Comparative Transcriptome Analysis of Streptomyces Clavuligerus in Response to Favorable and Restrictive Nutritional Conditions

 , and
, and
Abstract
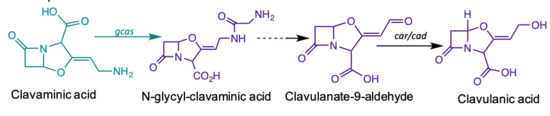
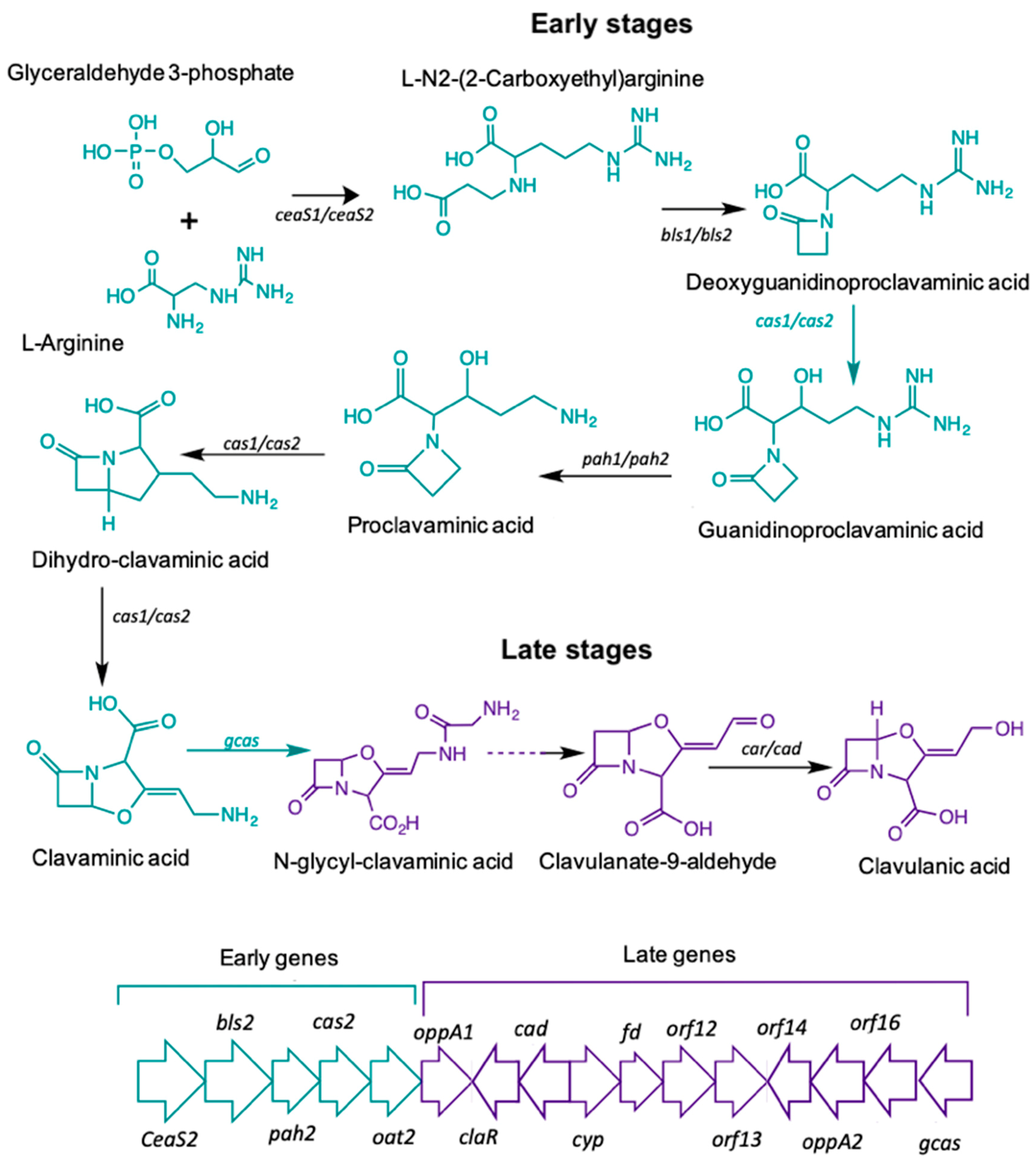
:
1. Introduction
2. Results
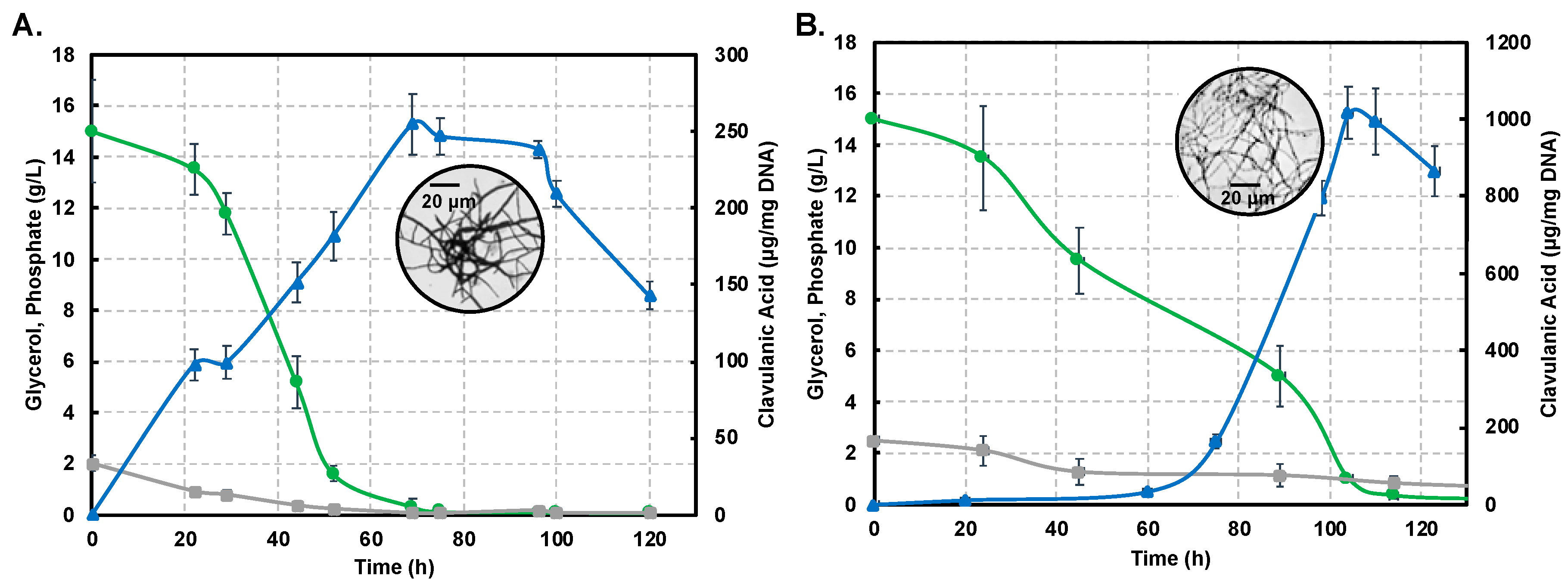
2.1. Shake-Flask Cultivations Under Different Culture Conditions
2.2. Identification of Differentially Expressed Genes Between SB-M and GSPG Conditions
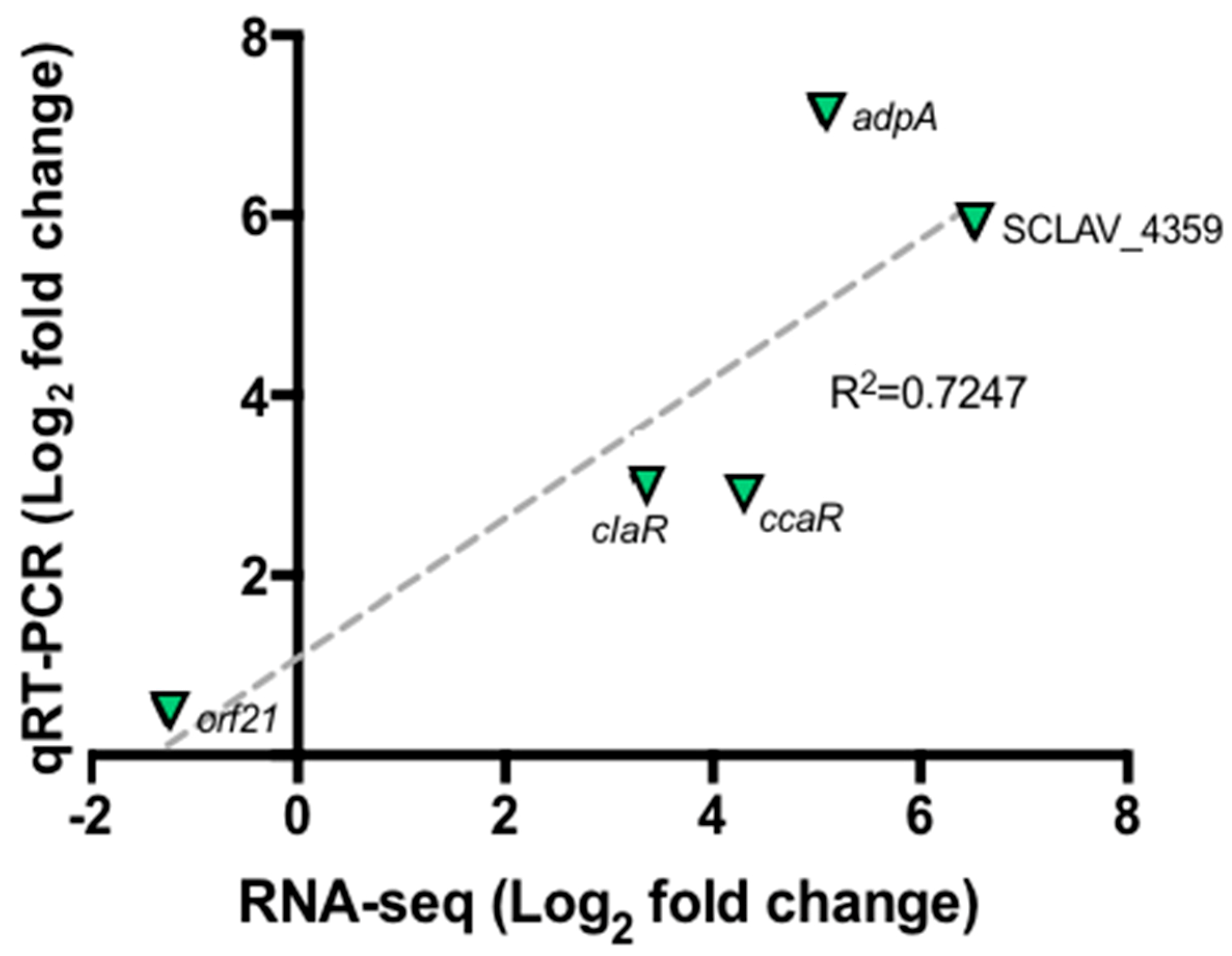
2.3. Validation of Differentially Expressed Genes by Quantitative qRT-PCR
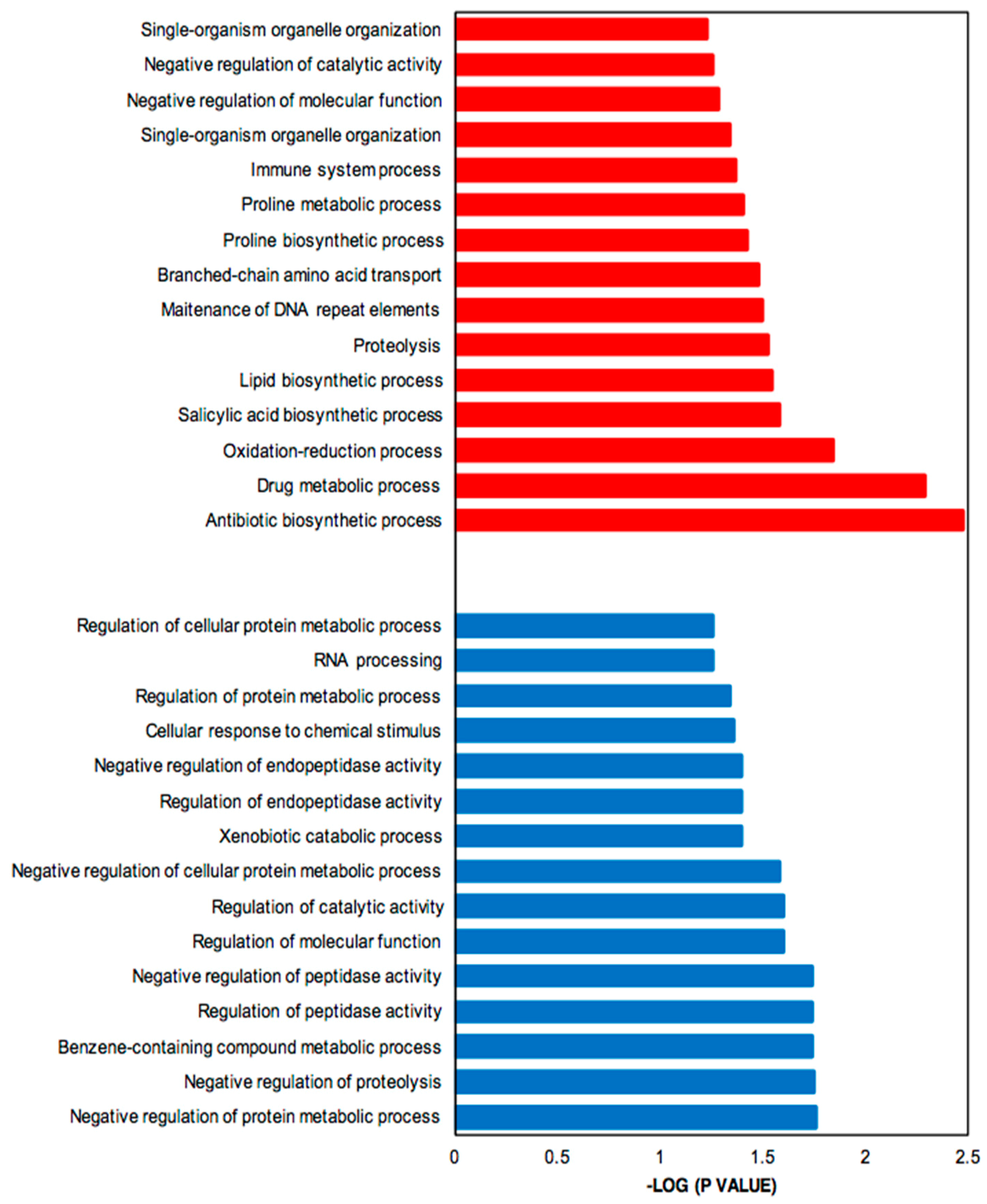
2.4. GO Term Enrichment Analysis
3. Discussion
4. Materials and Methods
4.1. Bacterial Strain and Cultivation
4.2. Analytical Techniques
4.3. RNA Extraction, Library Preparation, and Sequencing
4.4. Reads Mapping to Reference Genome and Differential Expression Analysis
4.5. RNA-seq Raw Sequencing Data Accession Information
4.6. Validation of Differentially Expressed Genes by Quantitative qRT-PCR
4.7. GO Term Enrichment Analysis of Differentially Expressed Genes
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ser, H.L.; Law, J.W.F.; Chaiyakunapruk, N.; Jacob, S.A.; Palanisamy, U.D.; Chan, K.G.; Goh, B.H.; Lee, L.H. Fermentation conditions that affect clavulanic acid production in Streptomyces clavuligerus: A systematic review. Front. Microbiol. 2016, 7, 522. [Google Scholar] [CrossRef] [PubMed]
- Liras, P.; Gomez-Escribano, J.P.; Santamarta, I. Regulatory mechanisms controlling antibiotic production in Streptomyces clavuligerus. J. Ind. Microbiol. Biotechnol. 2008, 35, 667–676. [Google Scholar] [CrossRef]
- Saudagar, P.S.; Survase, S.A.; Singhal, R.S. Clavulanic acid: A review. Biotechnol. Adv. 2008, 26, 335–351. [Google Scholar] [CrossRef] [PubMed]
- Reading, C.; Cole, M. Clavulanic acid: A beta-lactamase-inhiting beta-lactam from Streptomyces clavuligerus. Antimicrob. Agents Chemother. 1977, 11, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-H.; Yang, B.; Ren, J.; Dong, M.-L.; Liang, D.; Xu, A.-L. Optimization of medium composition for the production of clavulanic acid by Streptomyces clavuligerus. Process. Biochem. 2005, 40, 1161–1166. [Google Scholar] [CrossRef]
- Gouveia, E.R.; Baptista-Neto, A.; Badino, A.C.; Hokka, C.O. Optimisation of medium composition for clavulanic acid production by Streptomyces clavuligerus. Biotechnol. Lett. 2001, 23, 157–161. [Google Scholar] [CrossRef]
- Higo, A.; Hara, H.; Horinouchi, S.; Ohnishi, Y. Genome-wide distribution of AdpA, a global regulator for secondary metabolism and morphological differentiation in streptomyces, revealed the extent and complexity of the AdpA regulatory network. DNA Res. 2012, 19, 259–273. [Google Scholar] [CrossRef]
- Tahlan, K.; Anders, C.; Jensen, S.E. The paralogous pairs of genes involved in clavulanic acid and clavam metabolite biosynthesis are differently regulated in Streptomyces clavuligerus. J. Bacteriol. 2004, 186, 6286–6297. [Google Scholar] [CrossRef]
- Paradkar, A. Clavulanic acid production by Streptomyces clavuligerus: Biogenesis, regulation and strain improvement. J. Antibiot. (Tokyo) 2013, 66, 411–420. [Google Scholar] [CrossRef]
- Vivancos, A.P.; Guell, M.; Dohm, J.C.; Serrano, L.; Himmelbauer, H. Strand-specific deep sequencing of the transcriptome. Genome Res. 2010, 20, 989–999. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, S.C.A.; Hokka, C.O.; Badino, A.C. Utilization of soybean derivatives on clavulanic acid production by Streptomyces clavuligerus. Enzyme Microb. Technol. 2007, 40, 1071–1077. [Google Scholar] [CrossRef]
- Kirk, S.; Avignone-rossa, C.A.; Bushell, M.E. Growth limiting substrate affects antibiotic production and associated metabolic fluxes in Streptomyces clavuligerus. Biotechnol. Lett. 2000, 22, 1803–1809. [Google Scholar] [CrossRef]
- Medema, M.H.; Alam, M.T.; Breitling, R.; Takano, E. The future of industrial antibiotic production: From random mutagenesis to synthetic biology. Bioeng. Bugs 2011, 2, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Malule, H.; Junne, S.; Nicolás Cruz-Bournazou, M.; Neubauer, P.; Ríos-Estepa, R. Streptomyces clavuligerus shows a strong association between TCA cycle intermediate accumulation and clavulanic acid biosynthesis. Appl. Microbiol. Biotechnol. 2018, 102, 4009–4023. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Malule, H.; Restrepo, A.; Cardona, W.; Junne, S.; Neubauer, P.; Rios-Estepa, R. Inversion of the stereochemical configuration (3S, 5S)-clavaminic acid into (3R, 5R)-clavulanic acid: A computationally-assisted approach based on experimental evidence. J. Theor. Biol. 2016, 395, 40–50. [Google Scholar] [CrossRef]
- Toro, L.; Pinilla, L.; Avignone-Rossa, C.; Ríos-Estepa, R. An enhanced genome-scale metabolic reconstruction of Streptomyces clavuligerus identifies novel strain improvement strategies. Bioprocess Biosyst. Eng. 2018, 41, 657–669. [Google Scholar] [CrossRef]
- Ünsaldı, E.; Kurt-Kızıldoğan, A.; Voigt, B.; Becher, D.; Özcengiz, G. Proteome-wide alterations in an industrial clavulanic acid producing strain of Streptomyces clavuligerus. Synth. Syst. Biotechnol. 2017, 2, 39–48. [Google Scholar] [CrossRef]
- Rapaport, F.; Khanin, R.; Liang, Y.; Pirun, M.; Krek, A.; Zumbo, P.; Mason, C.E.; Socci, N.D.; Betel, D. Comprehensive evaluation of differential gene expression analysis methods for RNA-seq data. Genome Biol. 2013, 14, R95. [Google Scholar] [CrossRef]
- Gouveia, E.R.; Baptista-Neto, A.; Azevedo, A.G.; Badino, A.C.; Hokka, C.O. Improvement of clavulanic acid production by Streptomyces clavuligerus in medium containing soybean derivatives. World J. Microbiol. Biotechnol. 1999, 15, 623–627. [Google Scholar] [CrossRef]
- Pinilla, L.; Toro, L.F.; Avignone-Rossa, C.; Peñuela, M.; Rios-Estepa, R. Streptomyces clavuligerus strain selection for clavulanic acid biosynthesis: A study based on culture composition effects and statistical analysis. DYNA 2018, 85, 111–118. [Google Scholar] [CrossRef]
- Rioseras, B.; López-García, M.T.; Yagüe, P.; Sánchez, J.; Manteca, Á. Mycelium differentiation and development of Streptomyces coelicolor in lab-scale bioreactors: Programmed cell death, differentiation, and lysis are closely linked to undecylprodigiosin and actinorhodin production. Bioresour. Technol. 2014, 151, 191–198. [Google Scholar] [CrossRef]
- Denser Pamboukian, C.R.; Guimarães, L.M.; Facciotti, M.C.R. Applications of image analysis in the characterization of Streptomyces olindensis in submerged culture. Braz. J. Microbiol. 2002, 33, 17–21. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Burgo, Y.; Álvarez-Álvarez, R.; Rodríguez-García, A.; Liras, P. The Pathway-Specific Regulator ClaR of Streptomyces clavuligerus Has a Global Effect on the Expression of Genes for Secondary Metabolism and Differentiation. Appl. Environ. Microbiol. 2015, 81, 6637–6648. [Google Scholar] [CrossRef] [Green Version]
- Bignell, D.R.D.; Tahlan, K.; Colvin, K.R.; Jensen, S.E.; Leskiw, B.K. Expression of ccaR, encoding the positive activator of cephamycin C and clavulanic acid production in Streptomyces clavuligerus, is dependent on bldG. Antimicrob. Agents Chemother. 2005, 49, 1529–1541. [Google Scholar] [CrossRef]
- Álvarez-Álvarez, R.; Rodríguez-García, A.; Martínez-Burgo, Y.; Martín, J.F.; Liras, P. Transcriptional studies on a Streptomyces clavuligerus oppA2 deletion mutant: N -acetylglycyl-clavaminic acid is an intermediate of clavulanic acid biosynthesis. Appl. Environ. Microbiol. 2018, 84, e01701–e01718. [Google Scholar] [CrossRef]
- Tahlan, K.; Park, H.U.; Wong, A.; Beatty, P.H.; Jensen, S.E. Two sets of paralogous genes encode the enzymes involved in the early stages of clavulanic acid and clavam metabolite biosynthesis in Streptomyces clavuligerus. Antimicrob. Agents Chemother. 2004, 48, 930–939. [Google Scholar] [CrossRef]
- Rodríguez-García, A.; de la Fuente, A.; Pérez-Redondo, R.; Martín, J.F.; Liras, P. Characterization and expression of the arginine biosynthesis gene cluster of Streptomyces clavuligerus. J. Mol. Microbiol. Biotechnol. 2000, 2, 543–550. [Google Scholar]
- Kuo-Cheng, C.; Yun-Huin, L.; Jane-Yii, W.; Sz-Chwun, J.H. Enhancement of clavulanic acid production in Streptomyces clavuligerus with ornithine feeding. Enzyme Microb. Technol. 2003, 32, 152–156. [Google Scholar]
- Marsh, A.J.; O’Sullivan, O.; Ross, R.P.; Cotter, P.D.; Hill, C. In silico analysis highlights the frequency and diversity of type 1 lantibiotic gene clusters in genome sequenced bacteria. BMC Genom. 2010, 11, 679. [Google Scholar] [CrossRef]
- Imran, S.; Shaaban, K.A.; Hasnain, S. Identification, isolation and optimization of antifungal metabolites from the Streptomyces malachitofuscus CTF9. Braz. J. Microbiol. 2011, 42, 592–604. [Google Scholar]
- Freiberg, J.A.; Le Breton, Y.; Tran, B.Q.; Scott, A.J.; Harro, J.M.; Ernst, R.K.; Goo, Y.A.; Mongodin, E.F.; Goodlett, D.R.; McIver, K.S.; et al. Global analysis and comparison of the transcriptomes and proteomes of group A Streptococcus biofilms. mSystems 2016, 1, e00149-16. [Google Scholar] [CrossRef]
- Yagüe, P.; Rodríguez-García, A.; López-García, M.T.; Martín, J.F.; Rioseras, B.; Sánchez, J.; Manteca, A. Transcriptomic analysis of Streptomyces coelicolor differentiation in solid sporulating cultures: First compartmentalized and second multinucleated mycelia have different and distinctive transcriptomes. PLoS ONE 2013, 8, e60665. [Google Scholar] [CrossRef]
- Hesketh, A.; Bucca, G.; Laing, E.; Flett, F.; Hotchkiss, G.; Smith, C.P.; Chater, K.F. New pleiotropic effects of eliminating a rare tRNA from Streptomyces coelicolor, revealed by combined proteomic and transcriptomic analysis of liquid cultures. BMC Genom. 2007, 8, 261. [Google Scholar] [CrossRef]
- Castro-Melchor, M.; Charaniya, S.; Karypis, G.; Takano, E.; Hu, W.S. Genome-wide inference of regulatory networks in Streptomyces coelicolor. BMC Genom. 2010, 11, 578. [Google Scholar] [CrossRef]
- Alvarez-Álvarez, R.; Rodríguez-García, A.; Santamarta, I.; Pérez-Redondo, R.; Prieto-Domínguez, A.; Martínez-Burgo, Y.; Liras, P. Transcriptomic analysis of Streptomyces clavuligerus ΔccaR:tsr: Effects of the cephamycin C-clavulanic acid cluster regulator CcaR on global regulation. Microb. Biotechnol. 2014, 7, 221–231. [Google Scholar] [CrossRef]
- Kato, J.; Suzuki, A.; Yamazaki, H.; Ohnishi, Y.; Horinouchi, S. Control by A-factor of a metalloendopeptidase gene involved in aerial mycelium formation in Streptomyces griseus. J. Bacteriol. 2002, 184, 6016–6025. [Google Scholar] [CrossRef]
- Robles-Reglero, V.; Santamarta, I.; Álvarez-Álvarez, R.; Martín, J.F.; Liras, P. Transcriptional analysis and proteomics of the holomycin gene cluster in overproducer mutants of Streptomyces clavuligerus. J. Biotechnol. 2013, 163, 69–76. [Google Scholar] [CrossRef]
- Medema, M.H.; Alam, M.T.; Heijne, W.H.M.; van den Berg, M.A.; Müller, U.; Trefzer, A.; Bovenberg, R.A.L.; Breitling, R.; Takano, E. Genome-wide gene expression changes in an industrial clavulanic acid overproduction strain of Streptomyces clavuligerus. Microb. Biotechnol. 2011, 4, 300–305. [Google Scholar] [CrossRef]
- Maranesi, G.L.; Baptista-Neto, A.; Hokka, C.O.; Badino, A.C. Utilization of vegetable oil in the production of clavulanic acid by Streptomyces clavuligerus ATCC 27064. World J. Microbiol. Biotechnol. 2005, 21, 509–514. [Google Scholar] [CrossRef]
- Hodgson, D.A. Primary metabolism and its control in streptomycetes: A most unusual group of bacteria. Adv. Microb. Physiol. 2000, 42, 47–238. [Google Scholar]
- Ives, P.R.; Bushell, M.E. Manipulation of the physiology of clavulanic acid production in Streptomyces clavuligerus. Microbiology 1997, 143, 3573–3579. [Google Scholar] [CrossRef] [Green Version]
- Belmar-Beiny, M.T.; Thomas, C.R. Morphology and clavulanic acid production of Streptomyces clavuligerus: Effect of stirrer speed in batch fermentations. Biotechnol. Bioeng. 1991, 37, 456–462. [Google Scholar] [CrossRef]
- Kim, Y.-M.; Kim, J. Formation and dispersion of mycelial pellets of Streptomyces coelicolor A3(2). J. Microbiol. 2004, 42, 64–67. [Google Scholar]
- Kyung, Y.S.; Hu, W.S.; Sherman, D.H. Analysis of temporal and spatial expression of the CcaR regulatory element in the cephamycin C biosynthetic pathway using green fluorescent protein. Mol. Microbiol. 2001, 40, 530–541. [Google Scholar] [CrossRef] [Green Version]
- Medema, M.H.; Trefzer, A.; Kovalchuk, A.; Van Den Berg, M.; Müller, U.; Heijne, W.; Wu, L.; Alam, M.T.; Ronning, C.M.; Nierman, W.C.; et al. The sequence of a 1.8-Mb bacterial linear plasmid reveals a rich evolutionary reservoir of secondary metabolic pathways. Genome Biol. Evol. 2010, 2, 212–224. [Google Scholar] [CrossRef]
- Zhou, Z.; Gu, J.; Du, Y.-L.; Li, Y.-Q.; Wang, Y. The -omics era- toward a systems-level understanding of Streptomyces. Curr. Genom. 2011, 12, 404–416. [Google Scholar] [CrossRef]
- Borukhov, S.; Severinov, K. Role of the RNA polymerase sigma subunit in transcription initiation. Res. Microbiol. 2002, 153, 557–562. [Google Scholar] [CrossRef]
- Jnawali, H.N.; Liou, K.; Sohng, J.K. Role of σ-factor (orf21) in clavulanic acid production in Streptomyces clavuligerus NRRL3585. Microbiol. Res. 2011, 166, 369–379. [Google Scholar] [CrossRef]
- Santos, C.L.; Correia-Neves, M.; Moradas-Ferreira, P.; Mendes, M.V. A walk into the LuxR regulators of Actinobacteria: Phylogenomic distribution and functional diversity. PLoS ONE 2012, 7, e46758. [Google Scholar] [CrossRef]
- Jensen, S.E. Biosynthesis of clavam metabolites. J. Ind. Microbiol. Biotechnol. 2012, 39, 1407–1419. [Google Scholar] [CrossRef]
- Ozcengiz, G.; Demain, A.L. Recent advances in the biosynthesis of penicillins, cephalosporins and clavams and its regulation. Biotechnol. Adv. 2013, 31, 287–311. [Google Scholar] [CrossRef]
- López-García, M.T.; Santamarta, I.; Liras, P. Morphological differentiation and clavulanic acid formation are affected in a Streptomyces clavuligerus adpA-deleted mutant. Microbiology 2010, 156, 2354–2365. [Google Scholar] [CrossRef] [Green Version]
- Akanuma, G.; Hara, H.; Ohnishi, Y.; Horinouchi, S. Dynamic changes in the extracellular proteome caused by absence of a pleiotropic regulator AdpA in Streptomyces griseus. Mol. Microbiol. 2009, 73, 898–912. [Google Scholar] [CrossRef]
- Banbula, A.; Potempa, J.; Travis, J.; Fernandez-Catalán, C.; Mann, K.; Huber, R.; Bode, W.; Medrano, F. Amino-acid sequence and three-dimensional structure of the Staphylococcus aureus metalloproteinase at 1.72 A resolution. Structure 1998, 6, 1185–1193. [Google Scholar] [CrossRef]
- Meeske, A.J.; Rodrigues, C.D.A.; Brady, J.; Lim, H.C.; Bernhardt, T.G.; Rudner, D.Z. High-Throughput Genetic Screens Identify a Large and Diverse Collection of New Sporulation Genes in Bacillus subtilis. PLoS Biol. 2016, 14, e1002341. [Google Scholar] [CrossRef]
- Kim, D.W.; Kang, S.G.; Kim, I.S.; Lee, B.K.; Rho, Y.T.; Lee, K.J. Proteases and protease inhibitors produced in streptomycetes and their roles in morphological differentiation. J. Microbiol. Biotechnol. 2006, 16, 5–14. [Google Scholar]
- BLAST Nucleotide BLAST: Search Nucleotide Databases Using a Nucleotide Query. Available online: https://blast.ncbi.nlm.nih.gov/Blast.cgi (accessed on 12 June 2019).
- Ramos, J.L.; Martinez-Bueno, M.; Molina-Henares, A.J.; Teran, W.; Watanabe, K.; Zhang, X.; Gallegos, M.T.; Brennan, R.; Tobes, R. The TetR family of transcriptional repressors. Microbiol. Mol. Biol. Rev. 2005, 69, 326–356. [Google Scholar] [CrossRef]
- Ferguson, N.L.; Peña-Castillo, L.; Moore, M.A.; Bignell, D.R.D.; Tahlan, K. Proteomics analysis of global regulatory cascades involved in clavulanic acid production and morphological development in Streptomyces clavuligerus. J. Ind. Microbiol. Biotechnol. 2016, 43, 537–555. [Google Scholar] [CrossRef]
- Shepherd, M.D.; Kharel, M.K.; Bosserman, M.A.; Rohr, J. Laboratory maintenance of Streptomyces species. Curr. Protoc. Microbiol. 2010, 18, 10E.1.1–10E.1.8. [Google Scholar]
- Romero, J.; Liras, P.; Martín, J.F. Dissociation of cephamycin and clavulanic acid biosynthesis in Streptomyces clavuligerus. Appl. Microbiol. Biotechnol. 1984, 20, 318–325. [Google Scholar] [CrossRef]
- Gómez-Ríos, D.; Ramírez-Malule, H.; Neubauer, P.; Junne, S.; Ríos-Estepa, R. Degradation kinetics of clavulanic acid in fermentation broths at low temperatures. Antibiotics 2019, 8, 6. [Google Scholar] [CrossRef]
- Nakamura, G.R. Microdetermination of Phosphorus. Anal. Chem. 1952, 24, 1372. [Google Scholar] [CrossRef]
- Bok, S.H.; Demain, A.L. An improved colorimetric assay for polyols. Anal. Biochem. 1977, 81, 18–20. [Google Scholar] [CrossRef]
- Pederson, T. Use of diphenylamine as a colorimetric reagent for ribonucleic acid. Anal. Biochem. 1969, 28, 35–46. [Google Scholar] [CrossRef]
- Trapnell, C.; Hendrickson, D.G.; Sauvageau, M.; Goff, L.; Rinn, J.L.; Pachter, L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 2013, 31, 46–53. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 8 May 2018).
- HannonLab FASTX toolkit. Available online: http://hannonlab.cshl.edu/fastx_toolkit/ (accessed on 4 January 2017).
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Bauer, S.; Grossmann, S.; Vingron, M.; Robinson, P.N. Ontologizer 2.0—A multifunctional tool for GO term enrichment analysis and data exploration. Bioinformatics 2008, 24, 1650–1651. [Google Scholar] [CrossRef]
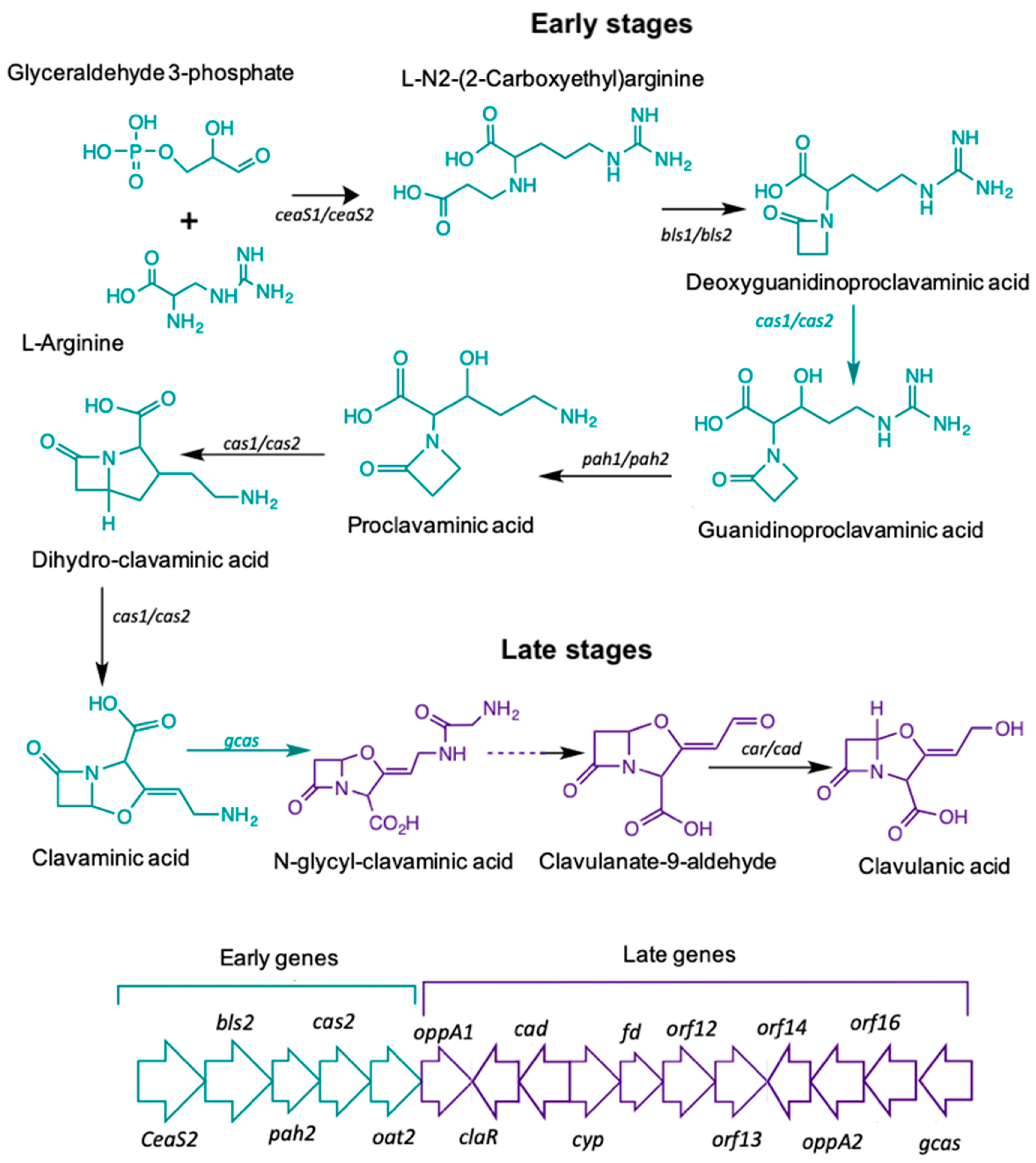

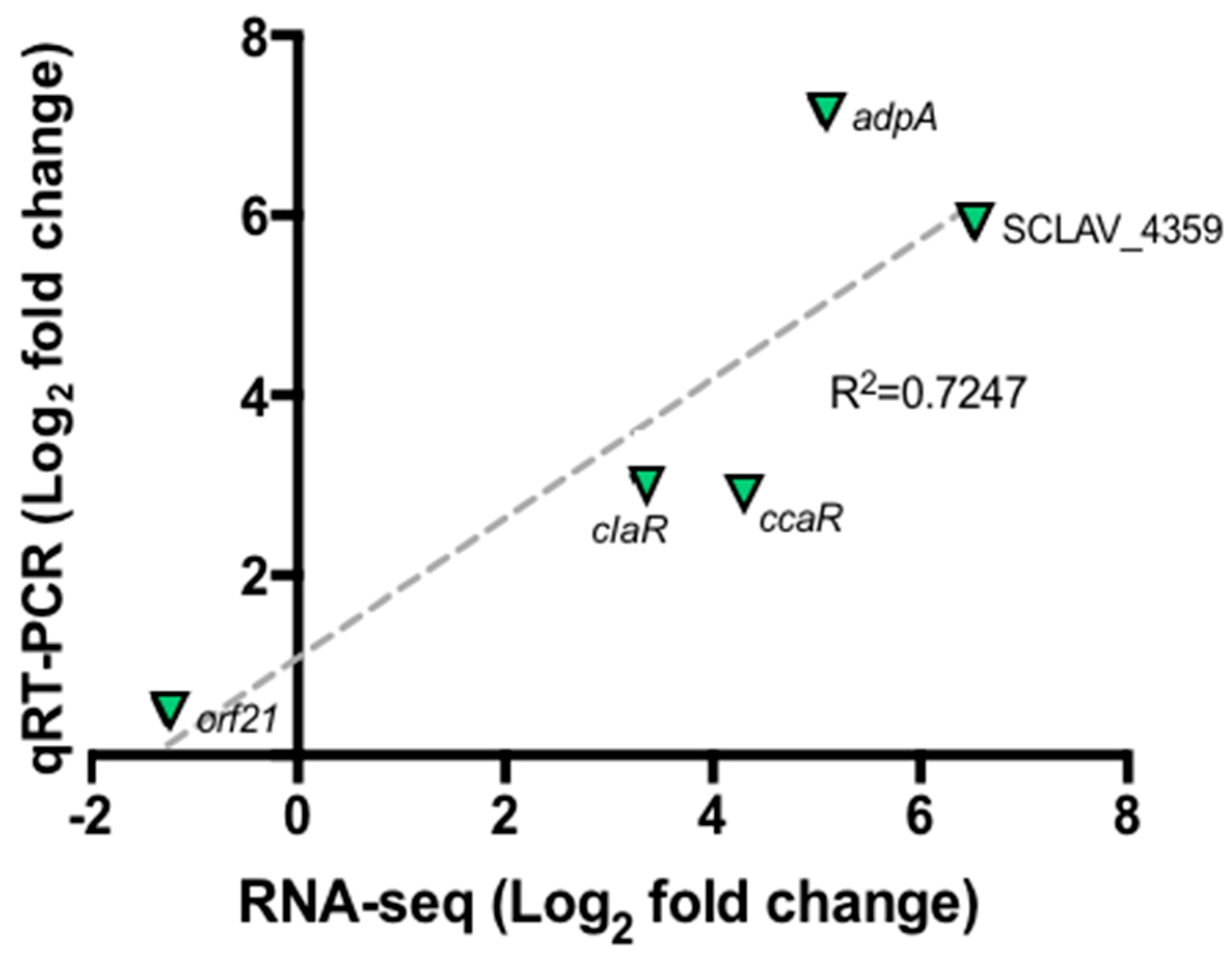


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID | Protein Name | log FC | p-Value | FDR |
|---|---|---|---|---|
| Up-regulated genes in SB-M condition | ||||
| Clavulanic acid biosynthesis | ||||
| SCLAV_4194 | Clavaminate synthase 2 (cas2) | 4.388 | 7.210 × 10−9 | 1.090 × 10−6 |
| SCLAV_4181 | N-glycyl-clavaminic acid synthetase (gcas) | 3.694 | 6.070 × 10−7 | 4.890 × 10−5 |
| SCLAV_4191 | Transcriptional activator (claR) | 3.393 | 2.960 × 10−5 | 1.307 × 10−3 |
| SCLAV_4195 | Proclavaminate amidinohydrolase (pah) | 3.303 | 5.560 × 10−6 | 3.266 × 10−4 |
| SCLAV_4196 | Carboxyethyl-arginine beta-lactam-synthase (bls) | 2.850 | 1.975 × 10−4 | 6.227 × 10−3 |
| SCLAV_4190 | Clavaldehyde dehydrogenase (car) | 2.739 | 5.920 × 10−5 | 2.268 × 10−3 |
| SCLAV_4197 | Carboxyethylarginine synthase (ceas) | 2.556 | 1.492 × 10−4 | 4.988 × 10−3 |
| Cephamycin C biosynthesis | ||||
| SCLAV_4206 | Deacetoxycephalosporin C hydroxylase (cefF) | 5.875 | 1.030 × 10−11 | 3.460 × 10−9 |
| SCLAV_4208 | Cephalosporin hydroxylase (cmcI) | 5.400 | 6.900 × 10−12 | 2.610 × 10−9 |
| SCLAV_4211 | Deacetoxycephalosporin C synthetase (DAOC) | 5.104 | 3.480 × 10−11 | 1.050 × 10−8 |
| SCLAV_4205 | 3′-hydroxymethylcephem-O-carbamoyltransferase (cmcH) | 4.947 | 2.180 × 10−10 | 5.280 × 10−8 |
| SCLAV_4204 | Positive regulator (ccaR) | 4.226 | 5.870 × 10−9 | 9.330 × 10−7 |
| SCLAV_4199 | Isopenicillin N synthetase (IPNS) | 4.106 | 1.050 × 10−8 | 1.550 × 10−6 |
| SCLAV_4201 | L-lysine-epsilon aminotransferase (lat) | 4.015 | 3.810 × 10−7 | 3.390 × 10−5 |
| SCLAV_4207 | 7-alpha-cephem-methoxylase P8 chain (cmcJ) | 3.274 | 1.060 × 10−5 | 5.652 × 10−4 |
| SCLAV_4200 | N-5-amino-5-carboxypentanoyl-L-cysteinyl-D-valine synthase (pcbAB) | 3.504 | 5.750 × 10−7 | 4.710 × 10−5 |
| SCLAV_4210 | Isopenicillin N epimerase (cefD) | 3.225 | 5.050 × 10−5 | 2.031 × 10−3 |
| Miscellaneous genes | ||||
| SCLAV_4359 | Neutral zinc metalloprotease | 8.313 | 1.630 × 10−19 | 9.840 × 10−16 |
| SCLAV_4112 | Extracellular small neutral protease | 6.589 | 6.580 × 10−14 | 6.640 × 10−11 |
| SCLAV_4455 | Beta-lactamase inhibitory protein | 5.316 | 5.960 × 10−10 | 1.240 × 10−7 |
| SCLAV_4202 | BLP (Beta-Lactamase Inhibitory Protein) | 5.227 | 2.420 × 10−10 | 5.640 × 10−8 |
| SCLAV_p1319 | Putative transcriptional regulator AraC family | 5.113 | 2.340 × 10−9 | 4.080 × 10−7 |
| SCLAV_2754 | RNA polymerase sigma factor | 4.943 | 5.760 × 10−5 | 2.247 × 10−3 |
| SCLAV_p1007 | Beta-lactamase domain protein | 4.755 | 2.820 × 10−9 | 4.740 × 10−7 |
| SCLAV_4723 | Beta-lactamase inhibitory protein | 4.694 | 1.720 × 10−8 | 2.480 × 10−6 |
| SCLAV_p0769 | Sigma factor, σ70 type, group 4 | 4.637 | 2.243 × 10−4 | 6.856 × 10−3 |
| SCLAV_3577 | NADH-quinone oxidoreductase subunit N | 4.603 | 6.800 × 10−6 | 7.350 × 10−6 |
| SCLAV_p1321 | LysR family transcriptional regulator | 4.238 | 1.840 × 10−7 | 1.790 × 10−5 |
| SCLAV_4189 | Cytochrome P450-SU2 (cyp) | 3.523 | 5.630 × 10−6 | 3.277 × 10−4 |
| SCLAV_4464 | Transcriptional regulator, LuxR family | 3.086 | 4.304 × 10−4 | 1.160 × 10−2 |
| SCLAV_1957 | AraC family transcriptional regulator (adpA) | 2.835 | 3.971 × 10−4 | 1.108 × 10−2 |
| Carbon metabolism | ||||
| SCLAV_5677 | Aldehyde dehydrogenase | 5.151 | 9.470 × 10−12 | 3.370 × 10−9 |
| SCLAV_0665 | Acyl-CoA dehydrogenase | 2.943 | 2.290 × 10−4 | 6.964 × 10−3 |
| SCLAV_3742 | Malate dehydrogenase | 2.146 | 1.345 × 10−3 | 2.800 × 10−2 |
| SCLAV_1401 | Pyruvate dehydrogenase E1 component | 2.107 | 1.630 × 10−3 | 3.202 × 10−2 |
| Nitrogen metabolism | ||||
| SCLAV_2388 | N-acetylglutamate synthase | 3.786 | 2.454 × 10−3 | 4.380 × 10−2 |
| SCLAV_0800 | Arginine biosynthesis bifunctional protein (argJ) | 3.184 | 1.391 × 10−3 | 2.844 × 10−2 |
| SCLAV_0796 | Argininosuccinate synthase | 3.171 | 8.450 × 10−6 | 4.685 × 10−4 |
| SCLAV_4534 | Ammonium transporter | 2.896 | 2.420 × 10−5 | 1.087 × 10−3 |
| SCLAV_0798 | Acetylornithine aminotransferase (oat2) | 2.582 | 5.623 × 10−4 | 1.418 × 10−2 |
| SCLAV_0799 | Acetylglutamate kinase | 2.553 | 1.598 × 10−3 | 3.160 × 10−2 |
| Down-regulated genes in GSPG | ||||
| SCLAV_5442 | Putative transcriptional regulator | −6.728 | 2.427 × 10−3 | 4.380 × 10−2 |
| SCLAV_1087 | Putative penicillin-binding protein | −4.02 | 1.310 × 10−4 | 4.536 × 10−3 |
| SCLAV_2302 | TetR-family transcriptional regulator | −3.962 | 1.670 × 10−4 | 5.440 × 10−3 |
| SCLAV_4124 | Putative PadR-like family transcriptional regulator | −3.839 | 1.910 × 10−5 | 9.100 × 10−4 |
| SCLAV_5441 | Pyruvate phosphate dikinase | −3.701 | 5.920 × 10−4 | 1.469 × 10−2 |
| SCLAV_3293 | RNA polymerase ECF-subfamily sigma factor | −3.549 | 1.024 × 10−3 | 2.237 × 10−2 |
| SCLAV_3942 | Penicillin-binding protein | −3.525 | 4.230 × 10−4 | 1.155 × 10−2 |
| SCLAV_1943 | Acetyl/propionyl CoA carboxylase | −3.410 | 2.440 × 10−5 | 1.086 × 10−3 |
| SCLAV_3597 | Two-component system sensor kinase | −3.380 | 2.453 × 10−3 | 4.380 × 10−2 |
| SCLAV_p0423 | Transcriptional regulator, XRE family protein | −3.266 | 1.760 × 10−5 | 8.660 × 10−4 |
| SCLAV_5443 | Transcriptional regulator | −2.982 | 2.360 × 10−4 | 7.104 × 10−3 |
| SCLAV_p1581 | Transcriptional regulator, BadM/Rrf2 family | −2.875 | 4.930 × 10−4 | 1.272 × 10−2 |
| SCLAV_0603 | Putative PadR-like family transcriptional regulator | −2.718 | 1.960 × 10−4 | 6.213v10−3 |
| SCLAV_2046 | 4-hydroxyphenylpyruvate dioxygenase | −2.707 | 1.520 × 10−4 | 5.053 × 10−3 |
| SCLAV_2979 | Putative dihydrolipoamide acyltransferase | −2.679 | 8.140 × 10−4 | 1.873 × 10−2 |
| SCLAV_2974 | 3-hydroxybutyryl-CoA dehydrogenase | −2.659 | 4.310 × 10−4 | 1.160 × 10−2 |
| SCLAV_4588 | Aldehyde dehydrogenase | −2.492 | 2.513 × 10−3 | 4.447 × 10−2 |
| SCLAV_4386 | Putative transcriptional regulator | −2.254 | 9.460 × 10−4 | 2.113 × 10−2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pinilla, L.; Toro, L.F.; Laing, E.; Alzate, J.F.; Ríos-Estepa, R. Comparative Transcriptome Analysis of Streptomyces Clavuligerus in Response to Favorable and Restrictive Nutritional Conditions. Antibiotics 2019, 8, 96. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics8030096
Pinilla L, Toro LF, Laing E, Alzate JF, Ríos-Estepa R. Comparative Transcriptome Analysis of Streptomyces Clavuligerus in Response to Favorable and Restrictive Nutritional Conditions. Antibiotics. 2019; 8(3):96. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics8030096
Chicago/Turabian StylePinilla, Laura, León F. Toro, Emma Laing, Juan Fernando Alzate, and Rigoberto Ríos-Estepa. 2019. "Comparative Transcriptome Analysis of Streptomyces Clavuligerus in Response to Favorable and Restrictive Nutritional Conditions" Antibiotics 8, no. 3: 96. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics8030096