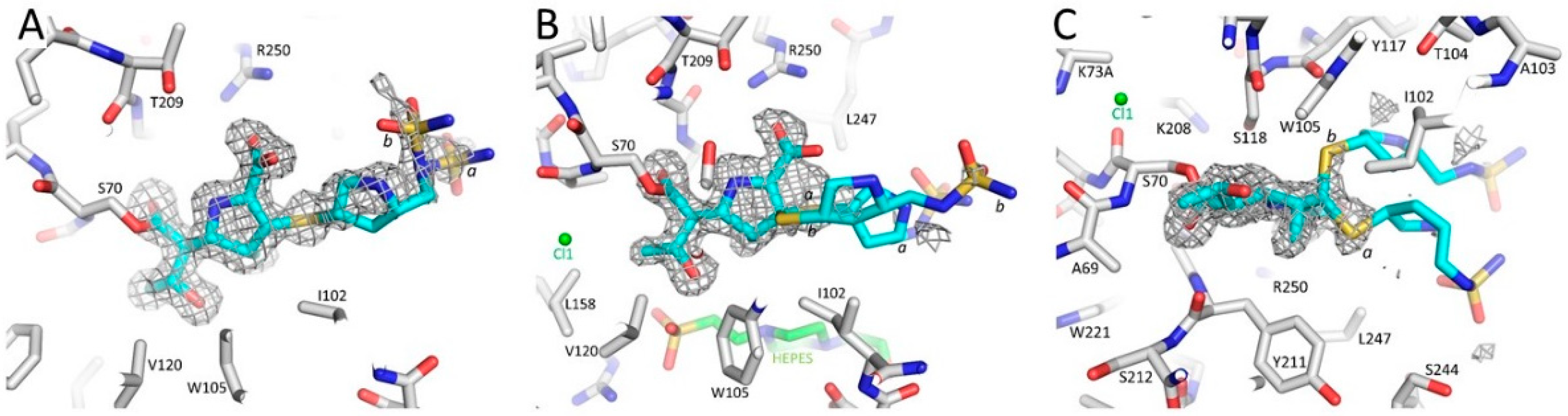
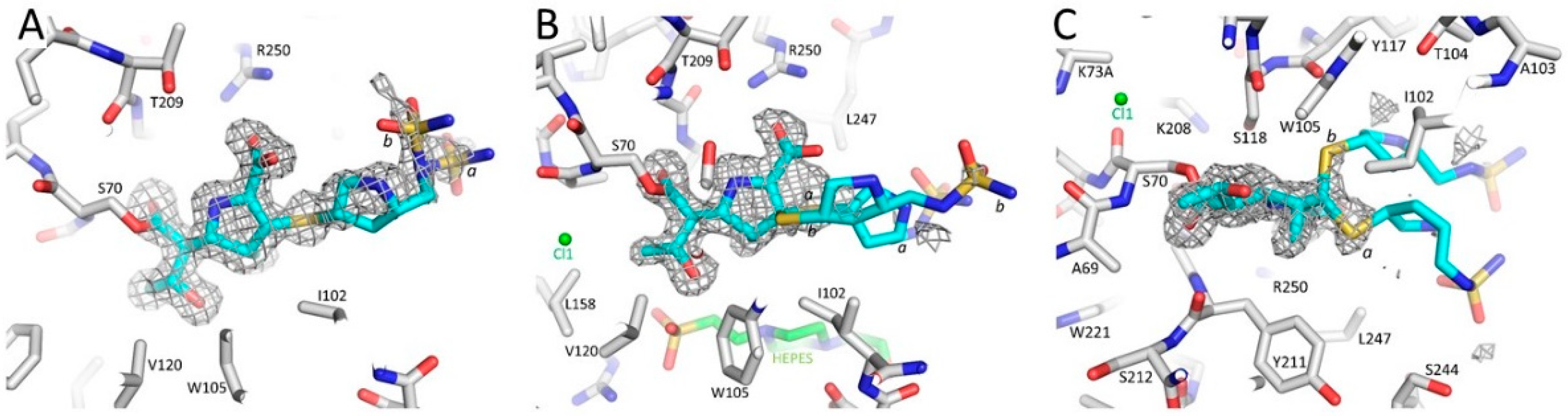
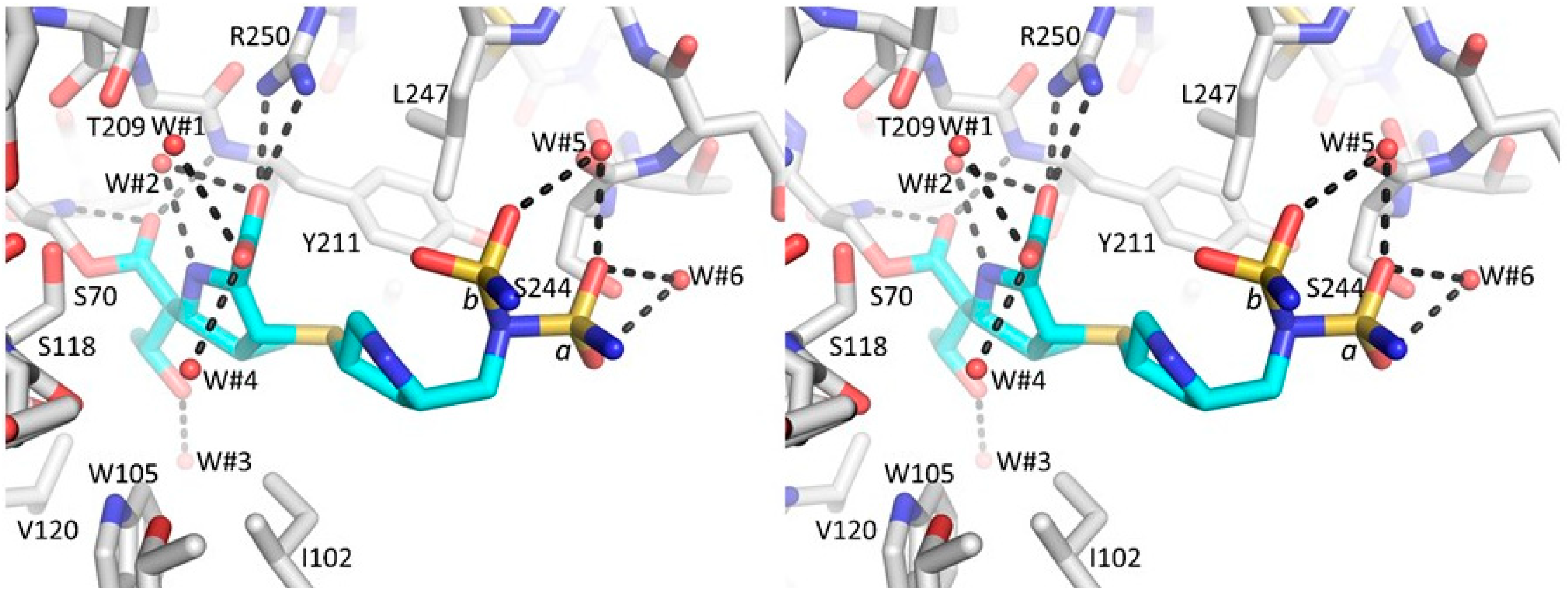
3.1. Summary of the OXA-48 K73A-Doripenem Crystal Structure
3.2. Major Interactions Observed Between Doripenem and Active Site Residues
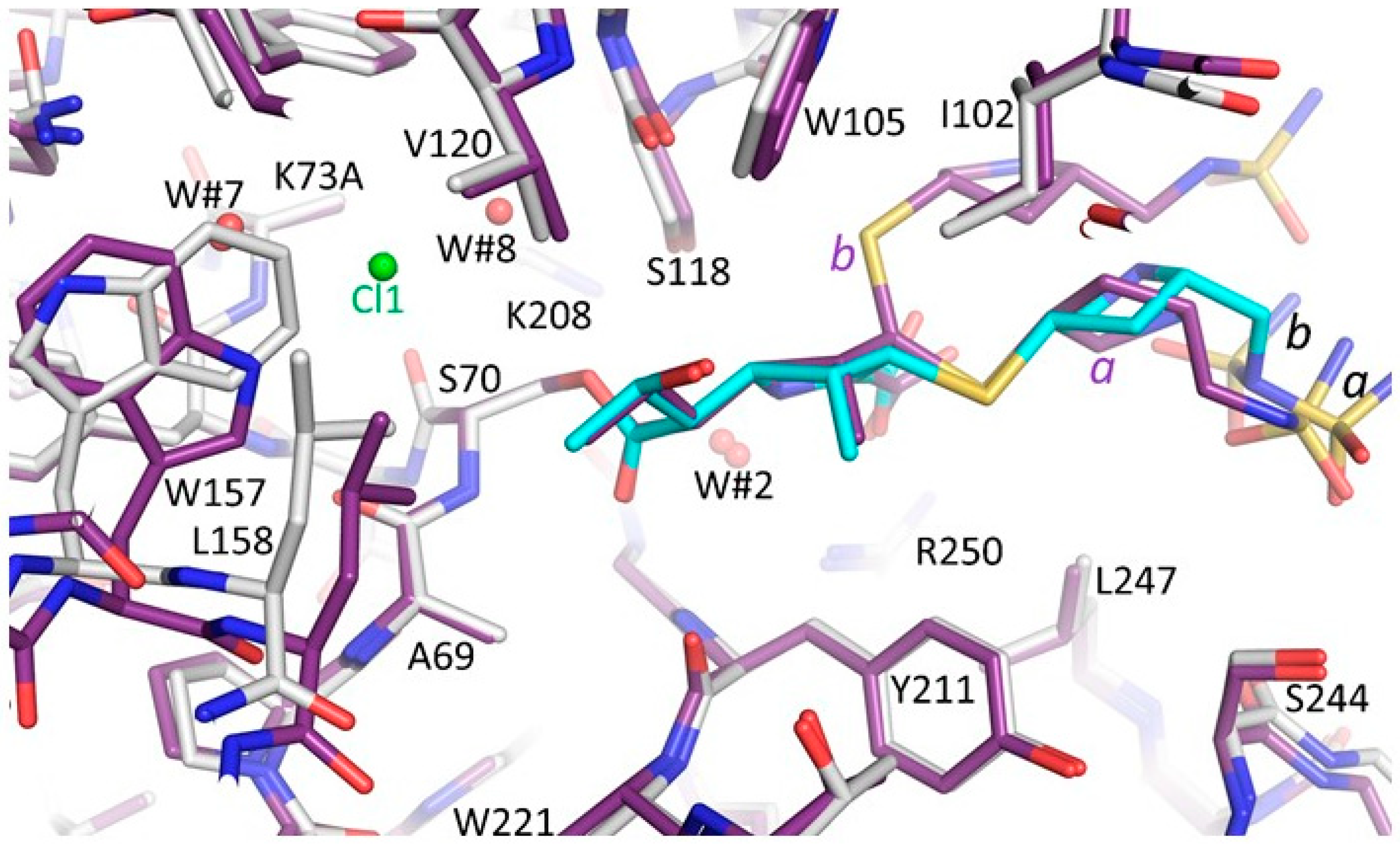
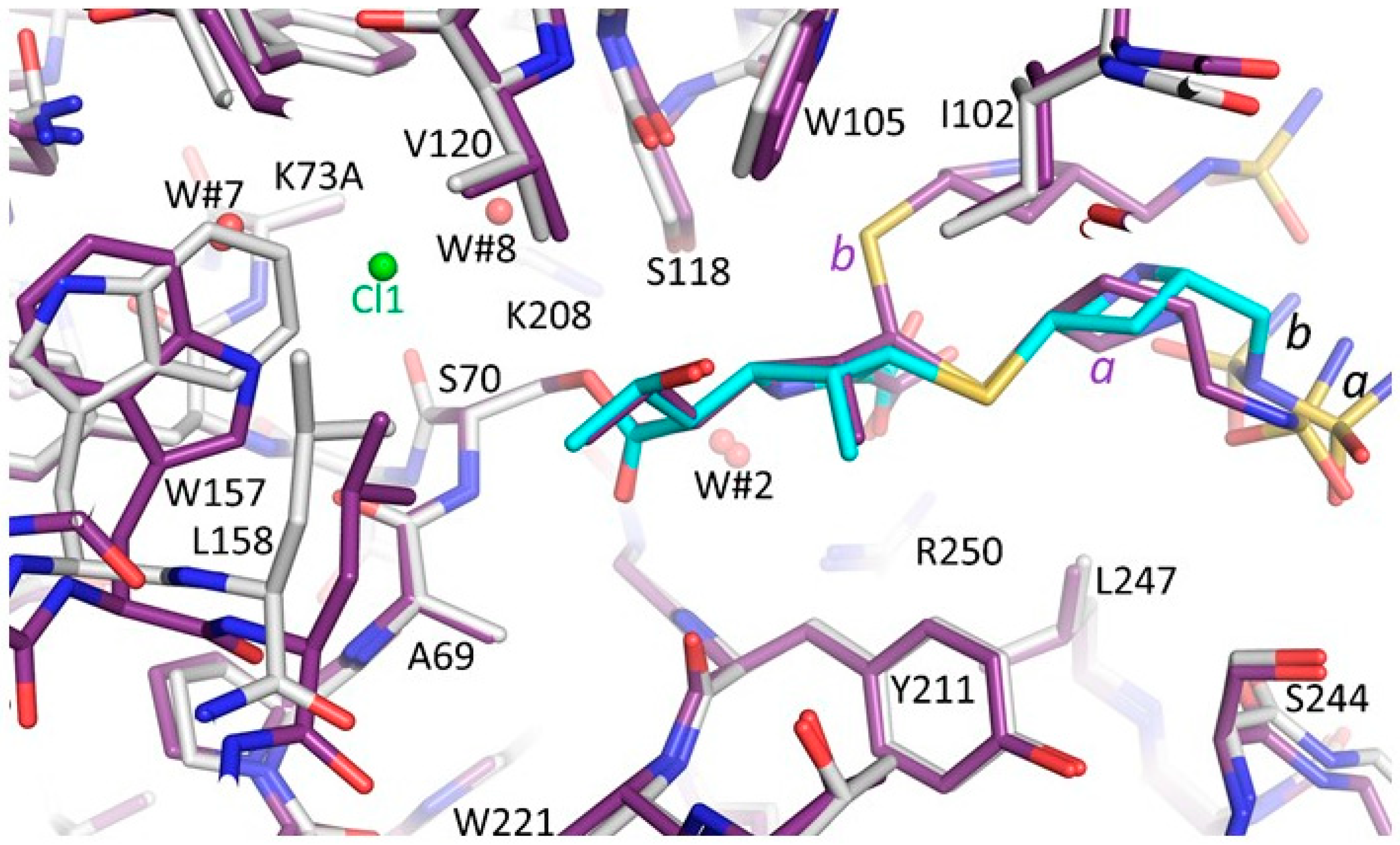
3.3. Comparison of the OXA-48 K73A-Doripenem Structure to the Wild-Type OXA-48-Doripenem and Wild-Type OXA-48-Imipenem Structures
3.4. Comparison of the OXA-48 K73A-Doripenem Structure to An OXA-51 CHDL Variant Bound to Doripenem
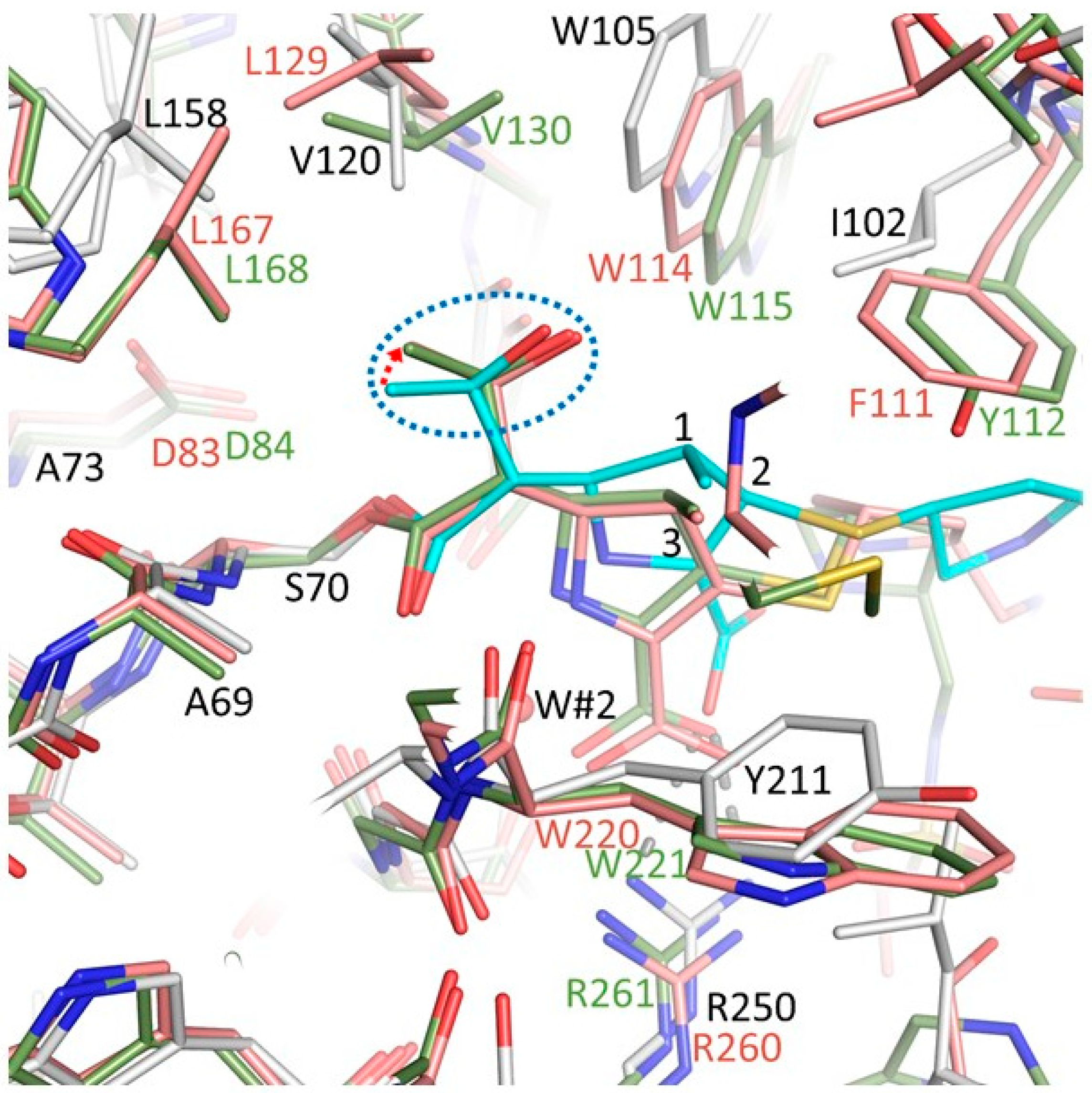
3.5. Comparisons of OXA-48 K73A-Doripenem Structure to OXA-23 and OXA-24/40 CHDL Variants Bound to Doripenem
3.6. Comparisons of OXA-48 K73A-Doripenem Structure to OXA-1, a Non-CHDL Bound to Doripenem
3.7. So Why is OXA-48 Unable to Hydrolyze Doripenem?
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cullmann, W.; Dick, W. Investigations on beta-lactamase stability of recently developed beta-lactam compounds: study of enzyme kinetics. Zentralbl Bakteriol Mikrobiol Hyg. A. 1983, 254, 413–422. [Google Scholar] [PubMed]
- Cartwright, S.J.; Waley, S.G. beta-Lactamase inhibitors. Med. Res. Rev. 1983, 3, 341–382. [Google Scholar] [CrossRef] [PubMed]
- Monks, J.; Waley, S.G. Imipenem as substrate and inhibitor of beta-lactamases. Biochem. J. 1988, 253, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Bethel, C.R.; Taracila, M.; Shyr, T.; Thomson, J.M.; Distler, A.M.; Hujer, K.M.; Hujer, A.M.; Endimiani, A.; Papp-Wallace, K.; Bonnet, R.; et al. Exploring the inhibition of CTX-M-9 by b-lactamase inhibitors and carbapenems. Antimicrob. Agents Chemother. 2011, 55, 3465–3475. [Google Scholar] [CrossRef] [PubMed]
- Papp-Wallace, K.M.; Endimiani, A.; Taracila, M.A.; Bonomo, R.A. Carbapenems: past, present, and future. Antimicrob. Agents Chemother. 2011, 55, 4943–4960. [Google Scholar] [CrossRef] [PubMed]
- Moellering, R.C., Jr.; Eliopoulos, G.M.; Sentochnik, D.E. The carbapenems: new broad spectrum beta-lactam antibiotics. J. Antimicrob. Chemother. 1989, 24, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Charnas, R.L.; Knowles, J.R. Inhibition of the RTEM beta-lactamase from Escherichia coli. Interaction of enzyme with derivatives of olivanic acid. Biochemistry 1981, 20, 2732–2737. [Google Scholar] [CrossRef] [PubMed]
- Easton, C.J.; Knowles, J.R. Inhibition of the RTEM beta-lactamase from Escherichia coli. Interaction of the enzyme with derivatives of olivanic acid. Biochemistry 1982, 21, 2857–2862. [Google Scholar] [CrossRef]
- Smith, C.A.; Antunes, N.T.; Stewart, N.K.; Toth, M.; Kumarasiri, M.; Chang, M.; Mobashery, S.; Vakulenko, S.B. Structural basis for carbapenemase activity of the OXA-23 beta-lactamase from Acinetobacter baumannii. Chem. Boil. 2013, 20, 1107–1115. [Google Scholar] [CrossRef]
- Stewart, N.K.; Smith, C.A.; Antunes, N.T.; Toth, M.; Vakulenko, S.B. Role of the Hydrophobic Bridge in the Carbapenemase Activity of Class D beta-Lactamases. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef]
- Global priority list of antibiotic-resistant bacteria to guide research, discovery, and development of new antibiotics. World Health Organization. Available online: https://www.who.int/medicines/publications/WHO-PPL-Short_Summary_25Feb-ET_NM_WHO.pdf. (accessed on 8 August 2019).
- Codjoe, F.S.; Donkor, E.S. Carbapenem Resistance: A Review. Med. Sci. (Basel) 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Papp-Wallace, K.M.; Bonomo, R.A. New beta-Lactamase Inhibitors in the Clinic. Infect Dis. Clin. N. Am. 2016, 30, 441–464. [Google Scholar] [CrossRef] [PubMed]
- Docquier, J.D.; Mangani, S. Structure-Function Relationships of Class D Carbapenemases. Curr. Drug Targets 2016, 17, 1061–1071. [Google Scholar] [CrossRef] [PubMed]
- Schneider, K.D.; Ortega, C.J.; Renck, N.A.; Bonomo, R.A.; Powers, R.A.; Leonard, D.A. Structures of the class D carbapenemase OXA-24 from Acinetobacter baumannii in complex with doripenem. J. Mol. Biol. 2011, 406, 583–594. [Google Scholar] [CrossRef] [PubMed]
- June, C.M.; Muckenthaler, T.J.; Schroder, E.C.; Klamer, Z.L.; Wawrzak, Z.; Powers, R.A.; Szarecka, A.; Leonard, D.A. The structure of a doripenem-bound OXA-51 class D beta-lactamase variant with enhanced carbapenemase activity. Protein sci. 2016, 25, 2152–2163. [Google Scholar] [CrossRef] [PubMed]
- Docquier, J.D.; Calderone, V.; De Luca, F.; Benvenuti, M.; Giuliani, F.; Bellucci, L.; Tafi, A.; Nordmann, P.; Botta, M.; Rossolini, G.M.; et al. Crystal structure of the OXA-48 beta-lactamase reveals mechanistic diversity among class D carbapenemases. Chem. Biol. 2009, 16, 540–547. [Google Scholar] [CrossRef] [PubMed]
- Bakthavatchalam, Y.D.; Anandan, S.; Veeraraghavan, B. Laboratory Detection and Clinical Implication of Oxacillinase-48 like Carbapenemase: The Hidden Threat. J. Glob. Infect. Dis. 2016, 8, 41–50. [Google Scholar] [PubMed]
- Poirel, L.; Potron, A.; Nordmann, P. OXA-48-like carbapenemases: the phantom menace. J. Antimicrob. Chemother. 2012, 67, 1597–1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oueslati, S.; Nordmann, P.; Poirel, L. Heterogeneous hydrolytic features for OXA-48-like beta-lactamases. J. Antimicrob. Chemother. 2015, 70, 1059–1063. [Google Scholar]
- Poirel, L.; Heritier, C.; Tolun, V.; Nordmann, P. Emergence of oxacillinase-mediated resistance to imipenem in Klebsiella pneumoniae. Antimicrob. Agents Chemother. 2004, 48, 15–22. [Google Scholar] [CrossRef]
- Mimoz, O.; Gregoire, N.; Poirel, L.; Marliat, M.; Couet, W.; Nordmann, P. Broad-spectrum beta-lactam antibiotics for treating experimental peritonitis in mice due to Klebsiella pneumoniae producing the carbapenemase OXA-48. Antimicrob. Agents Chemother. 2012, 56, 2759–2760. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.; Harris, P.; Henderson, A.; Paterson, D. Treatment of Infections by OXA-48-Producing Enterobacteriaceae. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef] [PubMed]
- Kasap, M.; Torol, S.; Kolayli, F.; Dundar, D.; Vahaboglu, H. OXA-162, a novel variant of OXA-48 displays extended hydrolytic activity towards imipenem, meropenem and doripenem. J. Enzyme Inhib. Med. Chem. 2013, 28, 990–996. [Google Scholar] [CrossRef] [PubMed]
- Stojanoski, V.; Chow, D.C.; Fryszczyn, B.; Hu, L.; Nordmann, P.; Poirel, L.; Sankaran, B.; Prasad, B.V.; Palzkill, T. Structural Basis for Different Substrate Profiles of Two Closely Related Class D beta-Lactamases and Their Inhibition by Halogens. Biochemistry 2015, 54, 3370–3380. [Google Scholar] [CrossRef] [PubMed]
- Poirel, L.; Castanheira, M.; Carrer, A.; Rodriguez, C.P.; Jones, R.N.; Smayevsky, J.; Nordmann, P. OXA-163, an OXA-48-related class D beta-lactamase with extended activity toward expanded-spectrum cephalosporins. Antimicrob. Agents Chemother. 2011, 55, 2546–2551. [Google Scholar] [CrossRef] [PubMed]
- De Luca, F.; Benvenuti, M.; Carboni, F.; Pozzi, C.; Rossolini, G.M.; Mangani, S.; Docquier, J.D. Evolution to carbapenem-hydrolyzing activity in noncarbapenemase class D beta-lactamase OXA-10 by rational protein design. Proc. Natl. Acad. Sci. USA 2011, 108, 18424–18429. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.A.; Stewart, N.K.; Toth, M.; Vakulenko, S.B. Structural Insights into the Mechanism of Carbapenemase Activity of the OXA-48 beta-Lactamase. Antimicrob. Agents Chemother. 2019. [Google Scholar] [CrossRef]
- Papp-Wallace, K.M.; Becka, S.A.; Taracila, M.A.; Winkler, M.L.; Gatta, J.A.; Rholl, D.A.; Schweizer, H.P.; Bonomo, R.A. Exposing a beta-Lactamase "Twist": the Mechanistic Basis for the High Level of Ceftazidime Resistance in the C69F Variant of the Burkholderia pseudomallei PenI beta-Lactamase. Antimicrob. Agents Chemother. 2016, 60, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. (Ed.) Acta Crystallographica. Section D, Biological Crystallography; IUCr/Wiley: Chester, England, 2010; Volume 66, Pt 2, pp. 125–132. [Google Scholar]
- Vagin, A.; Teplyakov, A. Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 2010, 66 Pt 1, 22–25. [Google Scholar] [CrossRef]
- Emsley, P.; Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60 Pt 12 Pt 1, 2126–2132. [Google Scholar] [CrossRef]
- Akhter, S.; Lund, B.A.; Ismael, A.; Langer, M.; Isaksson, J.; Christopeit, T.; Leiros, H.S.; Bayer, A. A focused fragment library targeting the antibiotic resistance enzyme - Oxacillinase-48: Synthesis, structural evaluation and inhibitor design. Eur. J. Med. Chem. 2018, 145, 634–648. [Google Scholar] [CrossRef] [PubMed]
- Liebschner, D.; Afonine, P.V.; Moriarty, N.W.; Poon, B.K.; Sobolev, O.V.; Terwilliger, T.C.; Adams, P.D. Polder maps: improving OMIT maps by excluding bulk solvent. Acta. Crystallogr D. Struct. Biol. 2017, 73, 148–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, J.M.; Leonard, D.A. Common clinical substitutions enhance the carbapenemase activity of OXA-51-like class D beta-lactamases from Acinetobacter spp. Antimicrob. Agents Chemother. 2014, 58, 7015–7016. [Google Scholar] [CrossRef] [PubMed]
- Harper, T.M.; June, C.M.; Taracila, M.A.; Bonomo, R.A.; Powers, R.A.; Leonard, D.A. Multiple substitutions lead to increased loop flexibility and expanded specificity in Acinetobacter baumannii carbapenemase OXA-239. Biochem. J. 2018, 475, 273–288. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.M.; Clasman, J.R.; June, C.M.; Kaitany, K.C.; LaFleur, J.R.; Taracila, M.A.; Klinger, N.V.; Bonomo, R.A.; Wymore, T.; Szarecka, A.; et al. Structural basis of activity against aztreonam and extended spectrum cephalosporins for two carbapenem-hydrolyzing class D beta-lactamases from Acinetobacter baumannii. Biochemistry 2015, 54, 1976–1987. [Google Scholar] [CrossRef] [PubMed]
- King, D.T.; King, A.M.; Lal, S.M.; Wright, G.D.; Strynadka, N.C. Molecular mechanism of avibactam mediated beta-lactamase inhibition. ACS Infect. Dis. 2015, 1, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Schneider, K.D.; Karpen, M.E.; Bonomo, R.A.; Leonard, D.A.; Powers, R.A. The 1.4 A crystal structure of the class D beta-lactamase OXA-1 complexed with doripenem. Biochemistry 2009, 48, 11840–11847. [Google Scholar] [CrossRef] [PubMed]
- Buchman, J.S.; Schneider, K.D.; Lloyd, A.R.; Pavlish, S.L.; Leonard, D.A. Site-saturation mutagenesis of position V117 in OXA-1 beta-lactamase: effect of side chain polarity on enzyme carboxylation and substrate turnover. Biochemistry 2012, 51, 3143–3150. [Google Scholar] [CrossRef] [PubMed]
- Dabos, L.; Bogaerts, P.; Bonnin, R.A.; Zavala, A.; Sacre, P.; Iorga, B.I.; Huang, D.T.; Glupczynski, Y.; Naas, T. Genetic and Biochemical Characterization of OXA-519, a Novel OXA-48-Like beta-Lactamase. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef]
- Chen, T.L.; Lee, Y.T.; Kuo, S.C.; Hsueh, P.R.; Chang, F.Y.; Siu, L.K.; Ko, W.C.; Fung, C.P. Emergence and Distribution of Plasmids Bearing the blaOXA-51-like gene with an upstream ISAba1 in carbapenem-resistant Acinetobacter baumannii isolates in Taiwan. Antimicrob. Agents Chemother. 2010, 54, 4575–4581. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Collection | OXA-48 K73A Doripenem Complex |
|---|---|
| Space group | P21212 |
| Unit cell dimensions (Å, °) | 105.77 125.09 45.12 90 90 90 |
| Wavelength (Å) | 0.97946 |
| Resolution (Å) | 40.38-1.50 (1.53-1.50) |
| Redundancy | 13.2 (12.0) |
| Unique reflections | 96,537 (4,704) |
| <I>/<σ(I)> | 19.6 (5.0) |
| Mn(I) half-set correlation CC(1/2) | 0.99 (0.96) |
| Rmerge (%) | 7.7 (48.5) |
| Completeness (%) | 99.8 (99.7) |
| Refinement | |
| Resolution range (Å) | 40.42-1.50 |
| R-factor (%) | 15.1 |
| Rfree (%) | 17.2 |
| Estimated coordinate error ESU from Rfree (Å) | 0.06 |
| Number of protein atoms | 4030 (2 molecules in the asymmetric unit) |
| Number of water molecules | 603 |
| Ligands (number of atoms) | 2 doripenem (28 atoms each), 1 partial HEPES molecule (15 atoms), 2 chloride ions, 5 ethylene glycol molecules (4 atoms each) |
| Real-space CC of ligands | |
| Doripenem, mol A; conformation 1, 2 | 0.96, 0.96 |
| Doripenem mol B; conformation 1, 2 | 0.91, 0.92 |
| HEPES | 0.94 |
| Chloride ions | 0.99, 0.99 |
| RMSD deviation from ideality | |
| Bond length (Å) | 0.011 |
| Bond angles (°) | 1.74 |
| Ramachandran plot statistics (%) | |
| Preferred regions | 96.4 |
| Allowed regions | 3.6 |
| Outliers | 0.0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papp-Wallace, K.M.; Kumar, V.; Zeiser, E.T.; Becka, S.A.; van den Akker, F. Structural Analysis of The OXA-48 Carbapenemase Bound to A “Poor” Carbapenem Substrate, Doripenem. Antibiotics 2019, 8, 145. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics8030145
Papp-Wallace KM, Kumar V, Zeiser ET, Becka SA, van den Akker F. Structural Analysis of The OXA-48 Carbapenemase Bound to A “Poor” Carbapenem Substrate, Doripenem. Antibiotics. 2019; 8(3):145. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics8030145
Chicago/Turabian StylePapp-Wallace, Krisztina M., Vijay Kumar, Elise T. Zeiser, Scott A. Becka, and Focco van den Akker. 2019. "Structural Analysis of The OXA-48 Carbapenemase Bound to A “Poor” Carbapenem Substrate, Doripenem" Antibiotics 8, no. 3: 145. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics8030145