Actinomycete-Derived Polyketides as a Source of Antibiotics and Lead Structures for the Development of New Antimicrobial Drugs


Abstract
:1. Introduction
2. The Biosynthetic Assembly Lines
3. Clinically-Relevant Polyketide Derived Antibiotics
3.1. Erythromycin and Derivatives
3.2. Tylosin
3.3. Monensins
3.4. Tiacumicin
3.5. Rifamycin and Derivatives
3.6. Tetracyclines
3.7. Streptogramins
4. Other Clinically Relevant Polyketide-Derived Antimicrobials
4.1. Nystatin A1
4.2. Amphotericin B
4.3. Pimaricin/Natamycin
5. Strategies and Tools for the Discovery of Natural Products
6. Conclusions and Outlook
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| A | adenylation domain | MBC | minimum bactericidal concentration |
| ABC | ATP-binding cassette | MIC | minimum inhibitory concentration |
| ACP | acyl carrier protein | MLSB | macrolide–lincosamide–streptogramin B |
| AHBA | 3-amino 5-hydroxybenzoic acid | MOA | mode of action |
| ARO | aromatase | MRSA | methicillin-resistant Staphylococcus aureus |
| AT | acyltransferase | MT | methyltransferase domain |
| BGC | biosynthetic gene cluster | NADPH | nicotinamide adenine dinucleotide phosphate |
| C | condensation domain | NGST | Next Generation Sequencing Technologies |
| CABP | community-acquired bacterial pneumonia | NMR | nuclear magnetic resonance |
| CCR | crotonyl-CoA carboxylase/reductase | NRPS | nonribosomal peptide synthetase |
| CDI | Clostridioides difficile infection | NTG | N-methyl-N′-nitro-N-nitrosoguanidine |
| CLF | chain elongation factor | ORF | open reading frame |
| CoA | coenzyme A | PCP | peptidyl carrier protein |
| cryo EM | cryogenic electron microscopy | PCR | polymerase chain reaction |
| CYC | cyclase | PKS | polyketide synthase |
| DEBS | deoxyerythronolide B synthase | PTC | peptidyl transferase center |
| DH | dehydratase | RNA | ribonucleic acid |
| DNA | deoxyribonucleic acid | RNAP | RNA polymerase |
| dTDP | deoxythymidine diphosphate | rRNA | ribosomal RNA |
| E | epimerisation domain | SAM | S-adenosyl methionine |
| ER | enoyl reductase | SAR | structure activity relationship |
| ESBL | extended spectrum β-lactamase | SARP | Streptomyces antibiotic regulatory protein |
| FDA | Food and Drug Administration | Spp. | Species |
| GDP | guanosine diphosphate | TB | Tuberculosis |
| GI | gastrointestinal | TDP | thymidine diphosphate |
| Kb | kilobase | TE | Thioesterase |
| KR | ketoreductase | tRNA | transfer RNA |
| KS | ketosynthase | VRE | vancomycin-resistant Enterococci |
| LSAP | Lincosamide–streptogramin A–pleuromutilin | VRSA | vancomycin-resistant Staphylococcus aureus |
| MAC | Mycobacterium avium-intracellulare complex | WHO | World Health Organisation |
References
- Mohammadipanah, F.; Dehhaghi, M. Classification and taxonomy of actinobacteria. In Biology and Biotechnology of Actinobacteria; Wink, J., Mohammadipanah, F., Hamedi, J., Eds.; Springer: Berlin/Heidelberg, Germany, 2017; pp. 51–77. [Google Scholar]
- Barka, E.A.; Vatsa, P.; Sanchez, L.; Gaveau-Vaillant, N.; Jacquard, C.; Klenk, H.P.; Clément, C.; Ouhdouch, Y.; van Wezel, G.P. Taxonomy, physiology, and natural products of Actinobacteria. Microbiol. Mol. Biol. Rev. 2016, 80, 1–43. [Google Scholar] [CrossRef] [PubMed]
- García, J.C.; Patrão, B.; Almeida, L.; Pérez, J.; Menezes, P.; Dias, J.; Sanz, P.J. A natural interface for remote operation of underwater robots. IEEE Comput. Graph. Appl. 2015, 37, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Cook, T.B.; Pfleger, B.F. Leveraging synthetic biology for producing bioactive polyketides and non-ribosomal peptides in bacterial heterologous hosts. Medchemcomm 2019, 10, 668–681. [Google Scholar] [CrossRef] [PubMed]
- Abu-Melha, S. Design, Synthesis and DFT/DNP Modeling Study of New 2-Amino-5-arylazothiazole Derivatives as Potential Antibacterial Agents. Molecules 2018, 23, 434. [Google Scholar] [CrossRef] [PubMed]
- Lenci, E.; Trabocchi, A. Smart Design of Small-Molecule Libraries: When Organic Synthesis Meets Cheminformatics. ChemBioChem 2019, 20, 1115–1123. [Google Scholar] [CrossRef] [PubMed]
- Musiol-Kroll, E.; Wohlleben, W. Acyltransferases as tools for polyketide synthase engineering. Antibiotics 2018, 7, 62. [Google Scholar] [CrossRef]
- Tong, Y.; Robertsen, H.L.; Blin, K.; Weber, T.; Lee, S.Y. CRISPR-Cas9 toolkit for Actinomycete genome editing. In Synthetic Metabolic Pathways; Springer: Berlin/Heidelberg, Germany, 2018; pp. 163–184. [Google Scholar]
- Blin, K.; Shaw, S.; Steinke, K.; Villebro, R.; Ziemert, N.; Lee, S.Y.; Medema, M.H.; Weber, T. antiSMASH 5.0: Updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 2019, 47, W81–W87. [Google Scholar] [CrossRef]
- Komaki, H.; Sakurai, K.; Hosoyama, A.; Kimura, A.; Igarashi, Y.; Tamura, T. Diversity of nonribosomal peptide synthetase and polyketide synthase gene clusters among taxonomically close Streptomyces strains. Sci. Rep. 2018, 8, 6888. [Google Scholar] [CrossRef]
- Nett, M.; Ikeda, H.; Moore, B.S. Genomic basis for natural product biosynthetic diversity in the actinomycetes. Nat. Prod. Rep. 2009, 26, 1362–1384. [Google Scholar] [CrossRef]
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I.; et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 2017, 16, 19. [Google Scholar] [CrossRef]
- Wilson, D.N.; Harms, J.M.; Nierhaus, K.H.; Schlünzen, F.; Fucini, P. Species-specific antibiotic-ribosome interactions: Implications for drug development. Biol. Chem. 2005, 386, 1239–1252. [Google Scholar] [CrossRef]
- Koehbach, J.; Craik, D.J. The Vast Structural Diversity of Antimicrobial Peptides. Trends Pharmacol. Sci. 2019, 40, 517–528. [Google Scholar] [CrossRef]
- Tenover, F.C. Mechanisms of antimicrobial resistance in bacteria. Am. J. Med. 2006, 119 (Suppl. 1), S3–S10; discussion S62–S70. [Google Scholar] [CrossRef]
- Du Toit, A. Antimicrobials: Putting antibiotic action into context. Nat. Rev. Microbiol. 2016, 14, 725. [Google Scholar] [CrossRef]
- Levy, S.B.; Marshall, B. Antibacterial resistance worldwide: Causes, challenges and responses. Nat. Med. 2004, 10, S122. [Google Scholar] [CrossRef]
- Beckh, W.; Kulchar, G.V. Treatment-Resistant Syphilis: An Evaluation of the Causative Factors in Eighteen Cases. Arch. Derm. Syphilol. 1939, 40, 1–12. [Google Scholar] [CrossRef]
- Andersson, D.I.; Nicoloff, H.; Hjort, K. Mechanisms and clinical relevance of bacterial heteroresistance. Nat. Rev. Microbiol. 2019, 17, 479–496. [Google Scholar] [CrossRef]
- Hofer, U. The cost of antimicrobial resistance. Nat. Rev. Microbiol. 2019, 17, 3. [Google Scholar] [CrossRef]
- Eagle, H. The binding of penicillin in relation to its cytotoxic action: II. The reactivity with penicillin of resistant variants of Streptococci, Pneomocci, and Staphylococci. J. Exp. Med. 1954, 100, 103–115. [Google Scholar] [CrossRef]
- Stekel, D. First report of antimicrobial resistance pre-dates penicillin. Nature 2018, 562, 192. [Google Scholar] [CrossRef]
- Frieri, M.; Kumar, K.; Boutin, A. Antibiotic resistance. J. Infect. Public Health 2017, 10, 369–378. [Google Scholar] [CrossRef] [Green Version]
- Turner, N.A.; Sharma-Kuinkel, B.K.; Maskarinec, S.A.; Eichenberger, E.M.; Shah, P.P.; Carugati, M.; Holland, T.L.; Fowler, V.G., Jr. Methicillin-resistant Staphylococcus aureus: An overview of basic and clinical research. Nat. Rev. Microbiol. 2019, 17, 203–218. [Google Scholar] [CrossRef]
- Blair, J.M.A.; Webber, M.A.; Baylay, A.J.; Ogbolu, D.O.; Piddock, L.J.V. Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol. 2015, 13, 42. [Google Scholar] [CrossRef]
- Kaur, P.; Peterson, E. Antibiotic resistance mechanisms in bacteria: Relationships between resistance determinants of antibiotic producers, environmental bacteria, and clinical pathogens. Front. Microbiol. 2018, 9, 2928. [Google Scholar]
- Pambos, O.J.; Kapanidis, A.N. Tracking antibiotic mechanisms. Nat. Rev. Microbiol. 2019, 17, 201. [Google Scholar] [CrossRef]
- Alanis, A.J. Resistance to antibiotics: Are we in the post-antibiotic era? Arch. Med. Res. 2005, 36, 697–705. [Google Scholar] [CrossRef]
- Carroll, L.M.; Gaballa, A.; Guldimann, C.; Sullivan, G.; Henderson, L.O.; Wiedmann, M. Identification of Novel Mobilized Colistin Resistance Gene mcr-9 in a Multidrug-Resistant, Colistin-Susceptible Salmonella enterica Serotype Typhimurium Isolate. MBio 2019, 10, e00853-19. [Google Scholar] [CrossRef]
- World Health Organization. New report calls for urgent action to avert antimicrobial resistance crisis. Joint News Release. 29 April 2019. Available online: https://www.who.int/news-room/detail/29-04-2019-new-report-calls-for-urgent-action-to-avert-antimicrobial-resistance-crisis (accessed on 25 August 2019).
- World Health Organization. Antibiotic Resistance. 2019. Available online: https://www.who.int/news-room/fact-sheets/detail/antibiotic-resistance (accessed on 25 August 2019).
- Spellberg, B. The future of antibiotics. Crit. Care 2014, 18, 228. [Google Scholar] [CrossRef]
- Pew Charitable Trusts. Tracking the Pipeline of Antibiotics in Development. 2019. Available online: http://www.pewtrusts.org/en/research-and-analysis/issue-briefs/2014/03/12/tracking-the-pipeline-of-antibiotics-in-development (accessed on 25 August 2019).
- Owens, B. Solithromycin rejection chills antibiotic sector. Nat. Biotechnol. 2017, 35, 187–188. [Google Scholar] [CrossRef]
- Weissman, K. Chapter 1 Introduction to Polyketide Biosynthesis. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2009; Volume 459, pp. 3–16. [Google Scholar]
- Shen, B. Polyketide biosynthesis beyond the type I, II and III polyketide synthase paradigms. Curr. Opin. Chem. Biol. 2003, 7, 285–295. [Google Scholar] [CrossRef]
- Hertweck, C. The biosynthetic logic of polyketide diversity. Angew. Chemie Int. Ed. 2009, 48, 4688–4716. [Google Scholar] [CrossRef]
- Ridley, C.P.; Lee, H.Y.; Khosla, C. Evolution of polyketide synthases in bacteria. Proc. Natl. Acad. Sci. USA 2008, 105, 4595–4600. [Google Scholar] [CrossRef] [Green Version]
- Süssmuth, R.D.; Mainz, A. Nonribosomal peptide synthesis—Principles and prospects. Angew. Chem. Int. Ed. 2017, 56, 3770–3821. [Google Scholar] [CrossRef]
- Challis, G.L.; Wilkinson, B. Biosynthetic assembly lines themed issue. Nat. Prod. Rep. 2016, 33, 120–121. [Google Scholar] [CrossRef]
- Staunton, J.; Weissman, K.J. Polyketide biosynthesis: A millennium review. Nat. Prod. Rep. 2001, 18, 380–416. [Google Scholar] [CrossRef]
- Helfrich, E.J.N.; Piel, J. Biosynthesis of polyketides by trans-AT polyketide synthases. Nat. Prod. Rep. 2016, 33, 231–316. [Google Scholar] [CrossRef]
- Musiol, E.M.; Weber, T. Discrete acyltransferases involved in polyketide biosynthesis. Medchemcomm 2012, 3, 871–886. [Google Scholar] [CrossRef]
- Meurer, G.; Gerlitz, M.; Wendt-Pienkowski, E.; Vining, L.C.; Rohr, J.; Hutchinson, C.R. Iterative type II polyketide synthases, cyclases and ketoreductases exhibit context-dependent behavior in the biosynthesis of linear and angular decapolyketides. Chem. Biol. 1997, 4, 433–443. [Google Scholar] [CrossRef] [Green Version]
- Caffrey, P. Dissecting complex polyketide biosynthesis. Comput. Struct. Biotechnol. J. 2012, 3, e201210010. [Google Scholar] [CrossRef]
- Chen, A.; Re, R.N.; Burkart, M.D. Type II fatty acid and polyketide synthases: Deciphering protein–protein and protein–substrate interactions. Nat. Prod. Rep. 2018, 35, 1029–1045. [Google Scholar] [CrossRef]
- Herbst, D.A.; Townsend, C.A.; Maier, T. The architectures of iterative type I PKS and FAS. Nat. Prod. Rep. 2018, 35, 1046–1069. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Du, L. Iterative polyketide biosynthesis by modular polyketide synthases in bacteria. Appl. Microbiol. Biotechnol. 2016, 100, 541–557. [Google Scholar] [CrossRef]
- Weber, T. Antibiotics: Biosynthesis, Generation of Novel Compounds. Encycl. Ind. Biotechnol. 2010, 1–12. [Google Scholar] [CrossRef]
- Bloudoff, K.; Schmeing, T.M. Structural and functional aspects of the nonribosomal peptide synthetase condensation domain superfamily: Discovery, dissection and diversity. BBA Proteins Proteom. 2017, 1865, 1587–1604. [Google Scholar] [CrossRef]
- Miyanaga, A.; Kudo, F.; Eguchi, T. Protein–protein interactions in polyketide synthase–nonribosomal peptide synthetase hybrid assembly lines. Nat. Prod. Rep. 2018, 35, 1185–1209. [Google Scholar] [CrossRef]
- McGuire, J.M.; Bunch, R.L.; Anderson, R.C.; Boaz, H.E.; Flynn, E.H.; Powell, H.M.; Smith, J.W. Ilotycin, a new antibiotic. Antibiot. Chemother. (Northfield, III.) 1952, 2, 281–283. [Google Scholar]
- Wiley, P.F.; Gerzon, K.; Flynn, E.H.; Sigal, M.V., Jr.; Weaver, O.; Quarck, U.C.; Chauvette, R.R.; Monahan, R. Erythromycin. X. 1 Structure of Erythromycin. J. Am. Chem. Soc. 1957, 79, 6062–6070. [Google Scholar] [CrossRef]
- Harris, D.R.; McGeachin, S.G.; Mills, H.H. The structure and stereochemistry of erythromycin A. Tetrahedron Lett. 1965, 6, 679–685. [Google Scholar] [CrossRef]
- Cortes, J.; Haydock, S.F.; Roberts, G.A.; Bevitt, D.J.; Leadlay, P.F. An unusually large multifunctional polypeptide in the erythromycin-producing polyketide synthase of Saccharopolyspora erythraea. Nature 1990, 348, 176. [Google Scholar] [CrossRef]
- Summers, R.G.; Donadio, S.; Staver, M.J.; Wendt-Pienkowski, E.; Hutchinson, C.R.; Katz, L. Sequencing and mutagenesis of genes from the erythromycin biosynthetic gene cluster of Saccharopolyspora erythraea that are involved in L-mycarose and D-desosamine production. Microbiology 1997, 143, 3251–3262. [Google Scholar] [CrossRef]
- Donadio, S.; Staver, M.; McAlpine, J.; Swanson, S.J.; Katz, L. Modular Organization of Genes Required for Complex Polyketide Biosynthesis. Science 1991, 252, 675–679. [Google Scholar] [CrossRef]
- Oliynyk, M.; Samborskyy, M.; Lester, J.B.; Mironenko, T.; Scott, N.; Dickens, S.; Haydock, S.F.; Leadlay, P.F. Complete genome sequence of the erythromycin-producing bacterium Saccharopolyspora erythraea NRRL23338. Nat. Biotechnol. 2007, 25, 447. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, Y.; Wu, J.; Skalina, K.; Pfeifer, B.A. Complete biosynthesis of erythromycin A and designed analogs using E. coli as a heterologous host. Chem. Biol. 2010, 17, 1232–1240. [Google Scholar] [CrossRef]
- Weissman, K.J. Genetic engineering of modular PKSs: From combinatorial biosynthesis to synthetic biology. Nat. Prod. Rep. 2016, 33, 203–230. [Google Scholar] [CrossRef]
- Kibwage, I.O.; Hoogmartens, J.; Roets, E.; Vanderhaeghe, H.; Verbist, L.; Dubost, M.; Pascal, C.; Petitjean, P.; Levol, G. Antibacterial activities of erythromycins A, B, C, and D and some of their derivatives. Antimicrob. Agents Chemother. 1985, 28, 630–633. [Google Scholar] [CrossRef] [Green Version]
- Amsden, G.W. Erythromycin, clarithromycin, and azithromycin: Are the differences real? Clin. Ther. 1996, 18, 56–72. [Google Scholar] [CrossRef]
- Mazzei, T.; Mini, E.; Novelli, A.; Periti, P. Chemistry and mode of action of macrolides. J. Antimicrob. Chemother. 1993, 31, 1–9. [Google Scholar] [CrossRef]
- Vester, B.; Douthwaite, S. Macrolide resistance conferred by base substitutions in 23S rRNA. Antimicrob. Agents Chemother. 2001, 45, 1–12. [Google Scholar] [CrossRef]
- Tenson, T.; Lovmar, M.; Ehrenberg, M. The mechanism of action of macrolides, lincosamides and streptogramin B reveals the nascent peptide exit path in the ribosome. J. Mol. Biol. 2003, 330, 1005–1014. [Google Scholar] [CrossRef]
- Svetlov, M.S.; Plessa, E.; Chen, C.W.; Bougas, A.; Krokidis, M.G.; Dinos, G.P.; Polikanov, Y.S. High-resolution crystal structures of ribosome-bound chloramphenicol and erythromycin provide the ultimate basis for their competition. RNA 2019, 25, 600–606. [Google Scholar] [CrossRef]
- Sobin, B.A.; English, A.R.; Celmer, W.D. PA 105, a new antibiotic. In Antibiotics Annual; Welch, H., Marti-Ibannez, F., Eds.; Medical Encyclopedia Inc.: New York, NY, USA, 1955; pp. 827–830. [Google Scholar]
- English, A.R.; McBride, T.J.; Van Halsema, G.; Caklozzi, M. Biologic studies on PA 775, a combination of tetracycline and oleandomycin with synergistic activity. Antibiot. Chemother. 1956, 6, 511–522. [Google Scholar]
- Podolsky, S.H. The Antibiotic Era: Reform, Resistance, and the Pursuit of a Rational Therapeutics; JHU Press: Baltimore, MD, USA, 2015; pp. 1–328. [Google Scholar]
- Albouy, R.; Duchesnay, G.; Eloy, P.; Pestel, M.; Ravina, A.; Rey, M. A new French antibiotic: Spiramycin. Antibiot. Annu. 1955, 3, 223. [Google Scholar]
- Kellow, W.F.; Lepper, M.H.; Plaut, S.; Rosenthal, I.M.; Spies, H.W. Spiramycin in the treatment of infection. Antibiot. Annu. 1955, 3, 658. [Google Scholar]
- Sutherland, R. Spiramycin: A reappraisal of its antibacterial activity. Br. J. Pharmacol. Chemother. 1962, 19, 99–110. [Google Scholar] [CrossRef]
- Fernandes, P.; Martens, E.; Pereira, D. Nature nurtures the design of new semi-synthetic macrolide antibiotics. J. Antibiot. (Tokyo) 2016, 70, 527. [Google Scholar] [CrossRef]
- Barry, A.L.; Jones, R.N.; Thornsberry, C. In vitro activities of azithromycin (CP 62,993), clarithromycin (A-56268; TE-031), erythromycin, roxithromycin, and clindamycin. Antimicrob. Agents Chemother. 1988, 32, 752–754. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, Y.; Moritomo, S.; Adachi, T.; Kashimure, M.; Asaka, T. Chemical modification of erythromycin. IX. 1. J. Antibiot. (Tokyo) 1993, 46, 647–660. [Google Scholar] [CrossRef]
- Barlam, T.; Neu, H.C. In vitro comparison of the activity of RU 28965, a new macrolide, with that of erythromycin against aerobic and anaerobic bacteria. Antimicrob. Agents Chemother. 1984, 25, 529–531. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, J.H.; Redding, J.S.; Howell, A.W. In vitro activity of the new macrolide antibiotic roxithromycin (RU 28965) against clinical isolates of Haemophilus influenzae. Antimicrob. Agents Chemother. 1986, 29, 921–922. [Google Scholar] [CrossRef]
- Mutak, S. Azalides from azithromycin to new azalide derivatives. J. Antibiot. (Tokyo) 2007, 60, 85. [Google Scholar] [CrossRef]
- Retsema, J.; Girard, A.; Schelkly, W.; Manousos, M.; Anderson, M.; Bright, G.; Borovoy, R.; Brennan, L.; Mason, R. Spectrum and mode of action of azithromycin (CP-62,993), a new 15-membered-ring macrolide with improved potency against gram-negative organisms. Antimicrob. Agents Chemother. 1987, 31, 1939–1947. [Google Scholar] [CrossRef] [Green Version]
- Bryskier, A. Ketolides—Telithromycin, an example of a new class of antibacterial agents. Clin. Microbiol. Infect. 2000, 6, 661–669. [Google Scholar] [CrossRef]
- Ednie, L.M.; Jacobs, M.R.; Appelbaum, P.C. Comparative antianaerobic activities of the ketolides HMR 3647 (RU 66647) and HMR 3004 (RU 64004). Antimicrob. Agents Chemother. 1997, 41, 2019–2022. [Google Scholar] [CrossRef] [Green Version]
- Ross, D.B. The FDA and the case of Ketek. N. Engl. J. Med. 2007, 356, 1601–1604. [Google Scholar] [CrossRef]
- McGhee, P.; Clark, C.; Kosowska-Shick, K.M.; Nagai, K.; Dewasse, B.; Beachel, L.; Appelbaum, P.C. In vitro activity of CEM-101 against Streptococcus pneumoniae and Streptococcus pyogenes with defined macrolide resistance mechanisms. Antimicrob. Agents Chemother. 2010, 54, 230–238. [Google Scholar] [CrossRef]
- Rodvold, K.A.; Gotfried, M.H.; Chugh, R.; Gupta, M.; Friedland, H.D.; Bhatia, A. Comparison of plasma and intrapulmonary concentrations of nafithromycin (WCK 4873) in healthy adult subjects. Antimicrob. Agents Chemother. 2017, 61, e01096-17. [Google Scholar] [CrossRef]
- World Health Organisation. Antibacterial Agents in Clinical Development. 2018. Available online: https://apps.who.int/iris/handle/10665/275487 (accessed on 25 August 2019).
- McGuire, J.M.; Boniece, W.S.; Higgens, C.E.; Hoehn, M.M.; Stark, W.M.; Westhead, J.; Wolfe, R.N. Tylosin, a New Antibiotic: I. Microbiological Studies. Antibiot. Chemother. 1961, 11, 320–327. [Google Scholar]
- Cundliffe, E.; Bate, N.; Butler, A.; Fish, S.; Gandecha, A.; Merson-Davies, L. The tylosin-biosynthetic genes of Streptomyces fradiae. Antonie Van Leeuwenhoek 2001, 79, 229–234. [Google Scholar] [CrossRef]
- Baltz, R.H.; Seno, E.T. Genetics of Streptomyces fradiae and tylosin biosynthesis. Annu. Rev. Microbiol. 1988, 42, 547–574. [Google Scholar] [CrossRef]
- Tejedor, F.; Ballesta, J.P.G. Ribosome structure: Binding site of macrolides studied by photoaffinity labeling. Biochemistry 1985, 24, 467–472. [Google Scholar] [CrossRef]
- Ose, E.E. In vitro antibacterial properties of EL-870, a new semi-synthetic macrolide antibiotic. J. Antibiot. (Tokyo) 1987, 40, 190–194. [Google Scholar] [CrossRef]
- Debono, M.; Willard, K.E.; Kirst, H.A.; Wind, J.A.; Crouse, G.D.; Tao, E.V.; Vicenzi, J.T.; Counter, F.T.; Ott, J.L.; Ose, E.E.; et al. Synthesis and antimicrobial evaluation of 20-deoxo-20-(3,5-dimethylpiperidin-1-yl) desmycosin (tilmicosin, EL-870) and related cyclic amino derivatives. J. Antibiot. (Tokyo) 1989, 42, 1253–1267. [Google Scholar] [CrossRef]
- Michael, G.B.; Eidam, C.; Kadlec, K.; Meyer, K.; Sweeney, M.T.; Murray, R.W.; Watts, J.L.; Schwarz, S. Increased MICs of gamithromycin and tildipirosin in the presence of the genes erm (42) and msr(E)-mph(E) for bovine Pasteurella multocida and Mannheimia haemolytica. J. Antimicrob. Chemother. 2012, 67, 1555–1557. [Google Scholar] [CrossRef]
- Menge, M.; Rose, M.; Bohland, C.; Zschiesche, E.; Kilp, S.; Metz, W.; Allan, M.; Röpke, R.; Nürnberger, M. Pharmacokinetics of tildipirosin in bovine plasma, lung tissue, and bronchial fluid (from live, nonanesthetized cattle). J. Vet. Pharmacol. Ther. 2012, 35, 550–559. [Google Scholar] [CrossRef]
- Evans, N.A. Tulathromycin: An overview of a new triamilide antimicrobial for livestock respiratory disease. Vet. Ther. 2005, 6, 83. [Google Scholar]
- Huang, R.A.; Letendre, L.T.; Banav, N.; Fischer, J.; Somerville, B. Pharmacokinetics of gamithromycin in cattle with comparison of plasma and lung tissue concentrations and plasma antibacterial activity. J. Vet. Pharmacol. Ther. 2010, 33, 227–237. [Google Scholar] [CrossRef]
- Agtarap, A.; Chamberlin, J.W.; Pinkerton, M.; Steinrauf, L.K. Structure of monensic acid, a new biologically active compound. J. Am. Chem. Soc. 1967, 89, 5737–5739. [Google Scholar] [CrossRef]
- Chapman, H.D.; Jeffers, T.K.; Williams, R.B. Forty years of monensin for the control of coccidiosis in poultry. Poult. Sci. 2010, 89, 1788–1801. [Google Scholar] [CrossRef]
- Oliynyk, M.; Stark, C.B.; Bhatt, A.; Jones, M.A.; Hughes-Thomas, Z.A.; Wilkinson, C.; Oliynyk, Z.; Demydchuk, Y.; Staunton, J.; Leadlay, P.F. Analysis of the biosynthetic gene cluster for the polyether antibiotic monensin in Streptomyces cinnamonensis and evidence for the role of monB and monC genes in oxidative cyclization. Mol. Microbiol. 2003, 49, 1179–1190. [Google Scholar] [CrossRef]
- Goodrich, R.D.; Garrett, J.E.; Gast, D.R.; Kirick, M.A.; Larson, D.A.; Meiske, J.C. Influence of monensin on the performance of cattle. J. Anim. Sci. 1984, 58, 1484–1498. [Google Scholar] [CrossRef]
- Hochlowski, J.E.; Swanson, S.J.; Ranfranz, L.M.; Whittern, D.N.; Buko, A.M.; McAlpine, J.B. Tiacumicins, A Novel Complex of 18-Membered Macrolides. J. Antibiot. (Tokyo) 1987, 40, 575–588. [Google Scholar] [CrossRef]
- Xiao, Y.; Li, S.; Niu, S.; Ma, L.; Zhang, G.; Zhang, H.; Zhang, G.; Ju, J.; Zhang, C. Characterization of tiacumicin B biosynthetic gene cluster affording diversified tiacumicin analogues and revealing a tailoring dihalogenase. J. Am. Chem. Soc. 2011, 133, 1092–1105. [Google Scholar] [CrossRef]
- Swanson, R.N.; Hardy, D.J.; Shipkowitz, N.L.; Hanson, C.W.; Ramer, N.C.; Fernandes, P.B.; Clement, J.J. In vitro and in vivo evaluation of tiacumicins B and C against Clostridium difficile. Antimicrob. Agents Chemother. 1991, 35, 1108–1111. [Google Scholar] [CrossRef]
- Lin, W.; Das, K.; Degen, D.; Mazumder, A.; Duchi, D.; Wang, D.; Ebright, Y.W.; Ebright, R.Y.; Sineva, E.; Gigliotti, M.; et al. Structural Basis of Transcription Inhibition by Fidaxomicin (Lipiarmycin A3). Mol. Cell 2018, 70, 60–71.e15. [Google Scholar] [CrossRef] [Green Version]
- Prelog, V.; Oppolzer, W. Ansamycine, eine neuartige Klasse von mikrobiellen Stoffwechselprodukten. Helv. Chim. Acta 1973, 56, 2279–2287. [Google Scholar] [CrossRef]
- Floss, H.G.; Yu, T.W. Rifamycin mode of action, resistance, and biosynthesis. Chem. Rev. 2005, 105, 621–632. [Google Scholar] [CrossRef]
- Sensi, P.; Margalith, P.; Timbal, M.T. Rifomycin, a new antibiotic; preliminary report. Farm. Sci. 1959, 14, 146. [Google Scholar]
- August, P.R.; Tang, L.; Yoon, Y.J.; Ning, S.; Müller, R.; Yu, T.W.; Taylor, M.; Hoffmann, D.; Kim, C.G.; Zhang, X.; et al. Biosynthesis of the ansamycin antibiotic rifamycin: Deductions from the molecular analysis of the rif biosynthetic gene cluster of Amycolatopsis mediterranei S699. Chem. Biol. 1998, 5, 69–79. [Google Scholar] [CrossRef]
- Watanabe, K.; Rude, M.A.; Walsh, C.T.; Khosla, C. Engineered biosynthesis of an ansamycin polyketide precursor in Escherichia coli. Proc. Natl. Acad. Sci. USA 2003, 100, 9774–9778. [Google Scholar] [CrossRef]
- Hoy, S.M. Rifamycin SV MMX®: A Review in the Treatment of Traveller’s Diarrhoea. Clin. Drug Investig. 2019, 39, 691–697. [Google Scholar] [CrossRef]
- Maggi, N.; Pasqualucci, C.R.; Ballotta, R.; Sensi, P. Rifampicin: A new orally active rifamycin. Chemotherapy 1966, 11, 285–292. [Google Scholar] [CrossRef]
- Campbell, E.A.; Korzheva, N.; Mustaev, A.; Murakami, K.; Nair, S.; Goldfarb, A.; Darst, S.A. Structural mechanism for rifampicin inhibition of bacterial RNA polymerase. Cell 2001, 104, 901–912. [Google Scholar] [CrossRef]
- Della Bruna, C.; Schioppacassi, G.; Ungheri, D.; Jabès, D.; Morvillo, E.; Sanfilippo, A. LM 427, a new spiropiperidylrifamycin: In vitro and in vivo studies. J. Antibiot. (Tokyo) 1983, 36, 1502–1506. [Google Scholar] [CrossRef]
- Brogden, R.N.; Fitton, A. Rifabutin. Drugs 1994, 47, 983–1009. [Google Scholar] [CrossRef]
- Kunin, C.M. Antimicrobial activity of rifabutin. Clin. Infect. Dis. 1996, 22, S3–S14. [Google Scholar] [CrossRef]
- Jarvis, B.; Lamb, H.M. Rifapentine. Drugs 1998, 56, 607–616. [Google Scholar] [CrossRef]
- Scarpignato, C.; Pelosini, I. Rifaximin, a poorly absorbed antibiotic: Pharmacology and clinical potential. Chemotherapy 2005, 51, 36–66. [Google Scholar] [CrossRef]
- Koo, H.L.; DuPont, H.L. Rifaximin: A unique gastrointestinal-selective antibiotic for enteric diseases. Curr. Opin. Gastroenterol. 2010, 26, 17. [Google Scholar] [CrossRef]
- Fodor, A.A.; Pimentel, M.; Chey, W.D.; Lembo, A.; Golden, P.L.; Israel, R.J.; Carroll, I.M. Rifaximin is associated with modest, transient decreases in multiple taxa in the gut microbiota of patients with diarrhoea-predominant irritable bowel syndrome. Gut Microbes 2019, 10, 22–33. [Google Scholar] [CrossRef]
- Mosaei, H.; Molodtsov, V.; Kepplinger, B.; Harbottle, J.; Moon, C.W.; Jeeves, R.E.; Ceccaroni, L.; Shin, Y.; Morton-Laing, S.; Marrs, E.C.L.; et al. Mode of Action of Kanglemycin A, an Ansamycin Natural Product that Is Active against Rifampicin-Resistant Mycobacterium tuberculosis. Mol. Cell 2018, 72, 263–274.e5. [Google Scholar] [CrossRef]
- Peek, J.; Lilic, M.; Montiel, D.; Milshteyn, A.; Woodworth, I.; Biggins, J.B.; Ternei, M.A.; Calle, P.Y.; Danziger, M.; Warrier, T.; et al. Rifamycin congeners kanglemycins are active against rifampicin-resistant bacteria via a distinct mechanism. Nat. Commun. 2018, 9, 4147. [Google Scholar] [CrossRef]
- Finlay, A.C.; Hobby, G.L. Terramycin, a new antibiotic. Science 1950, 85–87. [Google Scholar] [CrossRef]
- Thomas, R.; Williams, D.J. Oxytetracycline biosynthesis: Origin of the carboxamide substituent. J. Chem. Soc. Chem. Commun. 1983, 677–679. [Google Scholar] [CrossRef]
- Chopra, I.; Roberts, M. Tetracycline antibiotics: Mode of action, applications, molecular biology, and epidemiology of bacterial resistance. Microbiol. Mol. Biol. Rev. 2001, 65, 232–260. [Google Scholar] [CrossRef]
- Brodersen, D.E.; Clemons, W.M., Jr.; Carter, A.P.; Morgan-Warren, R.J.; Wimberly, B.T.; Ramakrishnan, V. The structural basis for the action of the antibiotics tetracycline, pactamycin, and hygromycin B on the 30S ribosomal subunit. Cell 2000, 103, 1143–1154. [Google Scholar] [CrossRef]
- Duggar, B.M. Aureomycin: A product of the continuing search for new antibiotics. Ann. N. Y. Acad. Sci. 1948, 51, 177–181. [Google Scholar] [CrossRef]
- Zhu, T.; Cheng, X.; Liu, Y.; Deng, Z.; You, D. Deciphering and engineering of the final step halogenase for improved chlortetracycline biosynthesis in industrial Streptomyces aureofaciens. Metab. Eng. 2013, 19, 69–78. [Google Scholar] [CrossRef]
- Nguyen, F.; Starosta, A.L.; Arenz, S.; Sohmen, D.; Dönhöfer, A.; Wilson, D.N. Tetracycline antibiotics and resistance mechanisms. Biol. Chem. 2014, 395, 559–575. [Google Scholar] [CrossRef]
- Stephens, C.R.; Beereboom, J.J.; Rennhard, H.H.; Gordon, P.N.; Murai, K.; Blackwood, R.K.; von Wittenau, M.S. 6-Deoxytetracyclines. IV.1,2 Preparation, C-6 Stereochemistry, and Reactions. J. Am. Chem. Soc. 1963, 85, 2643–2652. [Google Scholar] [CrossRef]
- McCormick, J.R.D.; Sjolander, N.O.; Hirsch, U.; Jensen, E.R.; Doerschuk, A.P. A new family of antibiotics: The demethyltetracyclines. J. Am. Chem. Soc. 1957, 79, 4561–4563. [Google Scholar] [CrossRef]
- McCormick, J.R.D.; Hirsch, U.; Sjolander, N.O.; Doerschuk, A.P. Cosynthesis of tetracyclines by pairs of Streptomyces aureofaciens mutants. J. Am. Chem. Soc. 1960, 82, 5006–5007. [Google Scholar] [CrossRef]
- Martell, M.J.; Boothe, J.H. The 6-deoxytetracyclines. VII. Alkylated aminotetracyclines possessing unique antibacterial activity. J. Med. Chem. 1967, 10, 44–46. [Google Scholar] [CrossRef]
- Zakeri, B.; Wright, G.D. Chemical biology of tetracycline antibiotics. Biochem. Cell Biol. 2008, 86, 124–136. [Google Scholar] [CrossRef]
- Sum, P.E.; Lee, V.J.; Testa, R.T.; Hlavka, J.J.; Ellestad, G.A.; Bloom, J.D.; Gluzman, Y.; Tally, F.P. Glycylcyclines. 1. A new generation of potent antibacterial agents through modification of 9-aminotetracyclines. J. Med. Chem. 1994, 37, 184–188. [Google Scholar] [CrossRef]
- Petersen, P.J.; Jacobus, N.V.; Weiss, W.J.; Sum, P.E.; Testa, R.T. In vitro and in vivo antibacterial activities of a novel glycylcycline, the 9-t-butylglycylamido derivative of minocycline (GAR-936). Antimicrob. Agents Chemother. 1999, 43, 738–744. [Google Scholar] [CrossRef]
- Bergeron, J.; Ammirati, M.; Danley, D.; James, L.; Norcia, M.; Retsema, J.; Strick, C.A.; Su, W.G.; Sutcliffe, J.; Wondrack, L. Glycylcyclines bind to the high-affinity tetracycline ribosomal binding site and evade Tet(M)-and Tet(O)-mediated ribosomal protection. Antimicrob. Agents Chemother. 1996, 40, 2226–2228. [Google Scholar] [CrossRef]
- Nelson, M.L.; Ismail, M.Y.; McIntyre, L.; Bhatia, B.; Viski, P.; Hawkins, P.; Rennie, G.; Andorsky, D.; Messersmith, D.; Stapleton, K.; et al. Versatile and facile synthesis of diverse semisynthetic tetracycline derivatives via Pd-catalyzed reactions. J. Org. Chem. 2003, 68, 5838–5851. [Google Scholar] [CrossRef]
- Draper, M.P.; Weir, S.; Macone, A.; Donatelli, J.; Trieber, C.A.; Tanaka, S.K.; Levy, S.B. Mechanism of action of the novel aminomethylcycline antibiotic omadacycline. Antimicrob. Agents Chemother. 2014, 58, 1279–1283. [Google Scholar] [CrossRef]
- Dougherty, J.A.; Sucher, A.J.; Chahine, E.B.; Shihadeh, K.C. Omadacycline: A New Tetracycline Antibiotic. Ann. Pharmacother. 2019, 53, 486–500. [Google Scholar] [CrossRef]
- Sun, C.; Wang, Q.; Brubaker, J.D.; Wright, P.M.; Lerner, C.D.; Noson, K.; Charest, M.; Siegel, D.R.; Wang, Y.M.; Myers, A.G.; et al. A robust platform for the synthesis of new tetracycline antibiotics. J. Am. Chem. Soc. 2008, 130, 17913–17927. [Google Scholar] [CrossRef]
- Grossman, T.H.; Starosta, A.L.; Fyfe, C.; O’Brien, W.; Rothstein, D.M.; Mikolajka, A.; Wilson, D.N.; Sutcliffe, J.A. Target-and resistance-based mechanistic studies with TP-434, a novel fluorocycline antibiotic. Antimicrob. Agents Chemother. 2012, 56, 2559–2564. [Google Scholar] [CrossRef]
- Lee, Y.R.; Burton, C.E. Eravacycline, a newly approved fluorocycline. Eur. J. Clin. Microbiol. Infect. Dis. 2019, 1–8. [Google Scholar] [CrossRef]
- Zhanel, G.; Critchley, I.; Lin, L.Y.; Alvandi, N. Microbiological profile of sarecycline, a novel targeted spectrum tetracycline for the treatment of acne vulgaris. Antimicrob. Agents Chemother. 2019, 63, e01297-18. [Google Scholar] [CrossRef]
- Grossman, T.H.; Fyfe, C.; O’Brien, W.; Hackel, M.; Minyard, M.B.; Waites, K.B.; Dubois, J.; Murphy, T.M.; Slee, A.M.; Weiss, W.J.; et al. Fluorocycline TP-271 Is Potent against Complicated Community-Acquired Bacterial Pneumonia Pathogens. mSphere 2017, 2, e00004-17. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Myers, A.G. Development of a platform for the discovery and practical synthesis of new tetracycline antibiotics. Curr. Opin. Chem. Biol. 2016, 32, 48–57. [Google Scholar] [CrossRef] [Green Version]
- Tetraphase-Pharmaceuticals. Pipeline. Available online: https://www.tphase.com/products/pipeline/ (accessed on 25 August 2019).
- Preud’homme, J.; Tarridec, P.; Belloc, A. 90. Isolation, characterization and identification of the components of pristinamycin. Bull. Soc. Chim. Fr. 1968, 2, 585–591. [Google Scholar]
- Celmer, W.D.; Sobin, B.A. The isolation of two synergistic antibiotics from a single fermentation source. Antibiot. Annu. 1955, 3, 437–441. [Google Scholar]
- Mast, Y.; Weber, T.; Gölz, M.; Ort-Winklbauer, R.; Gondran, A.; Wohlleben, W.; Schinko, E. Characterization of the ‘pristinamycin supercluster’of Streptomyces pristinaespiralis. Microb. Biotechnol. 2011, 4, 192–206. [Google Scholar] [CrossRef]
- Cocito, C.; Di Giambattista, M.; Nyssen, E.; Vannuffel, P. Inhibition of protein synthesis by streptogramins and related antibiotics. J. Antimicrob. Chemother. 1997, 39 (Suppl. A), 7–13. [Google Scholar] [CrossRef] [Green Version]
- Bouanchaud, D.H. In vitro and in vivo antibacterial activity of quinupristin/dalfopristin. J. Antimicrob. Chemother. 1997, 39, 15–21. [Google Scholar] [CrossRef]
- Harms, J.M.; Schlünzen, F.; Fucini, P.; Bartels, H.; Yonath, A. Alterations at the peptidyl transferase centre of the ribosome induced by the synergistic action of the streptogramins dalfopristin and quinupristin. BMC Biol. 2004, 2, 4. [Google Scholar] [CrossRef] [PubMed]
- Barriere, J.C.; Bouanchaud, D.H.; Paris, J.M.; Rolin, O.; Harris, N.V.; Smith, C. Antimicrobial activity against Staphylococcus aureus of semisynthetic injectable streptogramins: RP 59500 and related compounds. J. Antimicrob. Chemother. 1992, 30, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Finch, R.G. Antibacterial Activity of Quinupristin/Dalfopristin. Drugs 1996, 51, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Politano, A.D.; Sawyer, R.G. NXL-103, a combination of flopristin and linopristin, for the potential treatment of bacterial infections including community-acquired pneumonia and MRSA. Curr. Opin. Investig. Drugs (Lond. UK 2000) 2010, 11, 225. [Google Scholar]
- Noeske, J.; Huang, J.; Olivier, N.B.; Giacobbe, R.A.; Zambrowski, M.; Cate, J.H.D. Synergy of streptogramin antibiotics occurs independently of their effects on translation. Antimicrob. Agents Chemother. 2014, 58, 5269–5279. [Google Scholar] [CrossRef]
- Hazen, E.L.; Brown, R. Fungicidin, an Antibiotic Produced by a Soil Actinomycete. Proc. Soc. Exp. Biol. Med. 1951, 76, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Hazen, E.L.; Brown, R.; Mason, A. Protective action of fungicidin (nystatin) in mice against virulence enhancing activity of oxytetracycline on Candida albicans. Antibiot. Chemother. (Northfield, III.) 1953, 3, 1125. [Google Scholar]
- Brautaset, T.; Sekurova, O.N.; Sletta, H.; Ellingsen, T.E.; Strøm, A.R.; Valla, S.; Zotchev, S.B. Biosynthesis of the polyene antifungal antibiotic nystatin in Streptomyces noursei ATCC 11455: Analysis of the gene cluster and deduction of the biosynthetic pathway. Chem. Biol. 2000, 7, 395–403. [Google Scholar] [CrossRef]
- Bolard, J. How do the polyene macrolide antibiotics affect the cellular membrane properties? BBA Rev. Biomembr. 1986, 864, 257–304. [Google Scholar] [CrossRef]
- Brautaset, T.; Sletta, H.; Nedal, A.; Borgos, S.E.; Degnes, K.F.; Bakke, I.; Volokhan, O.; Sekurova, O.N.; Treshalin, I.D.; Mirchink, E.P.; et al. Improved antifungal polyene macrolides via engineering of the nystatin biosynthetic genes in Streptomyces noursei. Chem. Biol. 2008, 15, 1198–1206. [Google Scholar] [CrossRef]
- Biosergen. BSG005 for Systemic Fungal Infections. Available online: http://biosergen.se/products-pipeline/bsg005-for-systemic-fungal-infections/ (accessed on 25 August 2019).
- Dutcher, J.D. The Discovery and Development of Amphotericin B. Dis. Chest 1968, 54, 296–298. [Google Scholar] [CrossRef] [PubMed]
- McNamara, C.; Crawforth, J.; Hickman, B.; Norwood, T.; Rawlings, B. Biosynthesis of amphotericin B. J. Chem. Soc. Perkin Trans. 1 1998, 83–88. [Google Scholar] [CrossRef] [Green Version]
- Brajtburg, J.; Powderly, W.G.; Kobayashi, G.S.; Medoff, G. Amphotericin B: Current understanding of mechanisms of action. Antimicrob. Agents Chemother. 1990, 34, 183. [Google Scholar] [CrossRef] [PubMed]
- Clemons, K.V.; Stevens, D.A. Comparative Efficacies of Four Amphotericin B Formulations—Fungizone, Amphotec (Amphocil), AmBisome, and Abelcet—Against Systemic Murine Aspergillosis. Antimicrob. Agents Chemother. 2004, 48, 1047–1050. [Google Scholar] [CrossRef] [PubMed]
- Struyk, A.P.; Drost, G.; Haisvisz, J.M.; van Eek, T.; Hoogerheide, J.C. Pimaricin, a new antifungal antibiotic. In Antibiotics Annual 1957–1958; Welch, H., Marti-Ibanez, F., Eds.; Medical Encylopedia, Inc.: New York, NY, USA, 1958; pp. 878–885. [Google Scholar]
- Aparicio, J.F.; Fouces, R.; Mendes, M.V.; Olivera, N.; Martín, J.F. A complex multienzyme system encoded by five polyketide synthase genes is involved in the biosynthesis of the 26-membered polyene macrolide pimaricin in Streptomyces natalensis. Chem. Biol. 2000, 7, 895–905. [Google Scholar] [CrossRef]
- Ansari, Z.; Miller, D.; Galor, A. Current thoughts in fungal keratitis: Diagnosis and treatment. Curr. Fungal Infect. Rep. 2013, 7, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Te Welscher, Y.M.; van Leeuwen, M.R.; de Kruijff, B.; Dijksterhuis, J.; Breukink, E. Polyene antibiotic that inhibits membrane transport proteins. Proc. Natl. Acad. Sci. USA 2012, 109, 11156–11159. [Google Scholar] [CrossRef] [Green Version]
- Ma, C.X.; Lv, W.; Li, Y.X.; Fan, B.Z.; Han, X.; Kong, F.S.; Tian, J.C.; Cushman, M.; Liang, J.H. Design, synthesis and structure-activity relationships of novel macrolones: Hybrids of 2-fluoro 9-oxime ketolides and carbamoyl quinolones with highly improved activity against resistant pathogens. Eur. J. Med. Chem. 2019, 169, 1–20. [Google Scholar] [CrossRef]
- Ma, C.; Ma, S. Various novel erythromycin derivatives obtained by different modifications: Recent advance in macrolide antibiotics. Mini Rev. Med. Chem. 2010, 10, 272–286. [Google Scholar] [CrossRef]
- Jelić, D.; Antolović, R. From erythromycin to azithromycin and new potential ribosome-binding antimicrobials. Antibiotics 2016, 5, 29. [Google Scholar] [CrossRef]
- Aronoff, S.C.; Laurent, C.; Jacobs, M.R. In vitro activity of erythromycin, roxithromycin and CP 62993 against common paediatric pathogens. J. Antimicrob. Chemother. 1987, 19, 275–276. [Google Scholar] [CrossRef] [PubMed]
- Schlünzen, F.; Harms, J.M.; Franceschi, F.; Hansen, H.A.S.; Bartels, H.; Zarivach, R.; Yonath, A. Structural basis for the antibiotic activity of ketolides and azalides. Structure 2003, 11, 329–338. [Google Scholar] [CrossRef]
- Ackermann, G.; Rodloff, A.C. Drugs of the 21st century: Telithromycin (HMR 3647)—The first ketolide. J. Antimicrob. Chemother. 2003, 51, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Bonnefoy, A.; Girard, A.M.; Agouridas, C.; Chantot, J.F. Ketolides lack inducibility properties of MLS (B) resistance phenotype. J. Antimicrob. Chemother. 1997, 40, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Douthwaite, S. Structure-activity relationships of ketolides vs. macrolides. Clin. Microbiol. Infect. 2001, 7, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Brockmann, H.; Henkel, W. Pikromycin, ein bitter schmeckendes Antibioticum aus Actinomyceten (Antibiotica aus Actinomyceten, VI. Mitteil. Chem. Ber. 1951, 84, 284–288. [Google Scholar] [CrossRef]
- Weinstein, M.J.; Wagman, G.H.; Marquez, J.A.; Testa, R.T.; Oden, E.; Waitz, J.A. Megalomicin, a new macrolide antibiotic complex produced by Micromonospora. J. Antibiot. (Tokyo) 1969, 22, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Garrod, L.P. The erythromycin group of antibiotics. Br. Med. J. 1957, 2, 57. [Google Scholar] [CrossRef] [PubMed]
- Pinnerts-Indico, S. Une nouvelle espèce de Streptomyces productrice d’antibiotiques: Streptomyces ambofaciens n. sp. Ann. L Inst. Pasteur 1954, 87, 702–707. [Google Scholar]
- Rubinstein, E.; Keller, N. Spiramycin renaissance. J. Antimicrob. Chemother. 1998, 42, 572–576. [Google Scholar] [CrossRef] [Green Version]
- Kanfer, I.; Skinner, M.F.; Walker, R.B. Analysis of macrolide antibiotics. J. Chromatogr. A 1998, 812, 255–286. [Google Scholar] [CrossRef]
- Washington, J.A.; Wilson, W.R. Erythromycin: A Microbial and Clinical Perspective after 30 Years of Clinical Use (Second of Two Parts). Mayo Clin. Proc. 1985, 60, 271–278. [Google Scholar] [CrossRef]
- Papich, M.G. Tylosin. In Saunders Handbook of Veterinary Drugs-E-Book: Small and Large Animal; Elsevier Health Sciences: St. Louis, MO, USA, 2015; pp. 826–827. [Google Scholar]
- Denny, C.B.; Sharpe, L.E.; Bohrer, C.W. Effects of tylosin and nisin on canned food spoilage bacteria. Appl. Microbiol. 1961, 9, 108. [Google Scholar] [PubMed]
- Denny, C.B.; Bohrer, C.W. Effect of antibiotics on the thermal death rate of spores of food spoilage organisms. J. Food Sci. 1959, 24, 247–252. [Google Scholar] [CrossRef]
- Hamill, R.L.; Haney, M.E., Jr.; Stamper, M.; Wlley, P.F. Tylosin, a New Antibiotic: II. Isolation, Properties, and Preparation of Pesmycosin, a Microbiologically Active Degradation Product. Antibiot. Chemother. 1961, 11, 328–334. [Google Scholar]
- Stark, W.M.; Daily, W.A.; McGuire, J.M. A fermentation study of the biosynthesis of tylosin in synthetic media. Sci. Rep. Ist. Super. Sanita 1961, 1, 340–354. [Google Scholar] [PubMed]
- Pape, H.; Brillinger, G.U. Metabolic products of microorganisms. 113. Biosynthesis of thymidine diphospho mycarose in a cell-free system from Streptomyces rimosus. Arch. Mikrobiol. 1973, 88, 25. [Google Scholar] [CrossRef]
- Jensen, A.L.; Darken, M.A.; Schultz, J.S.; Shay, A.J. Relomycin: Flask and Tank Fermentation Studies. Antimicrob. Agents Chemother. 1963, 161, 49–53. [Google Scholar]
- Hamill, R.L.; Stark, W.M. Macromicin, a new antibiotic, and Lactenocin, an active degradation product. J. Antibiot. (Tokyo) 1964, 17, 133–139. [Google Scholar]
- Whaley, H.A.; Patterson, E.L.; Dornbush, A.C.; Backus, E.J.; Bohonos, N. Isolation and characterization of relomycin, a new antibiotic. Antimicrob. Agents Chemother. 1963, 161, 45. [Google Scholar]
- Roets, E.; Beirinckx, P.; Quintens, I.; Hoogmartens, J. Quantitative analysis of tylosin by column liquid chromatography. J. Chromatogr. A 1993, 630, 159–166. [Google Scholar] [CrossRef]
- Loke, M.L.; Ingerslev, F.; Halling-Sørensen, B.; Tjørnelund, J. Stability of tylosin A in manure containing test systems determined by high performance liquid chromatography. Chemosphere 2000, 40, 759–765. [Google Scholar] [CrossRef]
- Cox, K.L.; Fishman, S.E.; Larson, J.L.; Stanzak, R.; Reynolds, P.A.; Yeh, W.K.; van Frank, R.M.; Birmingham, V.A.; Hershberger, C.L.; Seno, E.T. The use of recombinant DNA techniques to study tylosin biosynthesis and resistance in Streptomyces fradiae. J. Nat. Prod. 1986, 49, 971–980. [Google Scholar] [CrossRef]
- Merson-Davies, L.A.; Cundiiffe, E. Analysis of five tylosin biosynthetic genes from the tyllBA region of the Streptomyces fradiae genome. Mol. Microbiol. 1994, 13, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Gandecha, A.R.; Large, S.L.; Cundliffe, E. Analysis of four tylosin biosynthetic genes from the tylLM region of the Streptomyces fradiae genome. Gene 1997, 184, 197–203. [Google Scholar] [CrossRef]
- Fouces, R.; Mellado, E.; Diez, B.; Barredo, J.L. The tylosin biosynthetic cluster from Streptomyces fradiae: Genetic organization of the left region. Microbiology 1999, 145, 855–868. [Google Scholar] [CrossRef] [PubMed]
- Stratigopoulos, G.; Cundliffe, E. Expression Analysis of the Tylosin-Biosynthetic Gene Cluster: Pivotal Regulatory Role of the tylQ Product. Chem. Biol. 2002, 9, 71–78. [Google Scholar] [CrossRef]
- Baltz, R.H.; Seno, E.T. Properties of Streptomyces fradiae mutants blocked in biosynthesis of the macrolide antibiotic tylosin. Antimicrob. Agents Chemother. 1981, 20, 214–225. [Google Scholar] [CrossRef]
- Rodriguez, E.; Ward, S.; Fu, H.; Revill, W.P.; McDaniel, R.; Katz, L. Engineered biosynthesis of 16-membered macrolides that require methoxymalonyl-ACP precursors in Streptomyces fradiae. Appl. Microbiol. Biotechnol. 2004, 66, 85–91. [Google Scholar] [CrossRef]
- Castonguay, R.; Valenzano, C.R.; Chen, A.Y.; Keatinge-Clay, A.; Khosla, C.; Cane, D.E. Stereospecificity of ketoreductase domains 1 and 2 of the tylactone modular polyketide synthase. J. Am. Chem. Soc. 2008, 130, 11598–11599. [Google Scholar] [CrossRef]
- Butler, A.R.; Bate, N.; Cundliffe, E. Impact of thioesterase activity on tylosin biosynthesis in Streptomyces fradiae. Chem. Biol. 1999, 6, 287–292. [Google Scholar] [CrossRef]
- Butler, A.R.; Flint, S.A.; Cundliffe, E. Feedback control of polyketide metabolism during tylosin production. Microbiology 2001, 147, 795–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poulsen, S.M.; Kofoed, C.; Vester, B. Inhibition of the ribosomal peptidyl transferase reaction by the mycarose moiety of the antibiotics carbomycin, spiramycin and tylosin. J. Mol. Biol. 2000, 304, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.L.; Ippolito, J.A.; Ban, N.; Nissen, P.; Moore, P.B.; Steitz, T.A. The structures of four macrolide antibiotics bound to the large ribosomal subunit. Mol. Cell 2002, 10, 117–128. [Google Scholar] [CrossRef]
- Lyutskanova, D.G.; Stoilova-Disheva, M.M.; Peltekova, V.T. Increase in tylosin production by a commercial strain of Streptomyces fradiae. Appl. Biochem. Microbiol. 2005, 41, 165–168. [Google Scholar] [CrossRef]
- Haney, M.E.J.; Hoehn, M.M. Monensin, a new biologically active compound. I. Discovery and isolation. Antimicrob. Agents Chemother. 1967, 7, 349–352. [Google Scholar] [PubMed]
- Łowicki, D.; Huczyński, A. Structure and antimicrobial properties of monensin A and its derivatives: Summary of the achievements. BioMed Res. Int. 2013, 742149. [Google Scholar] [CrossRef]
- Duax, W.L.; Smith, G.D.; Strong, P.D. Complexation of metal ions by monensin. Crystal and molecular structure of hydrated and anhydrous crystal forms of sodium monensin. J. Am. Chem. Soc. 1980, 102, 6725–6729. [Google Scholar] [CrossRef]
- Barrans, Y.; Alleaume, M.; Jeminet, G. Complexe de sodium de l’ionophore monensine B monohydrate. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 1982, 38, 1144–1149. [Google Scholar] [CrossRef]
- Riddell, F.G.; Arumugam, S.; Cox, B.G. The monesin-mediated transport of Na+ and K+ through phospholipid bilayers studied by 23Na-and 39K-NMR. BBA Biomembr. 1988, 944, 279–284. [Google Scholar] [CrossRef]
- Nakazato, K.; Hatano, Y. Monensin-mediated antiport of Na+ and H+ across liposome membrane. BBA Biomembr. 1991, 1064, 103–110. [Google Scholar] [CrossRef]
- Sandeaux, R.; Seta, P.; Jeminet, G.; Alleaume, M.; Gavach, C. The influence of pH on the conductance of lipid bimolecular membranes in relation to the alkaline ion transport induced by carboxylic carriers grisorixin, alborixin and monensin. BBA Biomembr. 1978, 511, 499–508. [Google Scholar] [CrossRef]
- Antonenko, Y.N.; Yaguzhinsky, L.S. The ion selectivity of nonelectrogenic ionophores measured on a bilayer lipid membrane: Nigericin, monensin, A23187 and lasalocid A. BBA Biomembr. 1988, 938, 125–130. [Google Scholar] [CrossRef]
- Zhang, Y.; Lin, C.Y.; Li, X.M.; Tang, Z.K.; Qiao, J.; Zhao, G.R. DasR positively controls monensin production at two-level regulation in Streptomyces cinnamonensis. J. Ind. Microbiol. Biotechnol. 2016, 43, 1681–1692. [Google Scholar] [CrossRef] [PubMed]
- Harvey, B.M.; Hong, H.; Jones, M.A.; Hughes-Thomas, Z.A.; Goss, R.M.; Heathcote, M.L.; Bolanos-Garcia, V.M.; Kroutil, W.; Staunton, J.; Leadlay, P.F.; et al. Evidence that a Novel Thioesterase is Responsible for Polyketide Chain Release during Biosynthesis of the Polyether Ionophore Monensin. ChemBioChem 2006, 7, 1435–1442. [Google Scholar] [CrossRef] [PubMed]
- Hüttel, W.; Spencer, J.B.; Leadlay, P.F. Intermediates in monensin biosynthesis: A late step in biosynthesis of the polyether ionophore monensin is crucial for the integrity of cation binding. Beilstein J. Org. Chem. 2014, 10, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Hou, Y.; Zhang, H.; Chu, Y.; Xia, H.; Tian, Y. Regulatory genes and their roles for improvement of antibiotic biosynthesis in Streptomyces. 3 Biotech 2017, 7, 250. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.K.; Li, X.M.; Pang, A.P.; Lin, C.Y.; Zhang, Y.; Zhang, J.; Qiao, J.; Zhao, G.R. Characterization of three pathway-specific regulators for high production of monensin in Streptomyces cinnamonensis. Appl. Microbiol. Biotechnol. 2017, 101, 6083–6097. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Wang, H.; Kang, Q.; Liu, J.; Bai, L. Cloning and Characterization of the Polyether Salinomycin Biosynthesis Gene Cluster of Streptomyces albus XM211. Appl. Environ. Microbiol. 2012, 78, 994–1003. [Google Scholar] [CrossRef] [PubMed]
- Migita, A.; Watanabe, M.; Hirose, Y.; Watanabe, K.; Tokiwano, T.; Kinashi, H.; Oikawa, H. Identification of a gene cluster of polyether antibiotic lasalocid from Streptomyces lasaliensis. Biosci. Biotechnol. Biochem. 2009, 73, 169–176. [Google Scholar] [CrossRef]
- Roder, J.D. Ionophore Toxicity and Tolerance. Vet. Clin. N. Am. Food Anim. Pract. 2011, 27, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Russell, J.B.; Houlihan, A.J. Ionophore resistance of ruminal bacteria and its potential impact on human health. FEMS Microbiol. Rev. 2003, 27, 65–74. [Google Scholar] [CrossRef]
- Bergen, W.G.; Bates, D.B. Ionophores: Their effect on production efficiency and mode of action. J. Anim. Sci. 1984, 58, 1465–1483. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Wolin, M.J. Effect of monensin and lasalocid-sodium on the growth of methanogenic and rumen saccharolytic bacteria. Appl. Environ. Microbiol. 1979, 38, 72–77. [Google Scholar] [PubMed]
- Newbold, C.J.; Wallace, R.J.; Watt, N.D. Properties of ionophore-resistant Bacteroides rurninicola enriched by cultivation in the presence of tetronasin. J. Appl. Bacteriol. 1992, 72, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Callaway, T.R.; Adams, K.A.; Russell, J.B. The ability of “low G+C gram-positive” ruminal bacteria to resist monensin and counteract potassium depletion. Curr. Microbiol. 1999, 39, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Callaway, T.R.; Russell, J.B. Variations in the ability of ruminal gram-negative Prevotella species to resist monensin. Curr. Microbiol. 2000, 40, 185–189. [Google Scholar] [CrossRef]
- McAlpine, J.B. The ups and downs of drug discovery: The early history of Fidaxomicin. J. Antibiot. (Tokyo) 2017, 70, 492. [Google Scholar] [CrossRef]
- Niu, S.; Hu, T.; Li, S.; Xiao, Y.; Ma, L.; Zhang, G.; Zhang, H.; Yang, X.; Ju, J.; Zhang, C. Characterization of a Sugar-O-methyltransferase TiaS5 Affords New Tiacumicin Analogues with Improved Antibacterial Properties and Reveals Substrate Promiscuity. ChemBioChem 2011, 12, 1740–1748. [Google Scholar] [CrossRef]
- Erb, W.; Zhu, J. From natural product to marketed drug: The tiacumicin odyssey. Nat. Prod. Rep. 2013, 30, 161–174. [Google Scholar] [CrossRef]
- Koglin, A.; Löhr, F.; Bernhard, F.; Rogov, V.V.; Frueh, D.P.; Strieter, E.R.; Mofid, M.R.; Güntert, P.; Wagner, G.; Walsh, C.T.; et al. Structural basis for the selectivity of the external thioesterase of the surfactin synthetase. Nature 2008, 454, 907–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ōmura, S.; Imamura, N.; Oiwa, R.; Kuga, H.; Iwata, R.; Masuma, R.; Iwai, Y. Clostomicins, new antibiotics produced by Micromonospora echinospora subsp. armenica subsp. Nov. I Production, isolation and physico-chemical and biological properties. J. Antibiot. 1986, 39, 1407–1412. [Google Scholar]
- Coronelli, C.; White, R.J.; Lancini, G.C.; Parenti, F. Lipiarmycin, a new antibiotic from Actinoplanes. II. Isolation, chemical, biological and biochemical characterization. J. Antibiot. (Tokyo) 1975, 28, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Louie, T.J.; Emery, J.; Krulicki, W.; Byrne, B.; Mah, M. OPT-80 Eliminates Clostridium Difficile and Is Sparing of Bacteroides Species during Treatment of C. Difficile Infection. Antimicrob. Agents Chemother. 2009, 53, 261–263. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, C.M.; McDermott, L.A.; Tran, M.K.; Chang, J.; Jenkins, S.G.; Goldstein, E.J.C.; Patel, R.; Forbes, B.A.; Johnson, S.; Gerding, D.N.; et al. US-based National Surveillance for Fidaxomicin Susceptibility of Clostridioides difficile (formerly Clostridium) Associated Diarrheal Isolates from 2013–2016. Antimicrob. Agents Chemother. 2019, 63, e00391-19. [Google Scholar] [CrossRef] [PubMed]
- Talpaert, M.; Campagnari, F.; Clerici, L. Lipiarmycin: An antibiotic inhibiting nucleic acid polymerases. Biochem. Biophys. Res. Commun. 1975, 63, 328–334. [Google Scholar] [CrossRef]
- Zhang, H.; Tian, X.; Pu, X.; Zhang, Q.; Zhang, W.; Zhang, C. Tiacumicin Congeners with Improved Antibacterial Activity from a Halogenase-Inactivated Mutant. J. Nat. Prod. 2018, 81, 1219–1224. [Google Scholar] [CrossRef] [PubMed]
- Bartsch, S.M.; Umscheid, C.A.; Fishman, N.; Lee, B.Y. Is fidaxomicin worth the cost? An economic analysis. Clin. Infect. Dis. 2013, 57, 555–561. [Google Scholar] [CrossRef]
- Lechevalier, M.P.; Prauser, H.; Labeda, D.P.; Ruan, J.S. Two new genera of nocardioform actinomycetes: Amycolata gen. nov. and Amycolatopsis gen. nov. Int. J. Syst. Evol. Microbiol. 1986, 36, 29–37. [Google Scholar] [CrossRef]
- Mariani, R.; Maffioli, S.I. Bacterial RNA polymerase inhibitors: An organized overview of their structure, derivatives, biological activity and current clinical development status. Curr. Med. Chem. 2009, 16, 430–454. [Google Scholar] [CrossRef]
- Sensi, P. History of the Development of Rifampin. Clin. Infect. Dis. 1983, 5, S402–S406. [Google Scholar] [CrossRef] [PubMed]
- Oppolzer, W.; Prelog, V.; Sensi, P. Konstitution des Rifamycins B und verwandter Rifamycine. Experientia 1964, 20, 336–339. [Google Scholar] [CrossRef] [PubMed]
- Lancini, G.G.; Gallo, G.G.; Sartori, G.; Sensi, P. Isolation and structure of rifamycin L and its biogenetic relationship with other rifamycins. J. Antibiot. (Tokyo) 1969, 22, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Admiraal, S.J.; Walsh, C.T.; Khosla, C. The Loading Module of Rifamycin Synthetase Is an Adenylation−Thiolation Didomain with Substrate Tolerance for Substituted Benzoates. Biochemistry 2001, 40, 6116–6123. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.W.; Müller, R.; Müller, M.; Zhang, X.; Draeger, G.; Kim, C.G.; Leistner, E.; Floss, H.G. Mutational analysis and reconstituted expression of the biosynthetic genes involved in the formation of 3-amino-5-hydroxybenzoic acid, the starter unit of rifamycin biosynthesis in Amycolatopsis mediterranei S699. J. Biol. Chem. 2001, 276, 12546–12555. [Google Scholar] [CrossRef]
- Yu, T.W.; Shen, Y.; Doi-Katayama, Y.; Tang, L.; Park, C.; Moore, B.S.; Hutchinson, C.R.; Floss, H.G. Direct evidence that the rifamycin polyketide synthase assembles polyketide chains processively. Proc. Natl. Acad. Sci. USA 1999, 96, 9051–9056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doi-Katayama, Y.; Yoon, Y.J.; Choi, C.Y.; Yu, T.W.; Floss, H.G.; Hutchinson, C.R. Thioesterases and the premature termination of polyketide chain elongation in rifamycin B biosynthesis by Amycolatopsis mediterranei S699. J. Antibiot. (Tokyo) 2000, 53, 484–495. [Google Scholar] [CrossRef]
- Floss, H.G.; Yu, T.W. Lessons from the rifamycin biosynthetic gene cluster. Curr. Opin. Chem. Biol. 1999, 3, 592–597. [Google Scholar] [CrossRef]
- Xu, J.; Wan, E.; Kim, C.J.; Floss, H.G.; Mahmud, T. Identification of tailoring genes involved in the modification of the polyketide backbone of rifamycin B by Amycolatopsis mediterranei S699. Microbiology 2005, 151, 2515–2528. [Google Scholar] [CrossRef]
- White, R.J.; Martinelli, E.; Lancini, G. Ansamycin biogenesis: Studies on a novel rifamycin isolated from a mutant strain of Nocardia mediterranei. Proc. Natl. Acad. Sci. USA 1974, 71, 3260–3264. [Google Scholar] [CrossRef]
- Qi, F.; Lei, C.; Li, F.; Zhang, X.; Wang, J.; Zhang, W.; Fan, Z.; Li, W.; Tang, G.; Xiao, Y.; et al. Deciphering the late steps of rifamycin biosynthesis. Nat. Commun. 2018, 9, 2342. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Liu, X.; Lei, C.; Yan, H.; Shao, Z.; Wang, Y.; Zhao, G.; Wang, J.; Ding, X. RifZ (AMED_0655) is a pathway-specific regulator for rifamycin biosynthesis in Amycolatopsis mediterranei. Appl. Environ. Microbiol. 2017, 83, e03201-16. [Google Scholar] [CrossRef] [PubMed]
- Lei, C.; Wang, J.; Liu, Y.; Liu, X.; Zhao, G.; Wang, J. A feedback regulatory model for RifQ-mediated repression of rifamycin export in Amycolatopsis mediterranei. Microb. Cell Fact. 2018, 17, 14. [Google Scholar] [CrossRef] [PubMed]
- Absalon, A.E.; Fernández, F.J.; Olivares, P.X.; Barrios-González, J.; Campos, C.; Mejía, A. RifP; a membrane protein involved in rifamycin export in Amycolatopsis mediterranei. Biotechnol. Lett. 2007, 29, 951–958. [Google Scholar] [CrossRef] [PubMed]
- Wehrli, W. Rifampin: Mechanisms of action and resistance. Rev. Infect. Dis. 1983, 5, S407–S411. [Google Scholar] [CrossRef] [PubMed]
- Boucher, H.W.; Talbot, G.H.; Benjamin, D.K., Jr.; Bradley, J.; Guidos, R.J.; Jones, R.N.; Murray, B.E.; Bonomo, R.A.; Gilbert, D.; The Infectious Diseases Society of America. 10×’20 progress—Development of new drugs active against gram-negative bacilli: An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2013, 56, 1685–1694. [Google Scholar] [CrossRef] [PubMed]
- Ponziani, F.R.; Scaldaferri, F.; Petito, V.; Paroni Sterbini, F.; Pecere, S.; Lopetuso, L.R.; Palladini, A.; Gerardi, V.; Masucci, L.; Pompili, M.; et al. The role of antibiotics in gut microbiota modulation: The eubiotic effects of rifaximin. Dig. Dis. 2016, 34, 269–278. [Google Scholar] [CrossRef]
- Weber, D.; Oefner, P.J.; Dettmer, K.; Hiergeist, A.; Koestler, J.; Gessner, A.; Weber, M.; Stämmler, F.; Hahn, J.; Wolff, D.; et al. Rifaximin preserves intestinal microbiota balance in patients undergoing allogeneic stem cell transplantation. Bone Marrow Transplant. 2016, 51, 1087. [Google Scholar] [CrossRef] [PubMed]
- Aristoff, P.A.; Garcia, G.A.; Kirchhoff, P.D.; Showalter, H.D. Rifamycins-obstacles and opportunities. Tuberculosis (Edinb.) 2010, 90, 94–118. [Google Scholar] [CrossRef]
- Conover, L.H.; Moreland, W.T.; English, A.R.; Stephens, C.R.; Pilgrim, F.J.; Terramycin, X.I. Tetracycline. J. Am. Chem. Soc. 1953, 75, 4622–4623. [Google Scholar] [CrossRef]
- Nelson, M.L.; Levy, S.B. The history of the tetracyclines. Ann. N. Y. Acad. Sci. 2011, 1241, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Ames, B.D.; Tsai, S.C.; Tang, Y. Engineered biosynthesis of a novel amidated polyketide, using the malonamyl-specific initiation module from the oxytetracycline polyketide synthase. Appl. Environ. Microbiol. 2006, 72, 2573–2580. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Gao, X.; Chooi, Y.H.; Deng, Z.; Tang, Y. Genetic characterization of enzymes involved in the priming steps of oxytetracycline biosynthesis in Streptomyces rimosus. Microbiology 2011, 157, 2401–2409. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Watanabe, K.; Wang, C.C.C.; Tang, Y. Heterologous biosynthesis of amidated polyketides with novel cyclization regioselectivity from oxytetracycline polyketide synthase. J. Nat. Prod. 2006, 69, 1633–1636. [Google Scholar] [CrossRef] [PubMed]
- Pickens, L.B.; Tang, Y. Oxytetracycline biosynthesis. J. Biol. Chem. 2010, 285, 27509–27515. [Google Scholar] [CrossRef] [PubMed]
- Petković, H.; Lukežič, T.; Šušković, J. Biosynthesis of oxytetracycline by Streptomyces rimosus: Past, present and future directions in the development of tetracycline antibiotics. Food Technol. Biotechnol. 2017, 55, 3–13. [Google Scholar] [CrossRef]
- Wang, P.; Bashiri, G.; Gao, X.; Sawaya, M.R.; Tang, Y. Uncovering the enzymes that catalyze the final steps in oxytetracycline biosynthesis. J. Am. Chem. Soc. 2013, 135, 7138–7141. [Google Scholar] [CrossRef]
- Ohnuki, T.; Katoh, T.; Imanaka, T.; Aiba, S. Molecular cloning of tetracycline resistance genes from Streptomyces rimosus in Streptomyces griseus and characterization of the cloned genes. J. Bacteriol. 1985, 161, 1010–1016. [Google Scholar]
- Lešnik, U.; Gormand, A.; Magdevska, V.; Fujs, Š.; Raspor, P.; Hunter, I.; Petković, H. Regulatory elements in tetracycline-encoding gene clusters: The otcG gene positively regulates the production of oxytetracycline in Streptomyces rimosus. Food Technol. Biotechnol. 2009, 47, 323–330. [Google Scholar]
- Yin, S.; Wang, W.; Wang, X.; Zhu, Y.; Jia, X.; Li, S.; Yuan, F.; Zhang, Y.; Yang, K. Identification of a cluster-situated activator of oxytetracycline biosynthesis and manipulation of its expression for improved oxytetracycline production in Streptomyces rimosus. Microb. Cell Fact. 2015, 14, 46. [Google Scholar] [CrossRef]
- Jenner, L.; Starosta, A.L.; Terry, D.S.; Mikolajka, A.; Filonava, L.; Yusupov, M.; Blanchard, S.C.; Wilson, D.N.; Yusupova, G. Structural basis for potent inhibitory activity of the antibiotic tigecycline during protein synthesis. Proc. Natl. Acad. Sci. USA 2013, 110, 3812–3816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, M.L.; Park, B.H.; Levy, S.B. Molecular requirements for the inhibition of the tetracycline antiport protein and the effect of potent inhibitors on the growth of tetracycline-resistant bacteria. J. Med. Chem. 1994, 37, 1355–1361. [Google Scholar] [CrossRef] [PubMed]
- Villano, S.; Steenbergen, J.; Loh, E. Omadacycline: Development of a novel aminomethylcycline antibiotic for treating drug-resistant bacterial infections. Future Microbiol. 2016, 11, 1421–1434. [Google Scholar] [CrossRef] [PubMed]
- Barriere, J.C.; Berthaud, N.; Beyer, D.; Dutka-Malen, S.; Paris, J.M.; Desnottes, J.F. Recent developments in streptogramin research. Curr. Pharm. Des. 1998, 4, 155. [Google Scholar] [PubMed]
- Mast, Y.; Wohlleben, W. Streptogramins—Two are better than one! Int. J. Med. Microbiol. 2014, 304, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Charney, J.; Fisher, W.P.; Curran, C.; Machlowitz, R.A.; Tytell, A.A. Streptogramin, a new antibiotic. Antibiot. Chemother. (Northfield, III.) 1953, 3, 1283–1286. [Google Scholar]
- Arai, M.; Karasawa, K.; Nakamura, S.; Yonehara, H.; Umezawa, H. Studies on mikamycin. I. J. Antibiot. (Tokyo) 1958, 11, 14–20. [Google Scholar]
- Watanabe, K. Studies on mikamycin. VII. Structure of mikamycin B. J. Antibiot. Ser. A 1961, 14, 14–17. [Google Scholar]
- Bartz, Q.R.; Standiford, J.; Mold, J.D.; Johannessen, D.W.; Ryder, A.; Maretski, A.; Haskell, T.H. Antibiotics Annual (1954–1955); Medical Encyclopedia Inc.: New York, NY, USA, 1954; pp. 777–783. [Google Scholar]
- De Somer, P.; Van Dijck, P. A preliminary report on antibiotic number 899, a streptogramin-like substance. Antibiot. Chemother. (Northfield, III.) 1955, 5, 632–639. [Google Scholar]
- Casewell, M.; Friis, C.; Marco, E.; McMullin, P.; Phillips, I. The European ban on growth-promoting antibiotics and emerging consequences for human and animal health. J. Antimicrob. Chemother. 2003, 52, 159–161. [Google Scholar] [CrossRef] [Green Version]
- Musiol, E.M.; Greule, A.; Härtner, T.; Kulik, A.; Wohlleben, W.; Weber, T. The AT2 domain of KirCI loads malonyl extender units to the ACPs of the kirromycin PKS. ChemBioChem 2013, 14, 1343–1352. [Google Scholar] [CrossRef]
- Weber, T.; Laiple, K.J.; Pross, E.K.; Textor, A.; Grond, S.; Welzel, K.; Pelzer, S.; Vente, A.; Wohlleben, W. Molecular analysis of the kirromycin biosynthetic gene cluster revealed β-alanine as precursor of the pyridone moiety. Chem. Biol. 2008, 15, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Mast, Y.; Guezguez, J.; Handel, F.; Schinko, E. A complex signaling cascade governs pristinamycin biosynthesis in Streptomyces pristinaespiralis. Appl. Environ. Microbiol. 2015, 81, 6621–6636. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Tian, J.; Li, L.; Ge, M.; Zhu, H.; Zheng, G.; Huang, H.; Ruan, L.; Jiang, W.; Lu, Y. Identification of two novel regulatory genes involved in pristinamycin biosynthesis and elucidation of the mechanism for AtrA-p-mediated regulation in Streptomyces pristinaespiralis. Appl. Microbiol. Biotechnol. 2015, 99, 7151–7164. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Feng, R.; Zheng, G.; Ge, M.; Mast, Y.; Wohlleben, W.; Gao, J.; Jiang, W.; Lu, Y. Improvement of pristinamycin I (PI) production in Streptomyces pristinaespiralis by metabolic engineering approaches. Synth. Syst. Biotechnol. 2017, 2, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhao, Y.; Ruan, L.; Yang, S.; Ge, M.; Jiang, W.; Lu, Y. A stepwise increase in pristinamycin II biosynthesis by Streptomyces pristinaespiralis through combinatorial metabolic engineering. Metab. Eng. 2015, 29, 12–25. [Google Scholar] [CrossRef]
- Nyssen, E.; Di Giambattista, M.; Cocito, C. Analysis of the reversible binding of virginiamycin M to ribosome and particle functions after removal of the antibiotic. Biochim. Biophys. Acta Gene Struct. Expr. 1989, 1009, 39–46. [Google Scholar] [CrossRef]
- Johnston, N.J.; Mukhtar, T.A.; Wright, G.D. Streptogramin antibiotics: Mode of action and resistance. Curr. Drug Targets 2002, 3, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Roberts, M.C. Environmental macrolide–lincosamide–streptogramin and tetracycline resistant bacteria. Front. Microbiol. 2011, 2, 40. [Google Scholar] [CrossRef] [PubMed]
- Roberts, M.C. Resistance to macrolide, lincosamide, streptogramin, ketolide, and oxazolidinone antibiotics. Mol. Biotechnol. 2004, 28, 47. [Google Scholar] [CrossRef]
- Poehlsgaard, J.; Douthwaite, S. The bacterial ribosome as a target for antibiotics. Nat. Rev. Microbiol. 2005, 3, 870. [Google Scholar] [CrossRef] [PubMed]
- Allignet, J.; Loncle, V.; Mazodier, P.; El Solh, N. Nucleotide sequence of a staphylococcal plasmid gene, vgb, encoding a hydrolase inactivating the B components of virginiamycin-like antibiotics. Plasmid 1988, 20, 271–275. [Google Scholar] [CrossRef]
- Allignet, J.; Loncle, V.; El Solh, N. Sequence of a staphylococcal plasmid gene, vga, encoding a putative ATP-binding protein involved in resistance to virginiamycin A-like antibiotics. Gene 1992, 117, 45–51. [Google Scholar] [CrossRef]
- Aparicio, J.F.; Caffrey, P.; Gil, J.A.; Zotchev, S.B. Polyene antibiotic biosynthesis gene clusters. Appl. Microbiol. Biotechnol. 2003, 61, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Fjærvik, E.; Zotchev, S.B. Biosynthesis of the polyene macrolide antibiotic nystatin in Streptomyces noursei. Appl. Microbiol. Biotechnol. 2005, 67, 436–443. [Google Scholar] [CrossRef] [PubMed]
- Hazen, E.L.; Brown, R. Two antifungal agents produced by a soil actinomycete. Science 1950, 112, 423. [Google Scholar] [PubMed]
- Mechlinski, W.; Schaffner, C.P. Separation of polyene antifungal antibiotics by high-speed liquid chromatography. J. Chromatogr. A 1974, 99, 619–633. [Google Scholar] [CrossRef]
- Matsuoka, M. Biological studies on antifungal substances produced by Streptomyces fungicidicus. J. Antibiot. (Tokyo) 1960, 13, 121–124. [Google Scholar]
- Veiga, M.; Fabregas, J. Tetrafungin, a new polyene macrolide antibiotic. I. Fermentation, isolation, characterization, and biological properties. J. Antibiot. (Tokyo) 1983, 36, 770–775. [Google Scholar] [CrossRef]
- Chong, C.N.; Rickards, R.W. Macrolide antibiotic studies. XVI. The structure of nystatin. Tetrahedron Lett. 1970, 11, 5145–5148. [Google Scholar] [CrossRef]
- Sletta, H.; Borgos, S.E.F.; Bruheim, P.; Sekurova, O.N.; Grasdalen, H.; Aune, R.; Ellingsen, T.E.; Zotchev, S.B. Nystatin biosynthesis and transport: nysH and nysG genes encoding a putative ABC transporter system in Streptomyces noursei ATCC 11455 are required for efficient conversion of 10-deoxynystatin to nystatin. Antimicrob. Agents Chemother. 2005, 49, 4576–4583. [Google Scholar] [CrossRef] [PubMed]
- Sekurova, O.N.; Brautaset, T.; Sletta, H.; Borgos, S.E.F.; Jakobsen, Ø.M.; Ellingsen, T.E.; Strøm, A.R.; Valla, S.; Zotchev, S.B. In vivo analysis of the regulatory genes in the nystatin biosynthetic gene cluster of Streptomyces noursei ATCC 11455 reveals their differential control over antibiotic biosynthesis. J. Bacteriol. 2004, 186, 1345–1354. [Google Scholar] [CrossRef] [PubMed]
- Gupte, M.; Kulkarni, P.; Ganguli, B. Antifungal antibiotics. Appl. Microbiol. Biotechnol. 2002, 58, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Hamilton-Miller, J.M. Chemistry and biology of the polyene macrolide antibiotics. Bacteriol. Rev. 1973, 37, 166. [Google Scholar] [PubMed]
- Lee, M.Y.; Myeong, J.S.; Park, H.J.; Han, K.; Kim, E.S. Isolation and partial characterization of a cryptic polyene gene cluster in Pseudonocardia autotrophica. J. Ind. Microbiol. Biotechnol. 2006, 33, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.G.; Lee, M.J.; Seo, J.; Hwang, Y.B.; Lee, M.Y.; Han, K.; Sherman, D.H.; Kim, E.S. Identification of functionally clustered nystatin-like biosynthetic genes in a rare actinomycetes, Pseudonocardia autotrophica. J. Ind. Microbiol. Biotechnol. 2009, 36, 1425. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Kong, D.; Han, K.; Sherman, D.H.; Bai, L.; Deng, Z.; Lin, S.; Kim, E.S. Structural analysis and biosynthetic engineering of a solubility-improved and less-hemolytic nystatin-like polyene in Pseudonocardia autotrophica. Appl. Microbiol. Biotechnol. 2012, 95, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Han, C.Y.; Park, J.S.; Oh, S.H.; Kang, S.H.; Choi, S.S.; Kim, J.M.; Kwak, J.H.; Kim, E.S. Nystatin-like Pseudonocardia polyene B1, a novel disaccharide-containing antifungal heptaene antibiotic. Sci. Rep. 2018, 8, 13584. [Google Scholar] [CrossRef] [PubMed]
- Caffrey, P.; Lynch, S.; Flood, E.; Finnan, S.; Oliynyk, M. Amphotericin biosynthesis in Streptomyces nodosus: Deductions from analysis of polyketide synthase and late genes. Chem. Biol. 2001, 8, 713–723. [Google Scholar] [CrossRef]
- Carmody, M.; Murphy, B.; Byrne, B.; Power, P.; Rai, D.; Rawlings, B.; Caffrey, P. Biosynthesis of amphotericin derivatives lacking exocyclic carboxyl groups. J. Biol. Chem. 2005, 280, 34420–34426. [Google Scholar] [CrossRef]
- Carmody, M.; Byrne, B.; Murphy, B.; Breen, C.; Lynch, S.; Flood, E.; Finnan, S.; Caffrey, P. Analysis and manipulation of amphotericin biosynthetic genes by means of modified phage KC515 transduction techniques. Gene 2004, 343, 107–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellis, D. Amphotericin B: Spectrum and resistance. J. Antimicrob. Chemother. 2002, 49, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Goldman, C.; Akiyama, M.J.; Torres, J.; Louie, E.; Meehan, S.A. Scedosporium apiospermum infections and the role of combination antifungal therapy and GM-CSF: A case report and review of the literature. Med. Mycol. Case Rep. 2016, 11, 40–43. [Google Scholar] [CrossRef] [PubMed]
- Volmer, A.A.; Szpilman, A.M.; Carreira, E.M. Synthesis and biological evaluation of amphotericin B derivatives. Nat. Prod. Rep. 2010, 27, 1329–1349. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zhang, H.; Zhou, Y.; Huang, K.; Liu, Z.; Zheng, Y. Improvement of amphotericin B production by a newly isolated Streptomyces nodosus mutant. Biotechnol. Appl. Biochem. 2018, 65, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Aparicio, J.F.; Barreales, E.G.; Payero, T.D.; Vicente, C.M.; de Pedro, A.; Santos-Aberturas, J. Biotechnological production and application of the antibiotic pimaricin: Biosynthesis and its regulation. Appl. Microbiol. Biotechnol. 2016, 100, 61–78. [Google Scholar] [CrossRef]
- Divekar, P.V.; Bloomer, J.L.; Eastham, J.F.; Holtman, D.F.; Shirley, D.A. The isolation of crystalline tennecetin and the comparison of this antibiotic with pimaricin. Antibiot. Chemother. (Northfield, III.) 1961, 11, 377. [Google Scholar]
- Burns, J. Tennecetin: A new antifungal antibiotic. Antibiot. Chemother. 1959, 9, 398–405. [Google Scholar]
- Sui, Q.; Liu, W.; Lu, C.; Liu, T.; Qiu, J.; Liu, X. Extraction and structural identification of the antifungal metabolite of Streptomyces lydicus A02. Chin. J. Biotechnol. 2009, 25, 840–846. [Google Scholar]
- Wang, T.J.; Shan, Y.M.; Li, H.; Dou, W.W.; Jiang, X.H.; Mao, X.M.; Liu, S.P.; Guan, W.J.; Li, Y.Q. Multiple transporters are involved in natamycin efflux in Streptomyces chattanoogensis L10. Mol. Microbiol. 2017, 103, 713–728. [Google Scholar] [CrossRef]
- Anton, N.; Santos-Aberturas, J.; Mendes, M.V.; Guerra, S.M.; Martin, J.F.; Aparicio, J.F. PimM, a PAS domain positive regulator of pimaricin biosynthesis in Streptomyces natalensis. Microbiology 2007, 153, 3174–3183. [Google Scholar] [CrossRef] [PubMed]
- Anton, N.; Mendes, M.V.; Martin, J.F.; Aparicio, J.F. Identification of PimR as a positive regulator of pimaricin biosynthesis in Streptomyces natalensis. J. Bacteriol. 2004, 186, 2567–2575. [Google Scholar] [CrossRef] [PubMed]
- Vicente, C.M.; Santos-Aberturas, J.; Guerra, S.M.; Payero, T.D.; Martin, J.F.; Aparicio, J.F. PimT, an amino acid exporter controls polyene production via secretion of the quorum sensing pimaricin-inducer PI-factor in Streptomyces natalensis. Microb. Cell Fact. 2009, 8, 33. [Google Scholar] [CrossRef] [PubMed]
- Mendes, M.V.; Recio, E.; Antón, N.; Guerra, S.M.; Santos-Aberturas, J.; Martín, J.F.; Aparicio, J.F. Cholesterol oxidases act as signaling proteins for the biosynthesis of the polyene macrolide pimaricin. Chem. Biol. 2007, 14, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Aparicio, J.F.; Martín, J.F. Microbial cholesterol oxidases: Bioconversion enzymes or signal proteins? Mol. Biosyst. 2008, 4, 804–809. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.L.; Chen, S.F.; Cheng, L.Y.; Shen, X.L.; Tian, Y.; Li, Y.Q. Identification of a novel Streptomyces chattanoogensis L10 and enhancing its natamycin production by overexpressing positive regulator ScnRII. J. Microbiol. 2009, 47, 506–513. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Liu, F.; Hou, Z.; Zong, G.; Zhu, X.; Ling, P. Enhancement of natamycin production on Streptomyces gilvosporeus by chromosomal integration of the Vitreoscilla hemoglobin gene (vgb). World J. Microbiol. Biotechnol. 2014, 30, 1369–1376. [Google Scholar] [CrossRef]
- Lee, K.M.; Lee, C.K.; Choi, S.U.; Park, H.R.; Kitani, S.; Nihira, T.; Hwang, Y.I. Cloning and in vivo functional analysis by disruption of a gene encoding the γ-butyrolactone autoregulator receptor from Streptomyces natalensis. Arch. Microbiol. 2005, 184, 249–257. [Google Scholar] [CrossRef]
- Qi, Z.; Kang, Q.; Jiang, C.; Han, M.; Bai, L. Engineered biosynthesis of pimaricin derivatives with improved antifungal activity and reduced cytotoxicity. Appl. Microbiol. Biotechnol. 2015, 99, 6745–6752. [Google Scholar] [CrossRef]
- Li, X.Z.; Plésiat, P.; Nikaido, H. The challenge of efflux-mediated antibiotic resistance in Gram-negative bacteria. Clin. Microbiol. Rev. 2015, 28, 337–418. [Google Scholar] [CrossRef]
- Coates, A.R.M.; Halls, G.; Hu, Y. Novel classes of antibiotics or more of the same? Br. J. Pharmacol. 2011, 163, 184–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spellberg, B.; Bartlett, J.G.; Gilbert, D.N. The future of antibiotics and resistance. N. Engl. J. Med. 2013, 368, 299–302. [Google Scholar] [CrossRef] [PubMed]
- Blunt, J.W.; Copp, B.R.; Munro, M.H.G.; Northcote, P.T.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2011, 28, 196–268. [Google Scholar] [CrossRef] [PubMed]
- Katz, L.; Baltz, R.H. Natural product discovery: Past, present, and future. J. Ind. Microbiol. Biotechnol. 2016, 43, 155–176. [Google Scholar] [CrossRef] [PubMed]
- Kealey, C.; Creaven, C.A.; Murphy, C.D.; Brady, C.B. New approaches to antibiotic discovery. Biotechnol. Lett. 2017, 39, 805–817. [Google Scholar] [CrossRef] [PubMed]
- Iorio, M.; Tocchetti, A.; Cruz, J.; Del Gatto, G.; Brunati, C.; Maffioli, S.; Sosio, M.; Donadio, S. Novel Polyethers from Screening Actinoallomurus spp. Antibiotics 2018, 7, 47. [Google Scholar] [CrossRef] [PubMed]
- Hug, J.; Bader, C.; Remškar, M.; Cirnski, K.; Müller, R. Concepts and methods to access novel antibiotics from actinomycetes. Antibiotics 2018, 7, 44. [Google Scholar] [CrossRef] [PubMed]
- Genilloud, O. Mining actinomycetes for novel antibiotics in the omics era: Are we ready to exploit this new paradigm? Antibiotics 2018, 7, 85. [Google Scholar] [CrossRef]
- Amoutzias, G.; Chaliotis, A.; Mossialos, D. Discovery strategies of bioactive compounds synthesized by nonribosomal peptide synthetases and type-I polyketide synthases derived from marine microbiomes. Mar. Drugs 2016, 14, 80. [Google Scholar] [CrossRef]
- Adnani, N.; Chevrette, M.; Adibhatla, S.N.; Zhang, F.; Yu, Q.; Braun, D.R.; Nelson, J.; Simpkins, S.W.; McDonald, B.R.; Myers, C.L.; et al. Coculture of marine invertebrate-associated bacteria and interdisciplinary technologies enable biosynthesis and discovery of a new antibiotic, keyicin. ACS Chem. Biol. 2017, 12, 3093–3102. [Google Scholar] [CrossRef]
- Goers, L.; Freemont, P.; Polizzi, K.M. Co-culture systems and technologies: Taking synthetic biology to the next level. J. R. Soc. Interface 2014, 11, 20140065. [Google Scholar] [CrossRef]
- Dashti, Y.; Grkovic, T.; Abdelmohsen, U.; Hentschel, U.; Quinn, R. Production of induced secondary metabolites by a co-culture of sponge-associated actinomycetes, Actinokineospora sp. EG49 and Nocardiopsis sp. RV163. Mar. Drugs 2014, 12, 3046–3059. [Google Scholar] [CrossRef]
- Piel, J. Metabolites from symbiotic bacteria. Nat. Prod. Rep. 2009, 26, 338–362. [Google Scholar] [CrossRef]
- Ling, L.L.; Schneider, T.; Peoples, A.J.; Spoering, A.L.; Engels, I.; Conlon, B.P.; Mueller, A.; Schäberle, T.F.; Hughes, D.E.; Epstein, S.; et al. A new antibiotic kills pathogens without detectable resistance. Nature 2015, 517, 455. [Google Scholar] [CrossRef]
- Nichols, D.; Cahoon, N.; Trakhtenberg, E.M.; Pham, L.; Mehta, A.; Belanger, A.; Kanigan, T.; Lewis, K.; Epstein, S.S. Use of ichip for high-throughput in situ cultivation of “uncultivable” microbial species. Appl. Environ. Microbiol. 2010, 76, 2445–2450. [Google Scholar] [CrossRef]
- Piddock, L.J.V. Teixobactin, the first of a new class of antibiotics discovered by iChip technology? J. Antimicrob. Chemother. 2015, 70, 2679–2680. [Google Scholar] [CrossRef]
- Sherpa, R.T.; Reese, C.J.; Aliabadi, H.M. Application of iChip to grow “uncultivable” microorganisms and its impact on antibiotic discovery. J. Pharm. Pharm. Sci. 2015, 18, 303–315. [Google Scholar] [CrossRef]
- Alessi, A.M.; Redeker, K.R.; Chong, J.P.J. A practical introduction to microbial molecular ecology through the use of isolation chips. Ecol. Evol. 2018, 8, 12286–12298. [Google Scholar] [CrossRef]
- Lodhi, A.F.; Zhang, Y.; Adil, M.; Deng, Y. Antibiotic discovery: Combining isolation chip (iChip) technology and co-culture technique. Appl. Microbiol. Biotechnol. 2018, 102, 7333–7341. [Google Scholar] [CrossRef]
- Mohana, N.C.; Rao, H.C.Y.; Rakshith, D.; Mithun, P.R.; Nuthan, B.R.; Satish, S. Omics based approach for biodiscovery of microbial natural products in antibiotic resistance era. J. Genet. Eng. Biotechnol. 2018, 16, 1–8. [Google Scholar] [CrossRef]
- Vijayakumar, S.; Conway, M.; Lió, P.; Angione, C. Optimization of multi-omic genome-scale models: Methodologies, hands-on tutorial, and perspectives. In Metabolic Network Reconstruction and Modeling; Springer: Berlin/Heidelberg, Germany, 2018; pp. 389–408. [Google Scholar]
- Franzosa, E.A.; Hsu, T.; Sirota-Madi, A.; Shafquat, A.; Abu-Ali, G.; Morgan, X.C.; Huttenhower, C. Sequencing and beyond: Integrating molecular’omics’ for microbial community profiling. Nat. Rev. Microbiol. 2015, 13, 360. [Google Scholar] [CrossRef]
- Palazzotto, E.; Weber, T. Omics and multi-omics approaches to study the biosynthesis of secondary metabolites in microorganisms. Curr. Opin. Microbiol. 2018, 45, 109–116. [Google Scholar] [CrossRef]
- Nikolouli, K.; Mossialos, D. Bioactive compounds synthesized by non-ribosomal peptide synthetases and type-I polyketide synthases discovered through genome-mining and metagenomics. Biotechnol. Lett. 2012, 34, 1393–1403. [Google Scholar] [CrossRef]
- Medema, M.H.; Fischbach, M.A. Computational approaches to natural product discovery. Nat. Chem. Biol. 2015, 11, 639. [Google Scholar] [CrossRef]
- Carbonell, P.; Currin, A.; Jervis, A.J.; Rattray, N.J.W.; Swainston, N.; Yan, C.; Takano, E.; Breitling, R. Bioinformatics for the synthetic biology of natural products: Integrating across the Design-Build-Test cycle. Nat. Prod. Rep. 2016, 33, 925–932. [Google Scholar] [CrossRef]
- Ito, T.; Masubuchi, M. Dereplication of microbial extracts and related analytical technologies. J. Antibiot. (Tokyo) 2014, 67, 353–360. [Google Scholar] [CrossRef]
- Seger, C.; Sturm, S.; Stuppner, H. Mass spectrometry and NMR spectroscopy: Modern high-end detectors for high resolution separation techniques--state of the art in natural product HPLC-MS, HPLC-NMR, and CE-MS hyphenations. Nat. Prod. Rep. 2013, 30, 970–987. [Google Scholar] [CrossRef]
- Perez-Victoria, I.; Martin, J.; Reyes, F. Combined LC/UV/MS and NMR Strategies for the Dereplication of Marine Natural Products. Planta Med. 2016, 82, 857–871. [Google Scholar] [CrossRef] [Green Version]
- Spraker, J.E.; Luu, G.T.; Sanchez, L.M. Imaging mass spectrometry for natural products discovery: A review of ionization methods. Nat. Prod. Rep. 2019. [Google Scholar] [CrossRef]
- Zhu, H.; Sandiford, S.K.; van Wezel, G.P. Triggers and cues that activate antibiotic production by actinomycetes. J. Ind. Microbiol. Biotechnol. 2014, 41, 371–386. [Google Scholar] [CrossRef]
- Rutledge, P.J.; Challis, G.L. Discovery of microbial natural products by activation of silent biosynthetic gene clusters. Nat. Rev. Microbiol. 2015, 13, 509–523. [Google Scholar] [CrossRef]
- Seyedsayamdost, M.R. High-throughput platform for the discovery of elicitors of silent bacterial gene clusters. Proc. Natl. Acad. Sci. USA 2014, 111, 7266–7271. [Google Scholar] [CrossRef] [Green Version]
- Rosen, P.C.; Seyedsayamdost, M.R. Though Much Is Taken, Much Abides: Finding New Antibiotics Using Old Ones. Biochemistry 2017, 56, 4925–4926. [Google Scholar] [CrossRef] [Green Version]
- Okada, B.K.; Seyedsayamdost, M.R. Antibiotic dialogues: Induction of silent biosynthetic gene clusters by exogenous small molecules. FEMS Microbiol. Rev. 2017, 41, 19–33. [Google Scholar] [CrossRef]
- Altaee, N.; Kadhim, M.J.; Hameed, I.H. Characterization of metabolites produced by E. coli and analysis of its chemical compounds using GC-MS. Int. J. Curr. Pharm. Rev. Res. 2017, 7, 13–19. [Google Scholar]
- Wexler, M.; Johnston, A.W.B. Wide host-range cloning for functional metagenomics. Methods Mol. Biol. 2010, 668, 77–96. [Google Scholar]
- Ongley, S.E.; Bian, X.; Neilan, B.A.; Muller, R. Recent advances in the heterologous expression of microbial natural product biosynthetic pathways. Nat. Prod. Rep. 2013, 30, 1121–1138. [Google Scholar] [CrossRef]
- Kallifidas, D.; Jiang, G.; Ding, Y.; Luesch, H. Rational engineering of Streptomyces albus J1074 for the overexpression of secondary metabolite gene clusters. Microb. Cell Fact. 2018, 17, 25. [Google Scholar] [CrossRef]
- Gomez-Escribano, J.P.; Bibb, M.J. Engineering Streptomyces coelicolor for heterologous expression of secondary metabolite gene clusters. Microb. Biotechnol. 2011, 4, 207–215. [Google Scholar] [CrossRef]
- Bonet, B.; Teufel, R.; Crusemann, M.; Ziemert, N.; Moore, B.S. Direct capture and heterologous expression of Salinispora natural product genes for the biosynthesis of enterocin. J. Nat. Prod. 2015, 78, 539–542. [Google Scholar] [CrossRef]
- Deng, Y.; Zhang, X.; Zhang, X. Recent advances in genetic modification systems for Actinobacteria. Appl. Microbiol. Biotechnol. 2017, 101, 2217–2226. [Google Scholar] [CrossRef]
- Dhakal, D.; Sohng, J.K.; Pandey, R.P. Engineering actinomycetes for biosynthesis of macrolactone polyketides. Microb. Cell Fact. 2019, 18, 137. [Google Scholar] [CrossRef]
- Baltz, R.H. Genetic manipulation of secondary metabolite biosynthesis for improved production in Streptomyces and other actinomycetes. J. Ind. Microbiol. Biotechnol. 2016, 43, 343–370. [Google Scholar] [CrossRef]
- Lee, N.; Hwang, S.; Lee, Y.; Cho, S.; Palsson, B.; Cho, B.K. Synthetic Biology Tools for Novel Secondary Metabolite Discovery in Streptomyces. J. Microbiol. Biotechnol. 2019, 29, 667–686. [Google Scholar] [CrossRef]
- Palazzotto, E.; Tong, Y.; Lee, S.Y.; Weber, T. Synthetic biology and metabolic engineering of actinomycetes for natural product discovery. Biotechnol. Adv. 2019, 37, 107366. [Google Scholar] [CrossRef]
- Genilloud, O. Actinomycetes: Still a source of novel antibiotics. Nat. Prod. Rep. 2017, 34, 1203–1232. [Google Scholar] [CrossRef]
- Mazzetti, C.; Ornaghi, M.; Gaspari, E.; Parapini, S.; Maffioli, S.; Sosio, M.; Donadio, S. Halogenated spirotetronates from Actinoallomurus. J. Nat. Prod. 2012, 75, 1044–1050. [Google Scholar] [CrossRef]










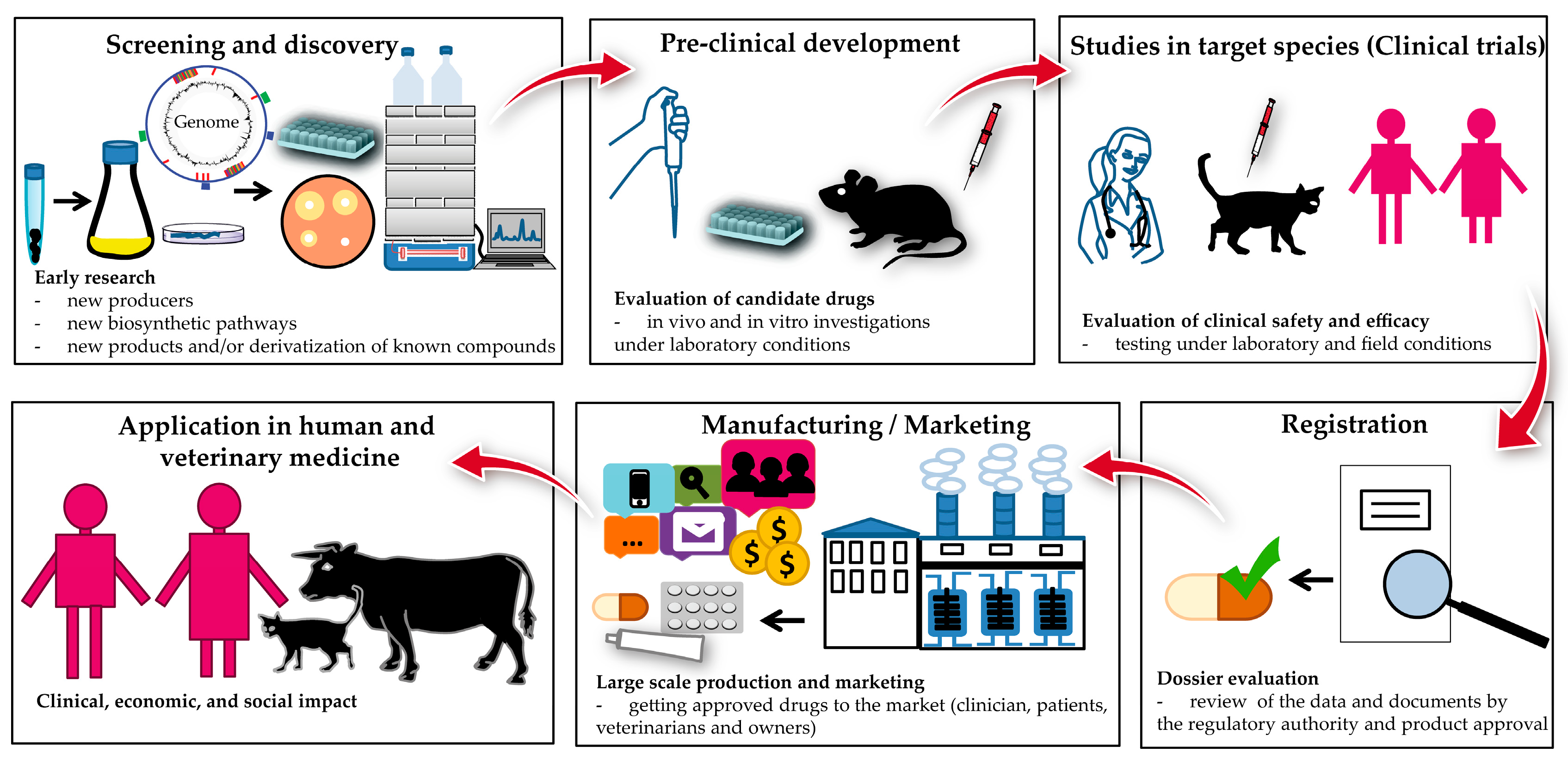{kind=link}
{kind=link}
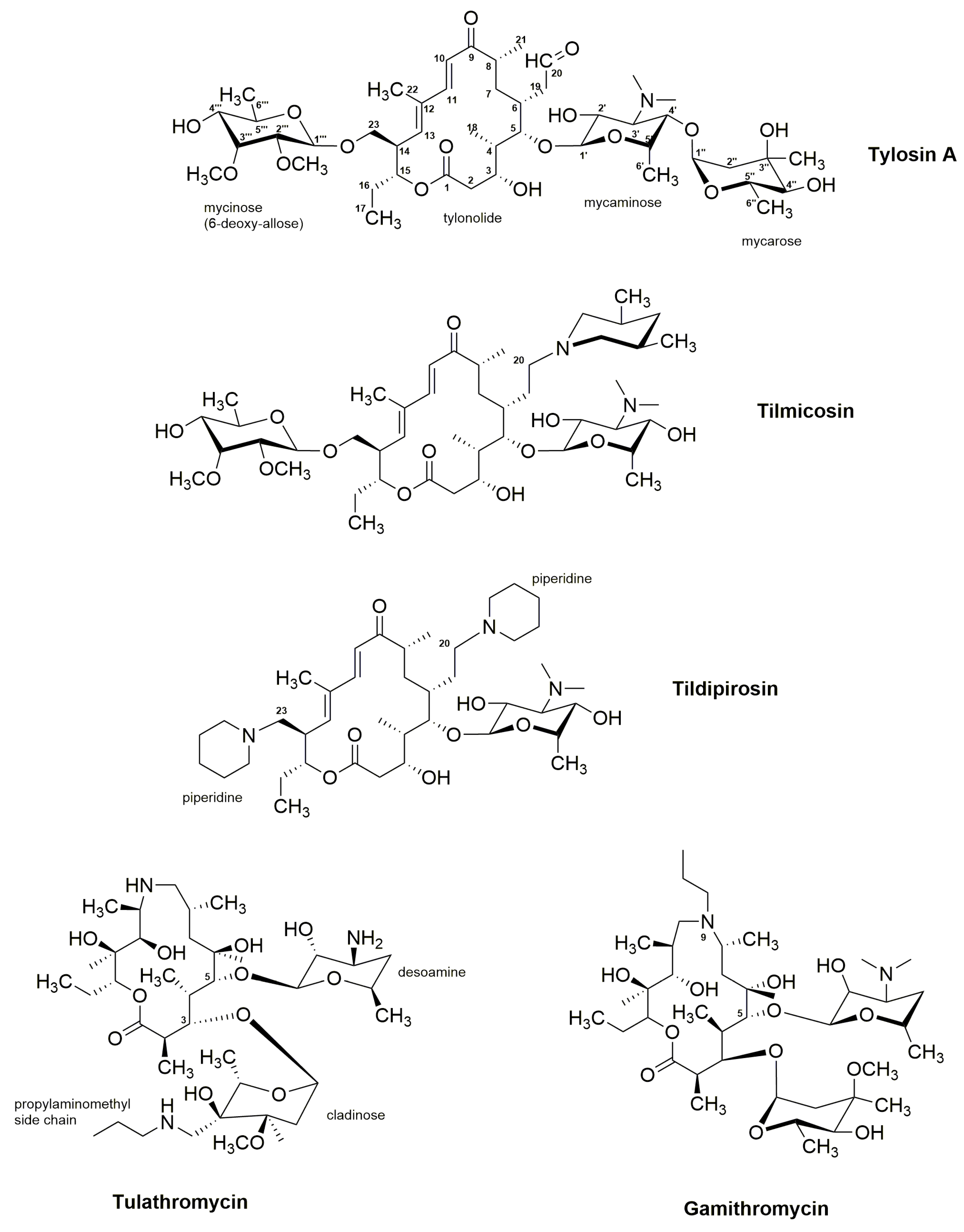{kind=link}
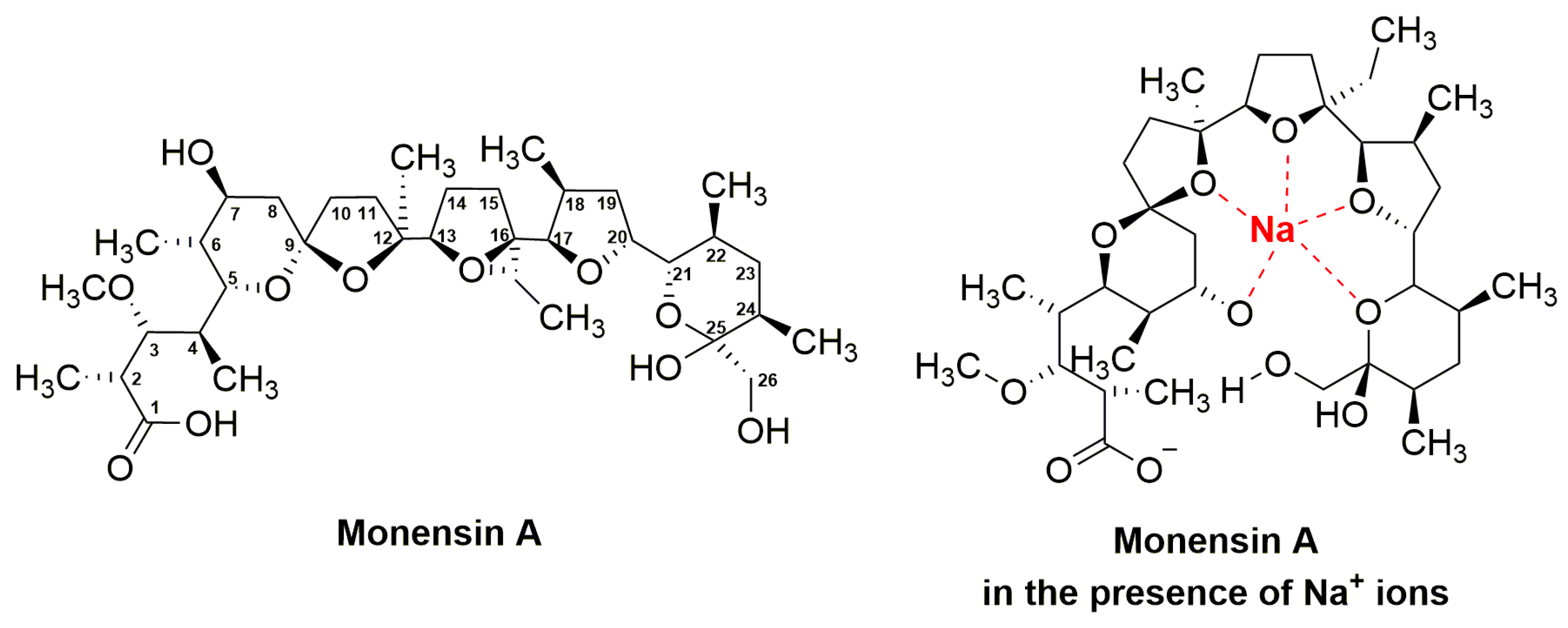{kind=link}
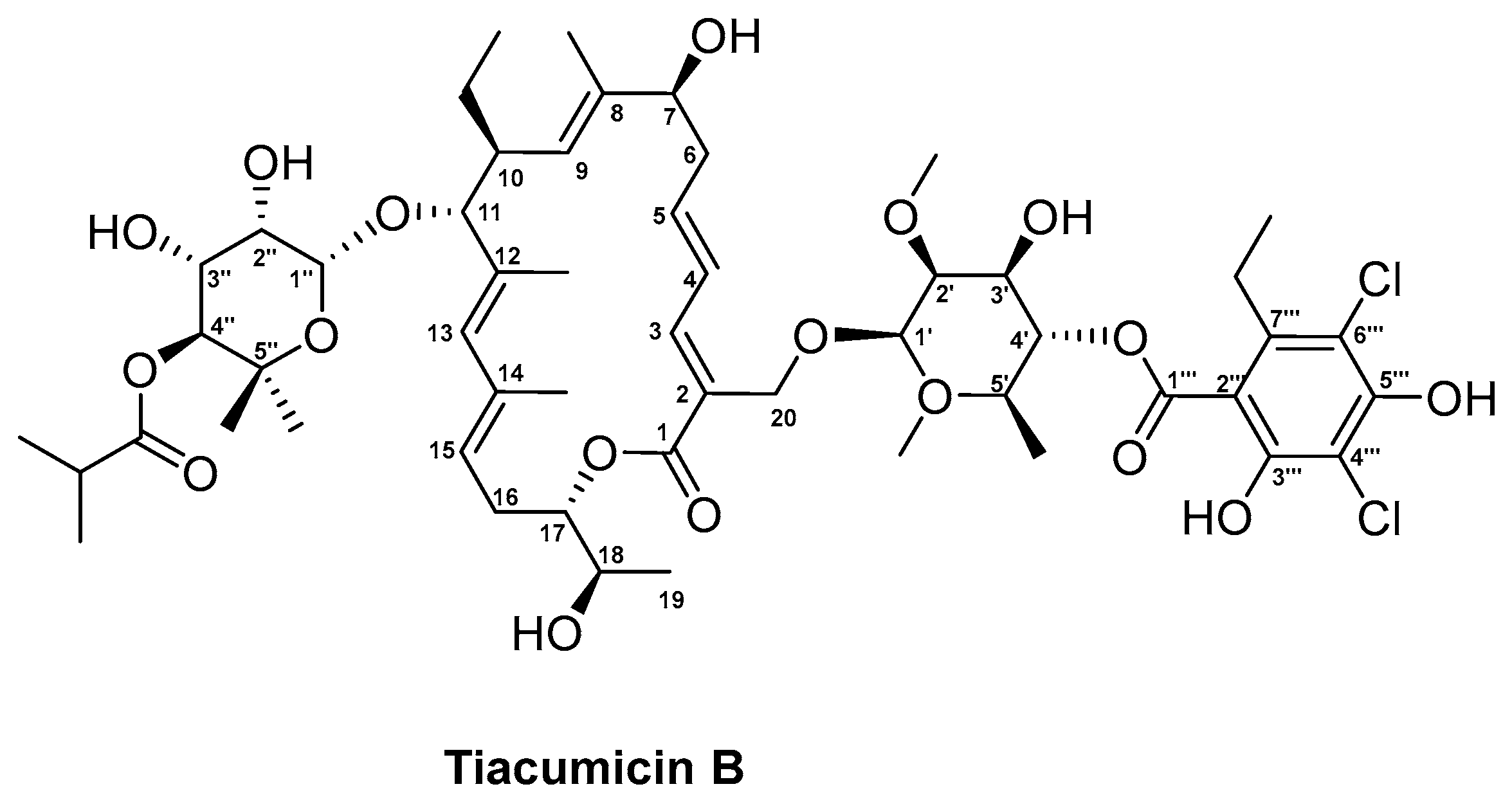{kind=link}
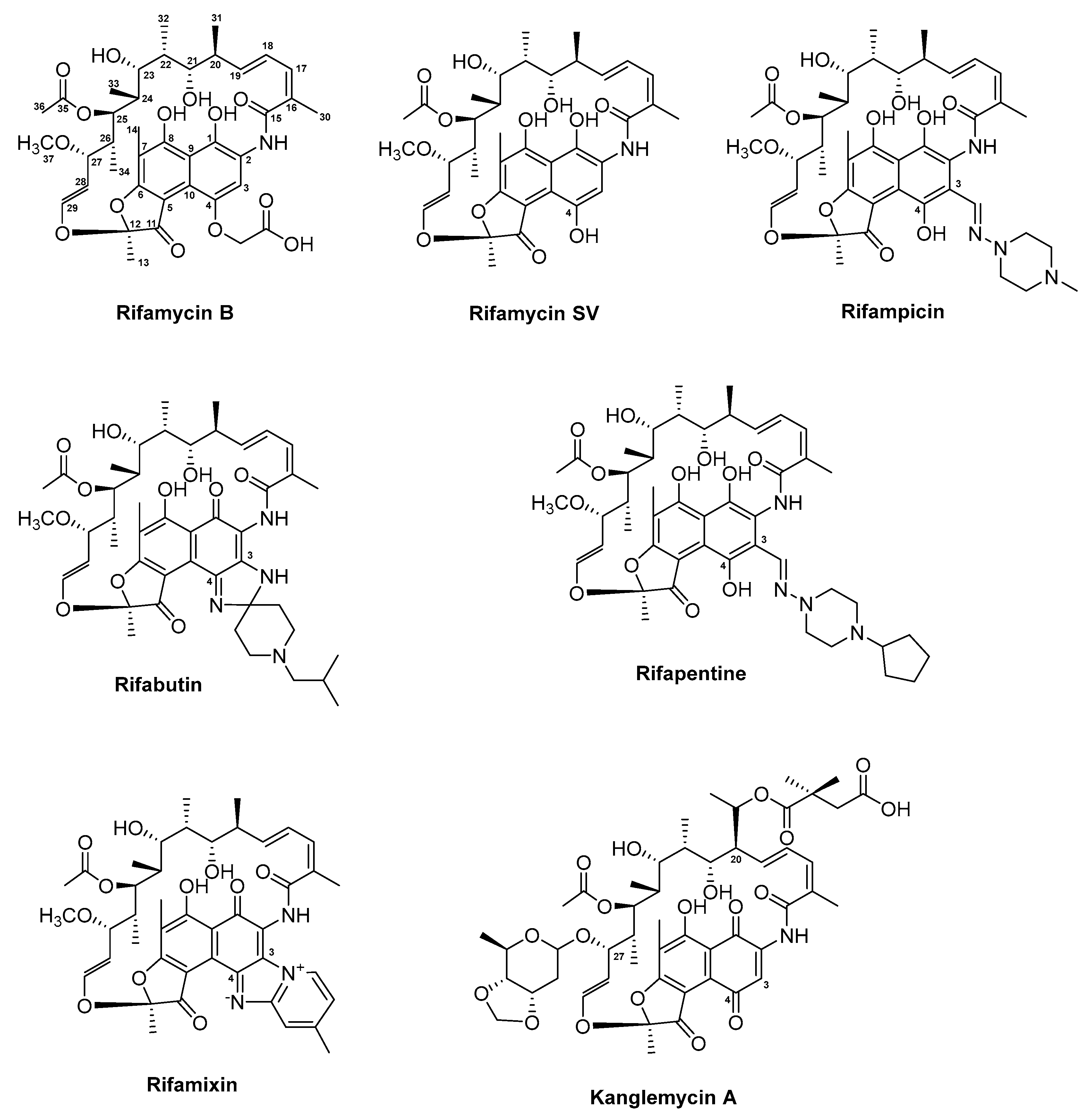{kind=link}
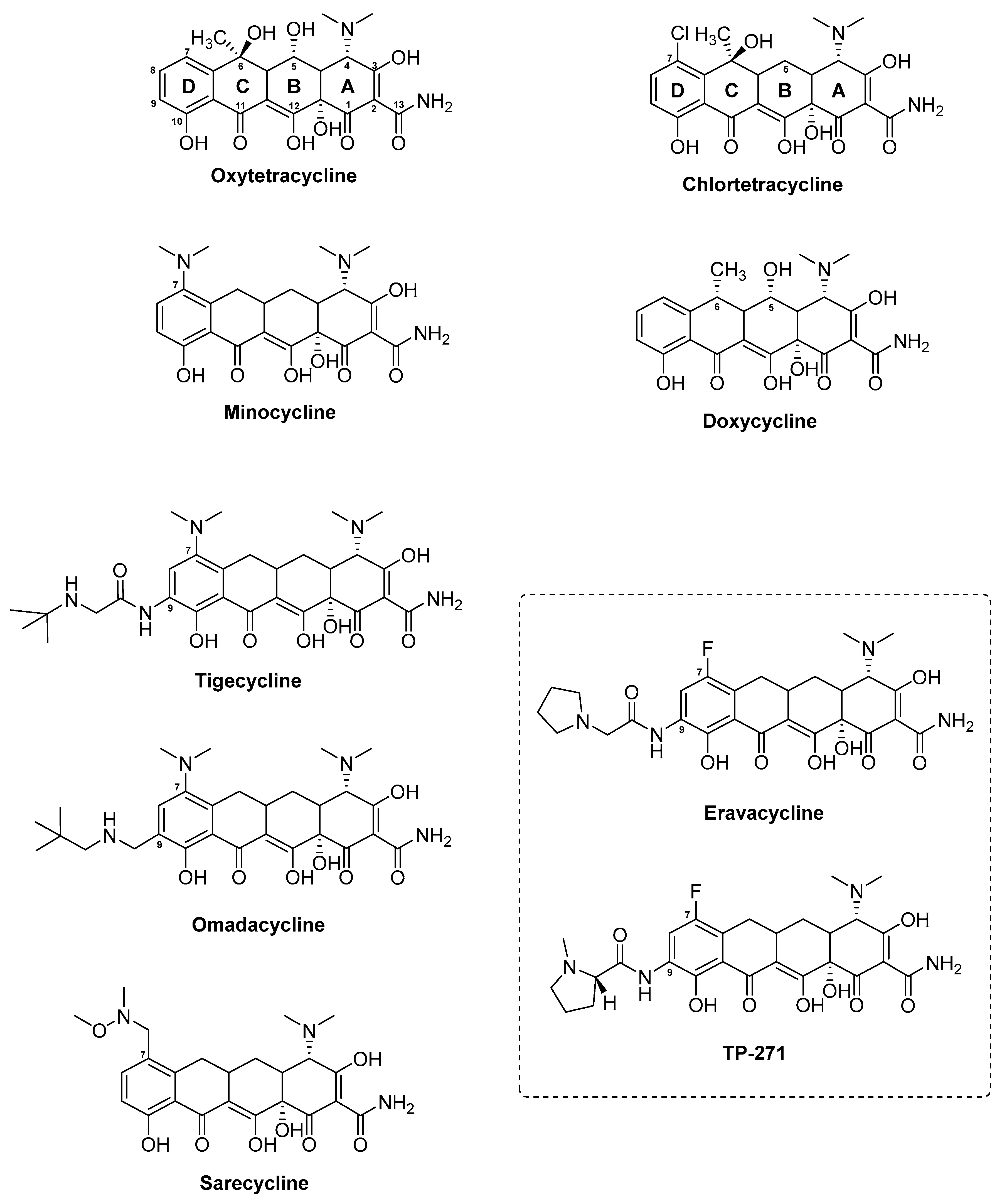{kind=link}
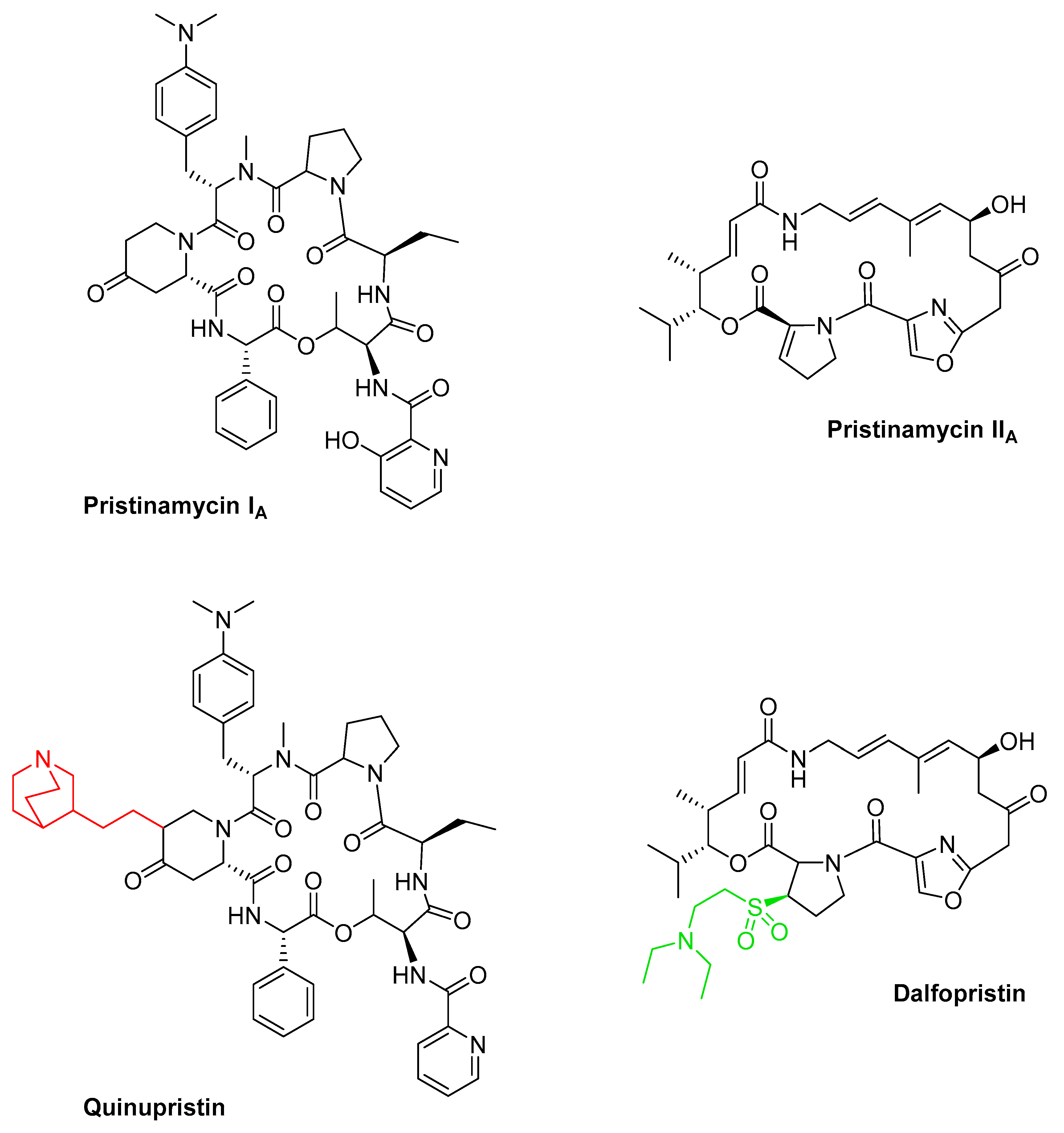{kind=link}
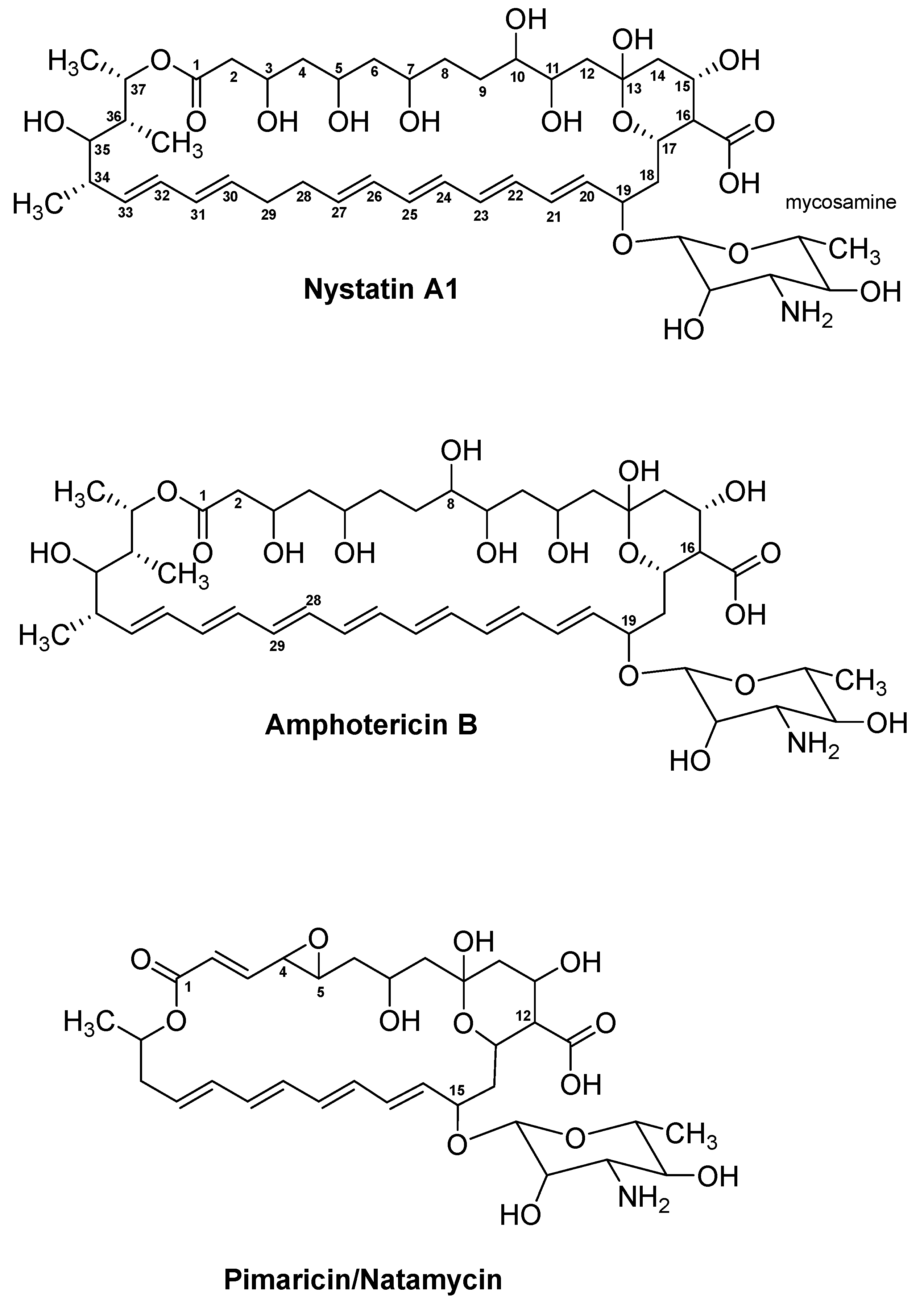{kind=link}
| Compound Class | Natural Product (NP) or Synthetic (S) | Compound Name | Trade Names | Original Producer Strain/Origin | Target/MOA | Most Relevant References |
|---|---|---|---|---|---|---|
| Erythromycin and related compounds | NP | Erythromycin A | Ilosone | Saccharopolyspora erythraea | Prokaryotic 50S ribosomal subunit | [52,55,58,61] |
| Oleandomycin | Sigmamycin (with tetracycline) | Streptomyces antibioticus | [67,68,69] | |||
| Spiramycin | Rovamycine | Streptomyces ambofaciens | [70,71,72] | |||
| S | Clarithromycin | Biaxin | Erythromycin A derivative | [62,73,74,75] | ||
| Roxithromycin | – | [76,77] | ||||
| Azithromycin | Zithromax | Erythromycin A (azalide) | [62,78,79] | |||
| Telithromycin | Ketek | Erythromycin A (ketolide) | [80,81,82] | |||
| Solithromycin | Solithera, CEM–101, T–4288 | [83,84] | ||||
| Nafithromycin | WCK 4873 | [83,84,85] | ||||
| Tylosin and derivatives | NP | Tylosin A | Tylocine, Tylan | Streptomyces fradiae | Prokaryotic 50S ribosomal subunit | [86,87,88,89] |
| S | Tilmicosin | Pulmotil, Micotil, Tilmovet | Tylosin A | [90,91] | ||
| Tildipirosin | Zuprevo | [92,93] | ||||
| Tulathromycin | Draxxin | [94] | ||||
| Gamithromycin | Zactran | [95,96] | ||||
| Monensin A | NP | Monensin A | Coban, Rumensin, Monensin | Streptomyces cinnamonensis | Ionophore (transport of Na+ ions) | [97,98,99,100] |
| Tiacumicin B | NP | Tiacumicin B | Dificid | Dactylosporangium aurantiacum subsp. hamdenensis | RNA polymerase σ factor | [101,102,103,104] |
| Rifamycin and derivatives | NP | Rifamycin SV | Aemcolo, Relafalk | Amycolatopsis mediterranei | Bacterial DNA–dependent RNA synthesis | [105,106,107,108,109] |
| S | Rifampicin | Rifadin, Rimactane | Rifamycin SV | [106,110,111] | ||
| Rifabutin | Mycobutin | [112,113,114] | ||||
| Rifapentine | Priftin | [115] | ||||
| Rifamixin | Normix, Rifacol, Xifacan | [116,117,118] | ||||
| NP | Kanglemycin A | – | Nocardia mediterranei var. kanglensis | [119,120] | ||
| Tetracyclines | NP | Oxytetracycline | Terracycline | Streptomyces rimosus | Prokaryotic 30S ribosomal subunit | [121,122,123,124] |
| Chlortetracycline | Aureomycin | Streptomyces aureofaciens | [123,124,125,126] | |||
| S | Doxycycline | Vibramycin | Chlortetracycline | [123,127,128] | ||
| Minocycline | Minocin | Oxytetracycline | [129,130,131] | |||
| Tigecycline | Tygacil | Minocycline | [132,133,134,135] | |||
| Omadacycline | Nuzyra | Minocycline | [133,136,137,138] | |||
| Eravacycline | Xerava | Fully synthetic | [139,140,141] | |||
| Sarecycline | Seysara | Tetracycline | [127,133,142] | |||
| TP–271 | TP–271 | Fully synthetic | [143,144,145] | |||
| Pristinamycins and derivatives | NP | Pristinamycin IA/IIA (PIA and PIIA) | Pyostacine | Streptomyces pristinaespiralis | Prokaryotic 50S ribosomal subunit | [146,147,148,149] |
| S | Quinupristin (30%)/Dalfopristin (70%) | Synercid | PIA and PIIA derivatives | [150,151,152,153] | ||
| Linopristin (30%)/Flopristin (70%) | NXL–103 | PIA and PIIA derivatives | [154,155] | |||
| Nystatin and derivative | NP | Nystatin A1 | Mycostatin, Nystop | Streptomyces noursei | Lipid receptor (ergosterol) | [156,157,158,159] |
| NP | BSG005 | – | Streptomyces noursei GG5073SP | [160,161] | ||
| Amphotericin | NP | Amphotericin B | Fungizone, Amphocin | Streptomyces nodosus | [162,163,164,165] | |
| Pimaricin/Natamycin | NP | Natamycin | Natacyn, E235 | Streptomyces natalensis | [166,167,168,169] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Robertsen, H.L.; Musiol-Kroll, E.M. Actinomycete-Derived Polyketides as a Source of Antibiotics and Lead Structures for the Development of New Antimicrobial Drugs. Antibiotics 2019, 8, 157. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics8040157
Robertsen HL, Musiol-Kroll EM. Actinomycete-Derived Polyketides as a Source of Antibiotics and Lead Structures for the Development of New Antimicrobial Drugs. Antibiotics. 2019; 8(4):157. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics8040157
Chicago/Turabian StyleRobertsen, Helene L., and Ewa M. Musiol-Kroll. 2019. "Actinomycete-Derived Polyketides as a Source of Antibiotics and Lead Structures for the Development of New Antimicrobial Drugs" Antibiotics 8, no. 4: 157. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics8040157