Natural Product Type III Secretion System Inhibitors

Department of Medicinal Chemistry, Virginia Commonwealth University, Richmond, VA 23284, USA
*
Author to whom correspondence should be addressed.
Antibiotics 2019, 8(4), 162; https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics8040162
Submission received: 10 September 2019
/
Revised: 18 September 2019
/
Accepted: 19 September 2019
/
Published: 24 September 2019
(This article belongs to the Special Issue Natural Compounds as Antimicrobial Agents)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Many known inhibitors of the bacterial type III secretion system (T3SS), a virulence factor used by pathogenic bacteria to infect host cells, are natural products. These compounds, produced by bacteria, fungi, and plants, may have developed as prophylactic treatments for potential attack by bacterial pathogens or as an attempt by symbiotic organisms to protect their hosts. Regardless, better understanding of the structures and mechanisms of action of these compounds may open opportunities for drug development against diseases caused by pathogens utilizing the T3SS. This review will cover selected known natural products of the T3SS and detail what is known of their origin and mechanism of action. These inhibitors highlight nature’s ability to modulate interactions between organisms at a cellular level.
1. Introduction
The type III secretion system (T3SS) is a virulence factor utilized by many Gram-negative pathogens to enable and perpetuate infection of a host [1,2,3]. Pathogens known to encode a T3SS include enteropathogenic and enterohemorrhagic Escherichia coli (EPEC and EHEC, respectively) [4,5,6], Salmonella enterica serovar Typhimurium [1], Chlamydia species [7], Yersinia pestis [2,8,9], Vibrio spp., Shigella spp., and Pseudomonas spp. [2]. The T3SS is indispensable in the ability of a pathogen to cause infection, with knockouts of the T3SS being avirulent [10]. Chemical inhibition of the T3SS has emerged as a strategy to combat these pathogens [11]. Inhibition of the T3SS results in an inability of a pathogen to cause infection in a host; in vivo studies in mice have shown that T3SS inhibitors allowed the host immune system to clear the infection better than a placebo [12,13,14]. In addition, a functioning T3SS is not necessary for bacterial cell viability, and inhibition of the T3SS is not toxic to the pathogen [15]. This removes the selective pressure for the formation of resistance to treatment [16].
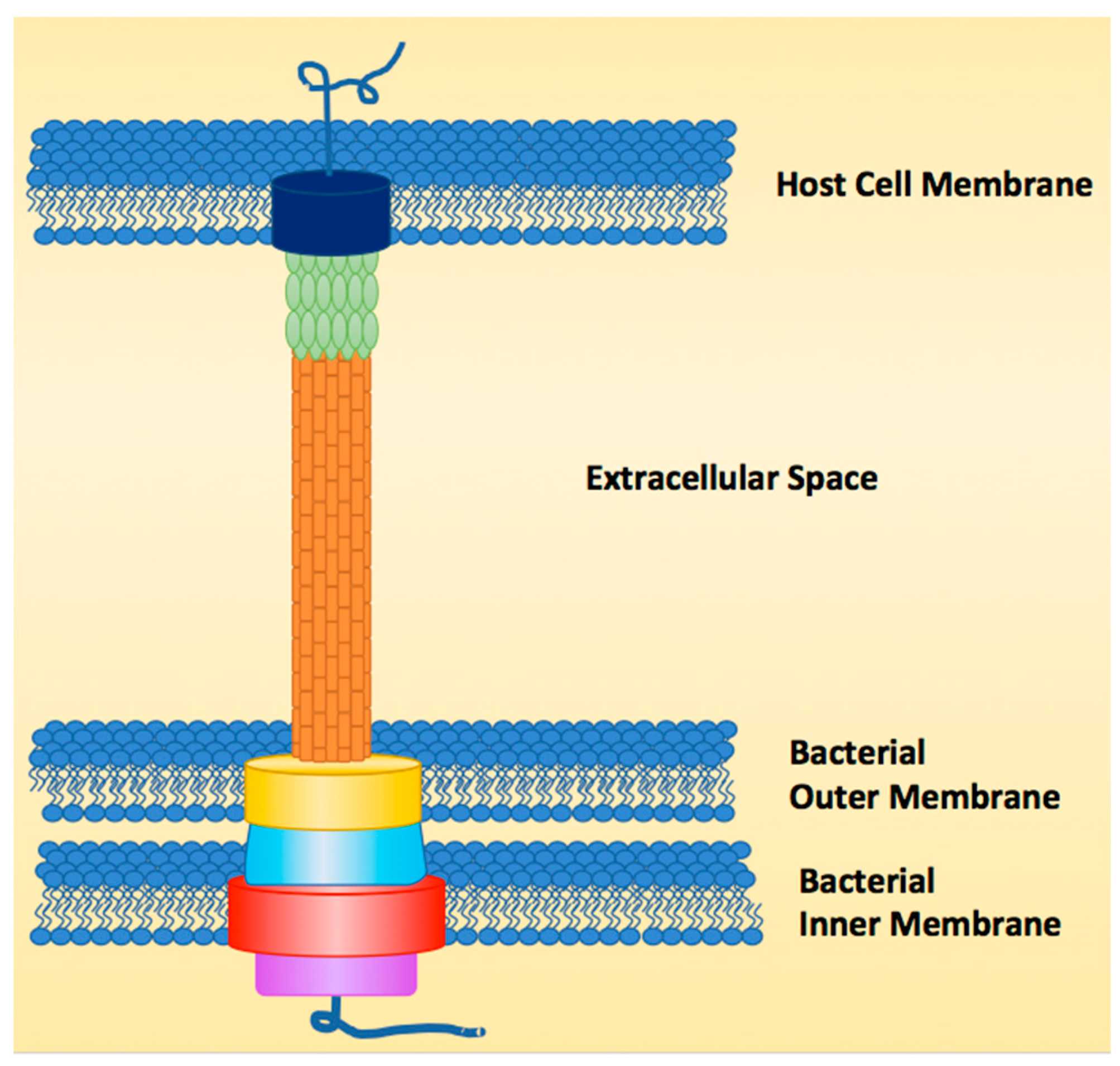
The T3SS functions like a molecular syringe by injecting or secreting effector proteins directly from the cytosol of the bacterial pathogen through the host cell membrane, earning it the nickname “the type III injectosome” [2,4,17,18,19,20]. The structure of the injectosome can be broken down into three major regions, the sorting platform, the basal body and the needle (Figure 1). The sorting platform, which is within the cytoplasmic region of the bacterial cell, contains an ATPase (purple) to power secretion of linear unfolded proteins, as folded proteins are too wide to go through the ~2.5 nm needle [21,22]. The ATPase also functions as the recognition domain for effectors and unfolds the effectors for secretion. The basal body (yellow, blue, red) is made of a dual ring system that spans the inner and outer bacterial membranes and anchors the needle to the bacterial cell surface. The needle is composed of helical monomers (orange) that form a tube-like structure after polymerization [20]. The needle differs in length from species to strain depending on the host that the pathogen has evolved to infect [23]. The tip of the needle (green) also varies in length from one species to another. In E. coli, for instance, the needle tip is longer than the needle itself, which is not the case for other T3SSs [20]. The final structural component to the injectosome is the pore (dark blue) that is formed in the host cell membrane, made up of two pore-forming proteins that allow effectors to pass into the host (for E. coli, EspB and D) [24,25,26,27].
Once the effectors pass into the host cell, they elicit specific responses from the host. In the case of E. coli, one such effector is translocated intimin receptor (Tir) [3,10,28,29,30]. Tir is secreted in an unfolded form into the host cell. Once there, it folds and presents itself on the surface of the host cell. Intimin, which is presented on the outer membrane of E. coli, recognizes the translocated Tir and binds. This forms an intimate attachment between the bacteria and the host cell. This attachment is necessary for the propagation of infection [26]. Other activities elicited by effector proteins include mimicking host signaling proteins and enzymes to hijack host cell machinery, inducing host cell death directly, or evading immune response in the host cell. Up to 30 different effector proteins can be secreted by a single pathogen [31,32,33].
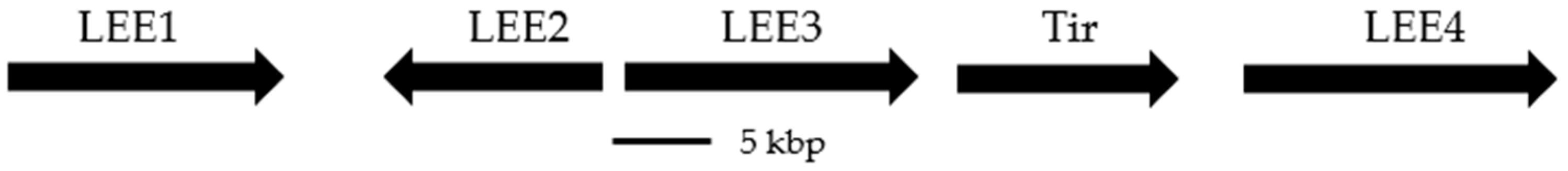
The T3SS and its components are typically encoded in pathogenicity islands (e.g., Salmonella pathogenicity islands, SPIs [34,35,36]); however, in the case of Yersinia species, the genes for the T3SS can also be found on a ~70 kbp virulence plasmid [8,37]. The ~35 kbp pathogenicity island that encodes the T3SS in E. coli is referred to as the locus of enterocyte effacement (LEE) [5,38,39,40], and encodes 41 different genes under the control of 5 promoter regions (LEE1–LEE4 and Tir, Figure 2). The T3SS is not constitutively active, and expression is tightly controlled by environmental signaling factors [8,34,36,37,39].
This review will cover a selection of natural product inhibitors of the T3SS, which were chosen to highlight the structural diversity of T3SS inhibitors as well as the diversity of bacteria, plants and fungi that make them. The discovery of natural products as inhibitors of the T3SS introduces new chemical scaffolds for exploration and development of drug-like compounds for clinical investigation, and understanding their mechanisms of action will create opportunity for rational drug design for more potent compounds.
2. Natural Products
2.1. Caminosides
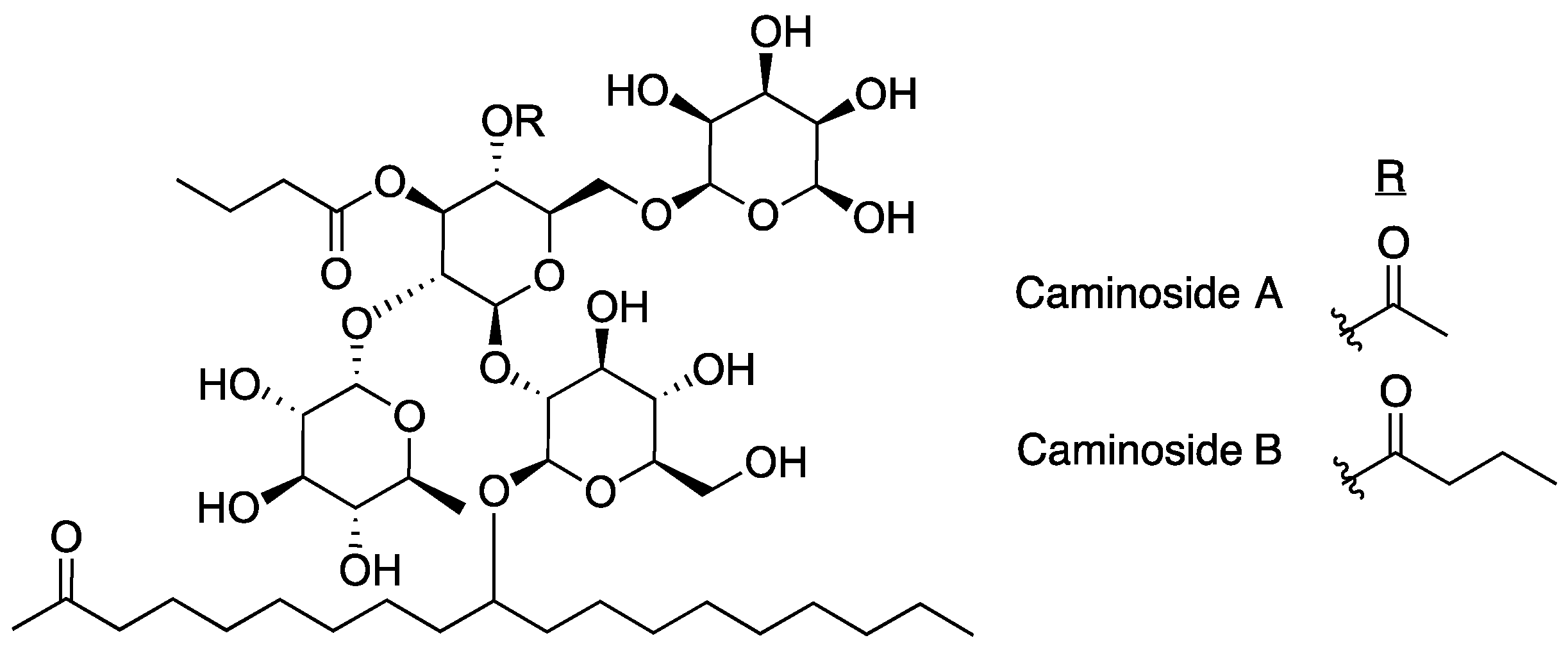
The first inhibitor of the T3SS discovered was caminoside A (Figure 3) [15]. The caminosides are glycolipids isolated from the marine sponge Caminus sphaeroconia found in the upper walls of the Toucari Caves on the island of Dominica. Marine invertebrates were collected and transported to the Anderson lab facilities in Canada, where they were extracted repetitively with methanol before screening for T3SS inhibitory activity in EPEC. The isolation of caminoside A was a result of a bioassay guided fractionation approach using a protocol designed to screen for T3SS inhibitors using sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and analyzing secretion of effector proteins (Esps). Samples having cytotoxic effects against EPEC were dropped from the study.
Caminoside A decreased the secretion of EspB, but not EspC. Since EspC is also secreted via the type IV secretion system this result indicates that caminoside A is specifically targeting the T3SS [41]. The structure of caminoside A was elucidated and potency was characterized (IC50 = 20 μm), although details on the mechanism of action are still not well understood [15]. Interestingly, although caminoside A has no cytotoxic effect against EPEC, it does have cytotoxic activity against Gram-positive methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant Enterococcus (MIC = 12 μg/mL for each). Since the discovery of the caminosides, full syntheses of caminoside A and B have been published [42,43], detailing a synthetic process of 33 and 25 steps, respectively. Despite the promising activity of the caminosides against EPEC, very little has been done since their discovery to further develop these natural products for use as T3SS inhibitors due to difficulties in production or synthesis of the compounds.
2.2. Aurodox
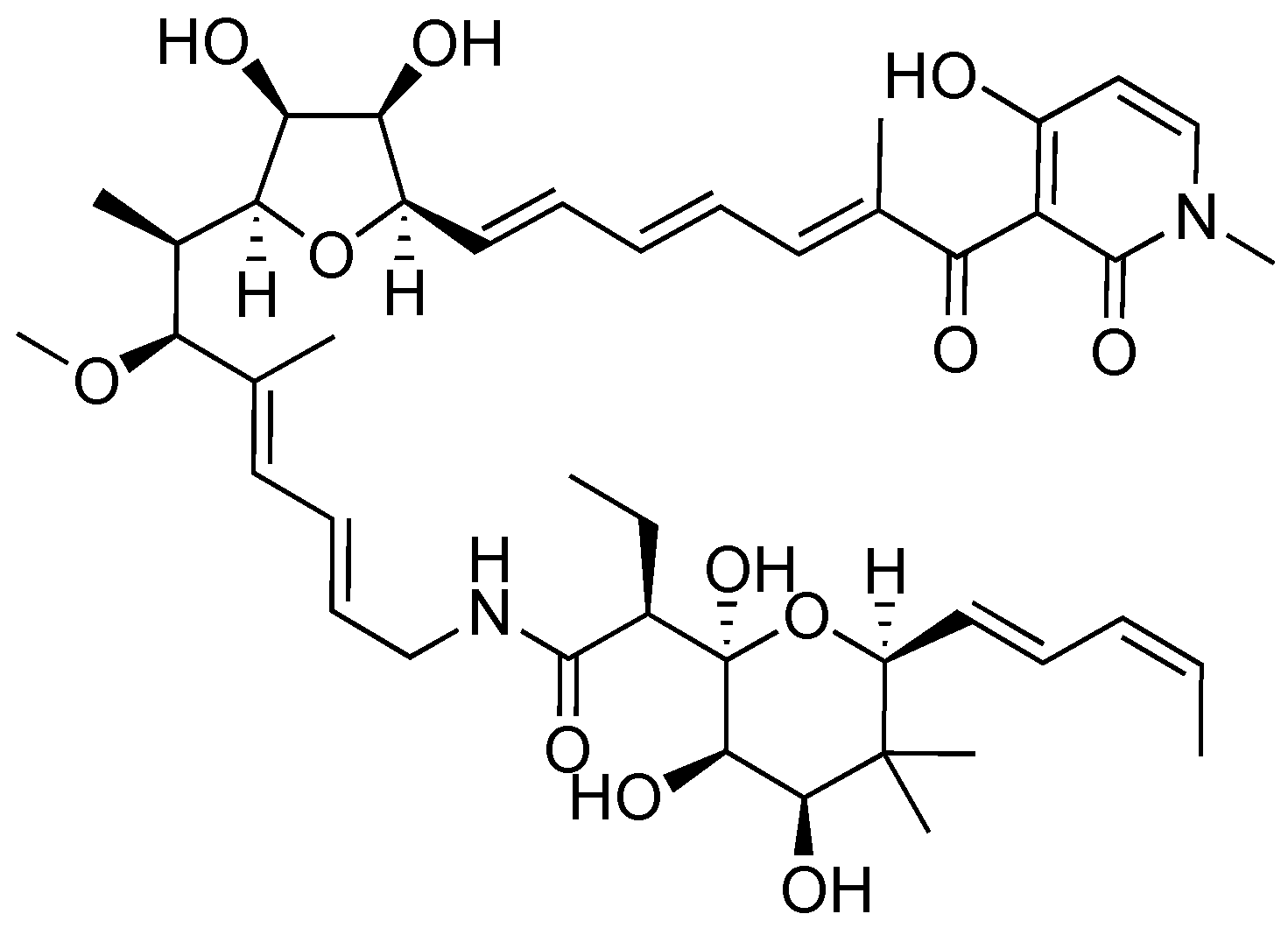
Aurodox, a polyketide produced by Streptomyces goldiniensis, was originally isolated and characterized as having antibiotic activity in 1973 (Figure 4) [44]. Aurodox was originally named antibiotic X-5108 by the Grunberg lab when they elucidated its structure and determined aurodox’s antibiotic activity against Gram-positive bacteria. The Chinali lab investigated the biological activity of aurodox by performing structure activity relationship (SAR) studies against the biological target elongation factor Tu (EF-Tu) [45].
Aurodox was identified as a T3SS inhibitor by the Abe group in 2011 after the development of a method to screen for inhibitors of the T3SS in EPEC known as EPEC-mediated hemolysis [14,46]. The molecular components of the translocon, EspB and EspD, typically form the end of the T3SS needle complex and allow for passage of effectors into the target host cell [24,25,26]; however, they also form pores on the surface of red blood cells (RBCs) [46,47]. Formation of these pores results in leakage of hemoglobin into the extracellular space. The supernatant may then be separated from cellular components and the concentration of hemoglobin may be measured indirectly by absorbance measurements. Thus, hemoglobin concentrations can be tied to T3SS expression.
A screen was performed on 13,300 biological extracts from actinomycetes, fungi, plants, and invertebrates [14]. After extracts from Streptomyces sp. K06-0806 showed potent inhibition of EPEC-mediated hemolysis of RBCs without significantly affecting bacterial growth, a large culture of Streptomyces sp. K06-0806 was fermented and aurodox was purified. Further testing with purified compound confirmed inhibitory potency of aurodox in the RBC assay (IC50 = 1.5 μg/mL). According to analysis by SDS-PAGE followed by Western blotting, aurodox reduces the amount of secreted proteins EspA, EspB, EspD, EspF and Map (an effector that targets and damages host cell mitochondria [48]) without significantly affecting overall protein levels. It was shown that T3SS inhibition (IC50 = 1.5 μg/mL) occurs at a concentration much lower than the concentration at which aurodox shows signs of toxicity against Gram-negative bacteria (~10 μg/mL) [14].
The Abe lab collected further data using an in vivo mouse model using Citrobacter rodentium [40] to analyze the effectiveness of aurodox on mitigating infection. C. rodentium is a commonly used model of EPEC infection in mice, due to a high identity of sequence between the EPEC LEE and the LEE in C. rodentium [14,40]. Mice were initially infected with C. rodentium, then treated either with 10% dimethyl sulfoxide as a control, a single dose of tetracycline (200 mg/kg), or aurodox (25 mg/kg) every 24 hours for four days. All of the mice that were treated with aurodox survived while none of those that were treated with tetracycline survived past day 13. These results show the power of T3SS inhibitors to protect against an otherwise lethal dose of pathogen.
A recent study has been published investigating the mechanism of action of aurodox [49]. The Roe lab showed that aurodox decreased the secretion of effector proteins via Western blotting. Aurodox was also shown to decrease infectability of epithelial cells by EHEC. Transcriptomal analysis on gene expression revealed that aurodox downregulates more than 100 genes cell-wide and downregulates 25 of 41 genes related to the T3SS. This suggests that the inhibitory activity of aurodox is a result of a change in gene expression, and not a result of physical manipulation of the T3SS needle complex. One of the genes downregulated by aurodox is ler, a major activator of the LEE [28]. In addition, aurodox downregulated the expression of EspG and NleB, which are non-LEE encoded effectors. Importantly, treatment with aurodox does not induce Shiga toxin production in EHEC, suggesting promise for the use of aurodox to treat EHEC infection. If the binding target of aurodox were identified, efforts could be made to strengthen that binding and increase the potency of aurodox further.
2.3. Piericidin A
Piericidin A was originally discovered for its insecticidal properties in 1963 (Figure 5) [50]. Soil samples were collected from Chiba Prefecture, a region encompassing the outskirts of Tokyo, and their microbial makeup was analyzed for their toxicity against a variety of larval species. The microorganism that exemplified the highest toxicity was Streptomyces sp. 16–22. Piericidin A was purified via bioactivity-guided fractionation approaches and its structure and chemical properties were characterized. Notably, this same study found that piericidin A presented limited cytotoxicity against Gram-negative bacteria such as E. coli and Xanthymonas oryzae [50]. In 1966, piericidin A was investigated for antibiotic properties against Gram-positive bacteria [51]. It was discovered that piericidin A targets nicotinamide adenine dinucleotide (NADH) dehydrogenase within complex I (100% inhibition at 0.03 nmol piericidin per mg protein), a complex important in mitochondrial electron transport. In 2010, the Svatos lab described a symbiotic relationship between beewolf digger wasps and certain strains of piericidin-producing Streptomyces [52]. The wasps cultivate Streptomyces on their antennae, and incorporate the cells into their larval cocoons as prophylaxis against pathogenic bacteria. This is just one example of nature’s purposeful use of natural products as a defensive mechanism against pathogens.
In 2014, the Auerbach lab performed a high-throughput screen to discover new inhibitors of the Yersinia pseudotuberculosis T3SS and uncovered piericidin A’s inhibitory activity [53]. Y. pseudotuberculosis possesses a unique ability to trigger Nf-κB signaling in HEK293T cells, a process that is dependent on YopB and YopD transport of effectors into the host cytosol [54]. T3SS function was measured using an Nf-κB luciferase reporter plasmid and changes in activity were monitored to identify potent inhibitors [53]. After eliminating compounds that produced cytotoxicity against either the mammalian or bacterial cells, the group identified piericidin A as one of their hit compounds. SDS-PAGE analysis indicated that secretion of YopE was decreased by 65% at 71 μm piericidin A. Piericidin A was also shown to potently inhibit translocation of YopM (75% decrease at 71 μm) into Chinese hamster ovary (CHO) cells.
A clue into the mechanism of action of piericidin A as a T3SS inhibitor has recently been discovered. Inhibition of the T3SS by piericidin A resulted in decreased formation of Ysc-type needle units on the surface of Y. pseudotuberculosis without interfering with gene expression, indicating the mechanism of action is related directly to needle assembly [54]. Although piericidin has a known antibacterial target (Complex I), an alternative Complex I inhibitor, rotenone, has no T3SS inhibitory activity, indicating the T3SS inhibitory activity of piericidin A may be independent of complex I inhibition [55]. Further work to find the binding partner to elicit T3SS inhibition by piericidin A would aid in the ability to rationally design more potent analogs that selectively inhibit the T3SS without producing antibiotic effects related to Complex I binding.
2.4. Cytosporone B
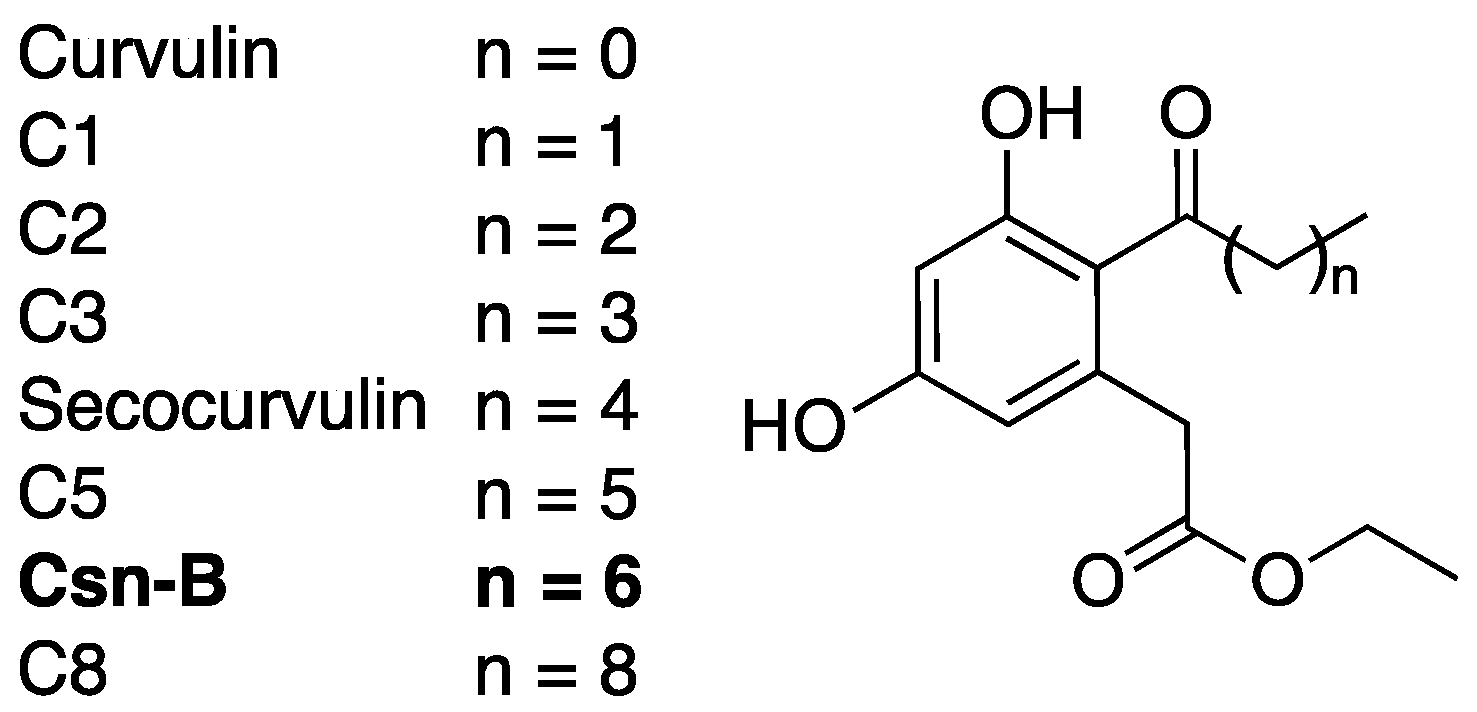
Cytosporone B (Csn-B) was originally identified as a naturally occurring substrate to nuclear orphan receptor Nur77 in 2008 [56]. This octaketide natural product (Figure 6), isolated from the endophytic fungus Dothiorella sp. HTF3, has since been extensively studied as a potential anti-cancer agent due to its ability to stimulate Nur77-mediated apoptosis in multiple cancer cell models [56,57,58]. Csn-B was later identified by the Shen lab as an inhibitor of the T3SS in Salmonella enterica serovar Typhimurium during a screen of Csn-B and analogues [59].
Western blots indicated that secocurvulin, C5, Csn-B, and C8 (Figure 6) were all capable of inhibiting secretion of SPI-1 effectors in S. enterica [60]. In addition, secocurvulin, C5, and Csn-B inhibited S. enterica invasion of HeLa cells, a process dependent on SPI-1, with Csn-B proving to be the most potent. General SAR analysis suggests that the inhibitory potency is maximized at n = 6 (Csn-B), and that the potency decreases with increased or decreased chain length. Csn-B also showed dose-dependent inhibition of SPI-1 effector secretion (IC50 = 6.25 μm) with no toxicity to bacterial cells. Although the molecular target for T3SS inhibition is unknown, Csn-B inhibition could be overcome by overexpression of HilA, a positive regulator of the S. enterica T3SS. This result suggests that Csn-B interferes with the HilA expression pathway. A route for Csn-B total synthesis was published in 2010 [61], but since then, very little has been done to further this compound as a T3SS inhibitor.
2.5. Guadinomines
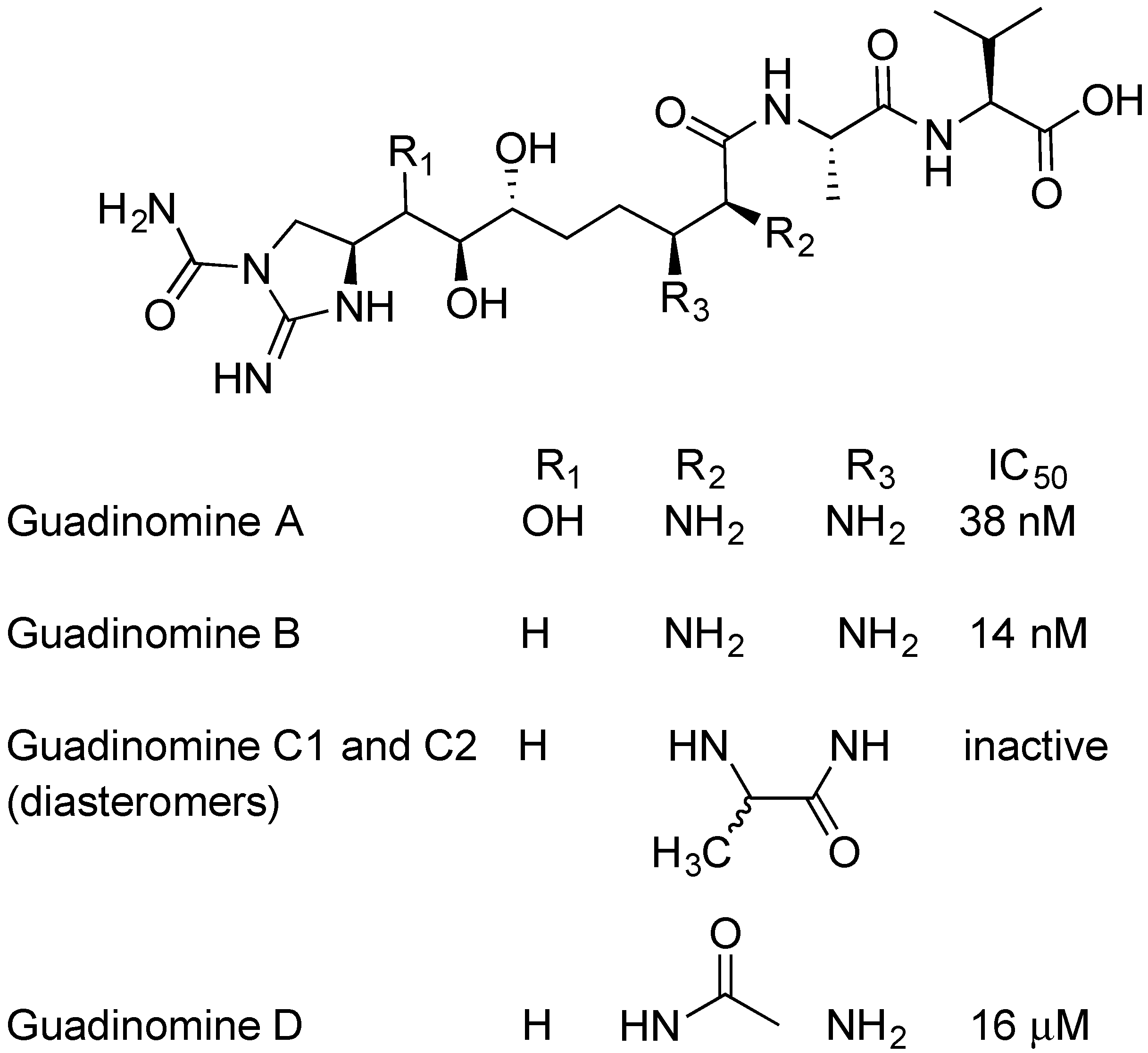
The guadinomines were discovered by Ōmura and colleagues using EPEC-mediated hemolysis to screen natural product extracts. [62,63]. Streptomyces sp. K01-0509 was found to produce potent inhibitors of RBC hemolysis [45,46]. Cultures were scaled up, and guadinomine A, B, C1, C2, and D were isolated, purified and analyzed (Figure 7) [62]. Guadinomines A and B are the most potent natural product TTSS inhibitors with IC50 = 0.007~0.01 μg/mL. The mechanism of action of the guadinomines is not well understood and yields of guadinomines from culture are low, making further research difficult. The lengthy total syntheses of guadinomine B and C2 have been published, with 33 steps in the longest linear synthetic sequence [64]. In 2012, a study on the biosynthetic pathway of guadinomine A was published by Khosla and coworkers [65]. Notably, guadinomine D, having an acylated amine at R2 (Figure 7), is 1000-fold less potent than guadinomine B. This shows the importance of the vicinal diamine to biological activity. The acyl group on guadinomine D may be installed by enzymes apart from the guadinomine synthetase, since no obvious acylation enzyme is part of the gene cluster. While guadinomines do not appear to produce any antimicrobial activity, Ōmura found that guadinomine B is cytotoxic to Jukat cells at a concentration 100 times higher than the IC50 for TTSS inhibition [62].
2.6. Butyric Acid
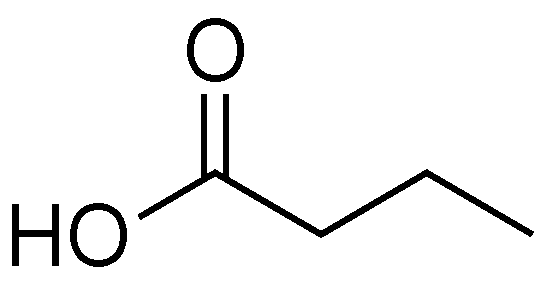
Initial studies relating to the biological effects of butyric acid predate knowledge of the T3SS (Figure 8). In the 1960s, William R. Martin and coworkers investigated how the infectious dose (ID50) of Salmonella enteritidis changed when mice were pretreated with antibiotics [66]. The authors showed that the ED50 went from 106 to <10 cells when a single dose of streptomycin was administered 24 hours before infection. The authors noted that treatment with antibiotic increased the gut pH and identified that butyric acid and acetic acid were being produced by the gut bacteria. The authors attributed the ability of the mice to tolerate Salmonella to a low gut pH and not a specific inhibitory effect caused by butyric acid. Since these experiments were performed before the discovery of the T3SS, the authors were unaware that they were completing initial studies on the effect that T3SS inhibitors have on severity of infection.
Butyric acid is classified as a short-chain fatty acid (SCFA), and is produced as a fermentation product by commensal Bacterioidetes species in mammals. In human intestines butyric acid is typically present at 10–20 mm [67,68,69]. Butyric acid is a major energy source for colonocytes, and the ability of colonic cells to absorb and utilize sodium butyrate is seen as a sign of good health [69]. Administration of butyric acid to the intestines of mice infected with C. rodentium results in decreased inflammation and increased mucus production from colonic cells.
Butyric acid interacts with the epigenetic modifier Lrp, a major regulator of gene expression in bacteria [5,66,67,68,69]. Lrp does not control the expression of genes in the same pattern from one organism to another. As a result, butyrate acts as a T3SS inhibitor for some organisms and as a T3SS activator in others [68,70,71,72]. A notable example involves LEE-encoding bacteria EPEC and C. rodentium. EPEC and C. rodentium have 90% sequence identity in their LEE pathogenicity islands [40]. Their T3SSs are so similar that C. rodentium is often used as a mouse model for EPEC infection [14]. Lrp is a non-LEE encoded T3SS regulator, and activation of Lrp has opposite responses in these two organisms. Activation of Lrp upregulates expression of the LEE in EPEC, while activation of Lrp downregulates expression of the LEE in C. rodentium [68,70,72].
Research into SCFAs as T3SS regulators has focused primarily on the effects of probiotics on infection [67,70,71]. By increasing the concentration of SCFA-producing bacteria in the gut, concentrations of a variety of SCFAs are altered. Depending on the pathogen attempting to infect the gut, differing ratios of SCFAs could have dramatically different results, from improving to worsening infection. Given the prevalence and widespread use of probiotics, this area requires further investigation.
2.7. Fusaric Acid
Fusaric acid is a toxin produced by fungal species Fusarium oxysporum, a common inhabitant of soil (Figure 9) [73,74]. Fusaric acid causes a variety of negative outcomes in plants, and it is thought to be a virulence factor in Fusarium wilt in banana, tomato, and cotton crops [73,74,75] and heavy decline disease in grapevine [76]. Fusaric acid was first studied as a potential inhibitor of the T3SS in S. enterica in 2014 as part of a screen of a small library [77]. In SDS-PAGE and Western blot analysis, fusaric acid potently inhibited secretion of SPI-1 effector proteins when cells were treated with 100 μm fusaric acid, without disrupting cell growth. Inhibition was dose-dependent with a calculated IC50 = 53.5 μm. In a gentamycin-protection assay, fusaric acid markedly inhibited Salmonella invasion into HeLa cells, with no toxicity toward the HeLa cells observed [77].
Some studies attempting to elucidate the mechanism of action of fusaric acid gave conclusive results that the inhibitory effect of fusaric acid cannot be overcome by overexpression of T3SS activator HilA, unlike the case of Csn-B [77]. Also, fusaric acid does not change the level of PgrH, an assembly protein for the needle complex, and does not interfere with the SicA/InfV transcriptional pathway for T3SS initiation. Further studies on the mechanism of action are needed in order to determine the pathway through which fusaric acid elicits an inhibitory response. The cytotoxicity of fusaric acid against plants and other organisms will need to be considered when moving forward with this compound as a potential T3SS inhibitor.
2.8. (-)-Hopeaphenol
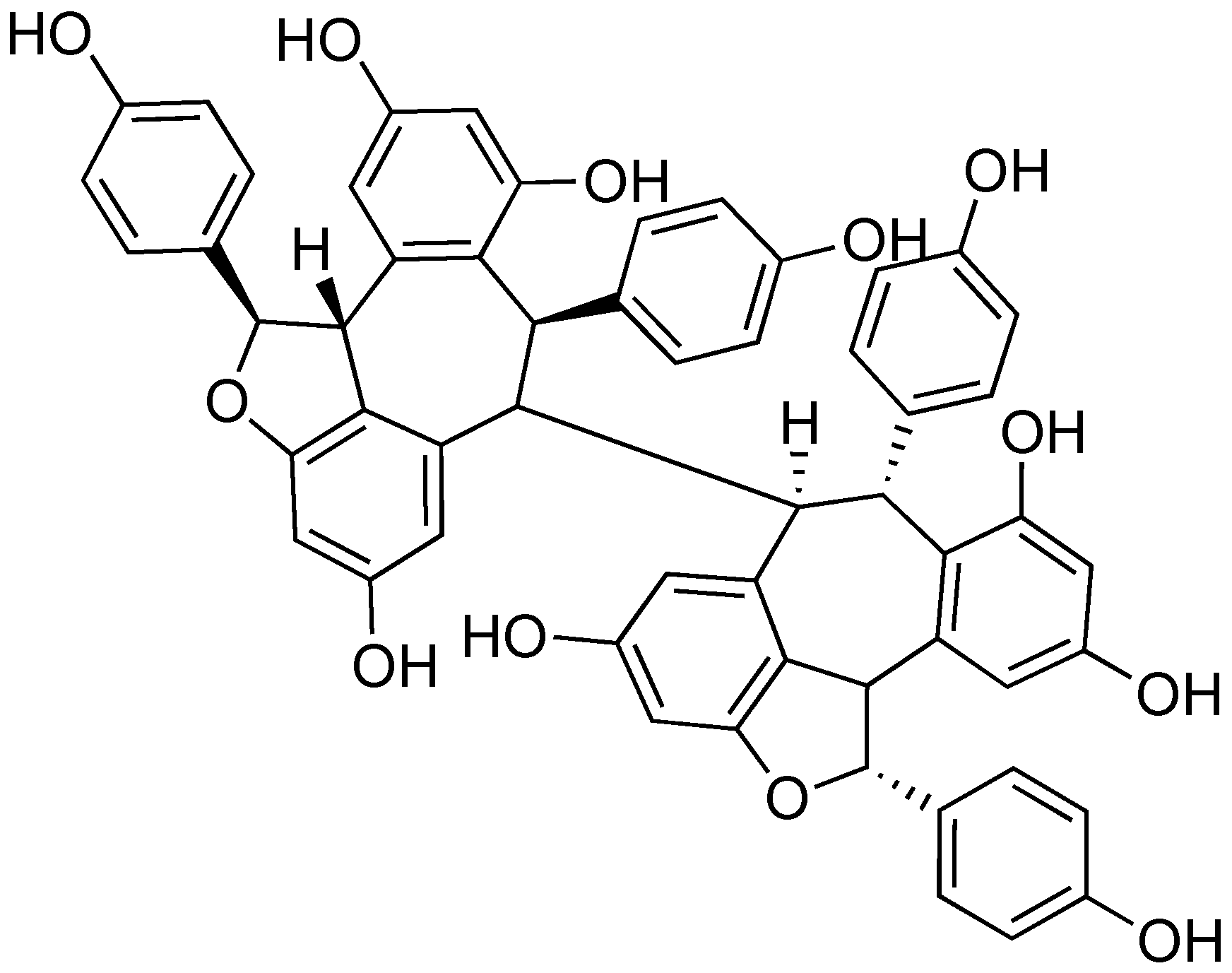
Many plants produce inhibitors to protect against infection caused by Gram-negative pathogens that utilize a T3SS. (-)-Hopeaphenol (Figure 10) was isolated as part of a bioassay-guided fractionation study to find natural product T3SS inhibitors from two rainforest plants from Papua New Guinea, Anisoptera thurifera and A. polyandra [78]. (-)-Hopeaphenol is a tetramer of resveratrol, a common building block used in nature to synthesize natural products. (-)-Hopeaphenol has also been explored for its anti-oxidant properties [79]. In their work, Eloffson and coworkers analyzed (-)-hopeaphenol for inhibition of T3SS in Y. pseudotuberculosis, P. aeruginosa, and C. trachomatis [78]. (-)-Hopeaphenol exhibited inhibition of YopE expression in a reporter gene assay and found the compound had inhibitory activity (IC50 = 6.6 μm). Western blot analysis showed dose-dependent inhibition of secretion and expression of YopD. When cells grown in the presence of (-)-hopeaphenol were moved to T3SS-inducing environments, they were incapable of expressing the T3SS, regardless of whether (-)-hopeaphenol was still present. This suggests an irreversible mechanism of inhibition.
(-)-Hopeaphenol was also found to inhibit expression and secretion of ExoS, an effector from the P. aeruginosa T3SS [79]. In infection model assays, (-)-hopeaphenol completely inhibited infection of HeLa cells by P. aeruginosa at a concentration of 100 μm. In addition, (-)-hopeaphenol was observed to inhibit intracellular growth of C. trachomatis in HeLa cells in a dose-dependent manner. When tested for cytotoxicity against a panel of Gram-positive and Gram-negative organisms, (-)-hopeaphenol did not affect cell growth or viability. Despite the promise of this structural class, there are large barriers for chemical synthesis. In addition, Anisoptera spp. that produce (-)-hopeaphenol are in danger of extinction; 6 of 10 within the genus are either endangered or critically endangered according to the International Union for Conservation of Nature (IUCN) Red List, with the remainder being vulnerable [80]. Without the ability to easily access samples of (-)-hopeaphenol and analogs to test for T3SS inhibitory activity, further development of this structural class as inhibitors will be difficult.
2.9. Sanguinarine Chloride
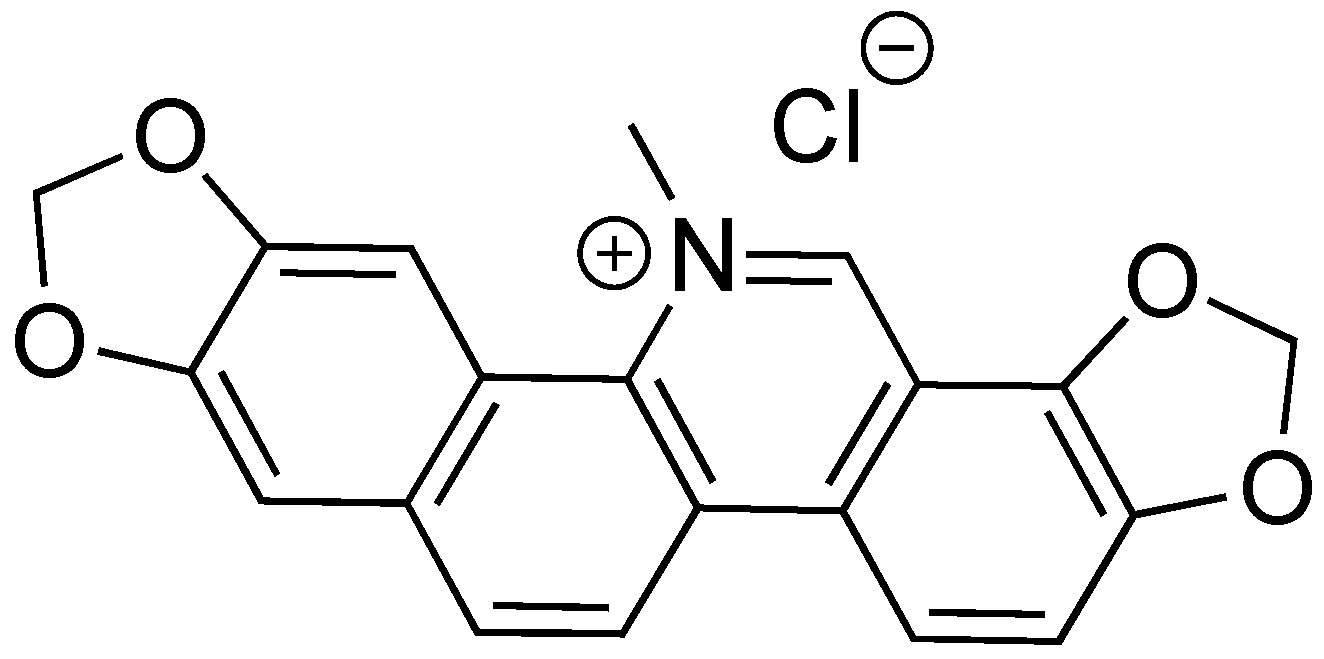
Sanguinarine chloride is a natural product isolated from the extracts of the bloodroot plant Sanguinaria canadensis (Figure 11) [81]. In the 1970s and 1980s, sanguinarine chloride was studied as a potential treatment for gingivitis due to its anti-inflammatory properties. It has since been studied as a chemotherapeutic agent [82]. Sanguinarine chloride was found to be a T3SS inhibitor against Salmonella enterica serovar Typhimurium [83]. It inhibited SipA-lactamase fusion translocation into HeLa cells at a concentration of 4 μm. In a gentamycin-protection assay, sanguinarine chloride was effective against pathogenic invasion of HeLa cells. Expression of SipA and SipB was also inhibited at 5 μm sanguinarine chloride. Overexpression of HilA overcomes the inhibitory effects of sanguinarine chloride, indicating a possible mechanism of action. While sanguinarine chloride shows promise as a T3SS inhibitor, efforts to reduce the cytotoxic affects that the compound has toward human cells must be made for this drug to move forward, as well as further characterizing its biological target.
2.10. Thymol
The Deng lab recently identified thymol during a study aimed at identifying T3SS inhibitors from traditional Chinese medicine (Figure 12) [84]. Thymol is a component of an essential oil derived from plants belonging to the Thymus genus [85]. The translocation of a SipA-lactamase fusion from Salmonella into HeLa cells in the presence or absence of thymol was monitored. At a concentration of 0.2 mm thymol, translocation was almost completely inhibited, while cytotoxicity was not observed until thymol concentrations reached 0.6 mm. In a gentamycin protection assay, preincubation with 0.2 mm thymol resulted in a 90% reduction of T3SS-dependent internalization of Salmonella by HeLa cells. Doses of 50 mg/kg thymol resulted in a 100% survival rate of mice administered lethal doses of Salmonella after a 10-day infection period. This dose also alleviated pathophysiology related to Salmonella infections to colonocytes. These promising results show that further investigations are needed into traditional medicines.
2.11. Cinnamic Acid and Derivatives
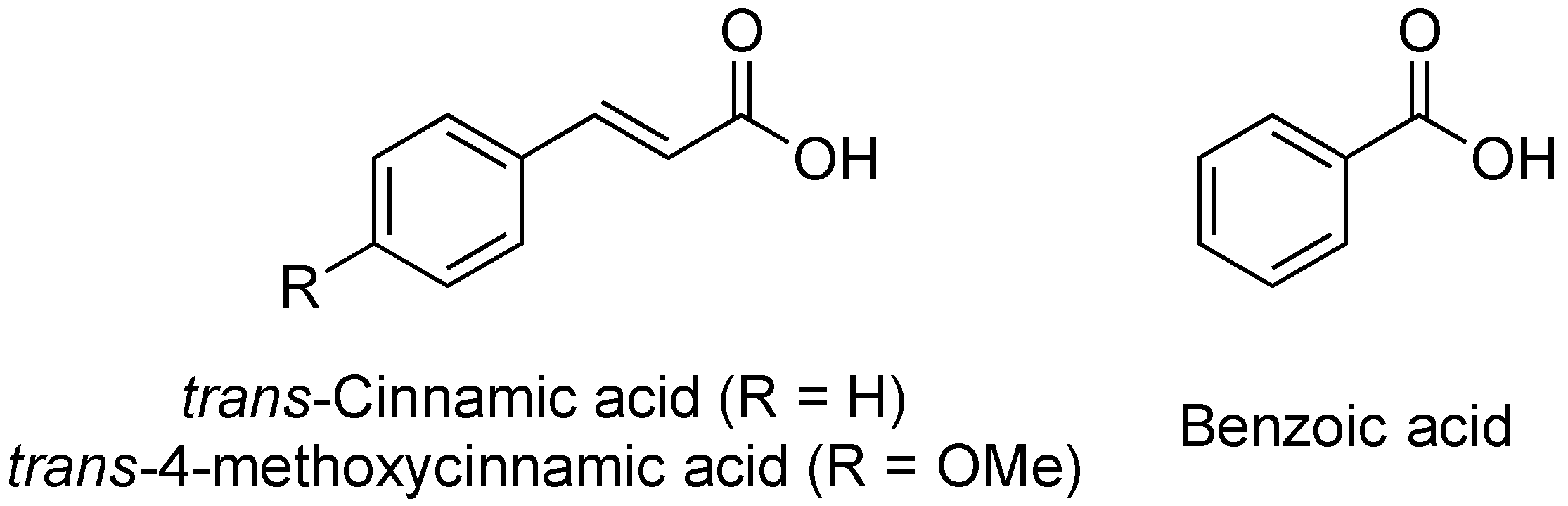
In 2008, the Yang lab investigated the influence of plant-derived compounds on the expression of the T3SS in Dickeya dadantii 3937 [86]. They chose to monitor change in expression of the T3SS via the two major regulatory pathways in D. dadantii 3937, the HrpX/HrpY-HrpS-HrpL and GacS/GacA-rsmB-RsmA pathways. The group found that trans-cinnamic acid (TCA, Figure 13) acts as an activator of the D. dadantii T3SS, increasing the expression of the gene hrpN, an indicator for the HrpX/HrpY-HrpS-HrpL pathway. Specifically, a 3-fold increase in expression of hrpN was observed at 5 μm TCA.
In later studies, the Yang lab investigated derivatives of TCA, including trans-4-methoxycinnamic acid (TMCA) and benzoic acid (BA, Figure 13) on T3SS expression in Erwinia amylovora, the fire blight pathogen [87]. Their results indicated that TMCA and BA act as inhibitors of T3SS expression in a fused-green fluorescence protein (GFP)-hrp reporter assay. TCA, TMCA and BA (100 μm) all decreased fluorescence to approximately 30%, 20%, and 3% of the control, respectively. Based on this assay, the IC50 concentrations for TCA (0.5 μm) and BA (1 μm) were approximated. Northern blot analysis indicated that TMCA inhibits the T3SS by reducing expression of rsmBEa and hrpN, while BA only inhibits the expression of hrpN, suggesting potential differences in their mechanisms of action. None of the compounds altered the expression of the regulatory genes hrpX/hrpY. Further analyses of this structural class, their mechanisms of action, and the differences in inhibitory/inducing behavior between pathogenic species will need to be conducted for this group of compounds to move forward as T3SS inhibitors.
3. Conclusions
Many natural products have been shown to possess T3SS inhibitory properties over the last decade. These compounds are made by a variety of different biological sources and cover a diverse range of chemical scaffolds. Notwithstanding these successes, most inhibitors of the T3SS have unknown biological targets. This complicates the rational design of new and more potent analogs. With a better understanding of their binding partners and mechanism of action, modern methods of analog design (e.g., computational modeling) can be employed effectively. More natural product T3SS inhibitors are still being discovered, indicating that there remains a lot to learn about how nature employs this strategy.
Author Contributions
Writing—original draft preparation, H.A.P. and A.E.M.; writing—review and editing, H.A.P. and A.E.M.
Funding
This research was funded by startup funds from the School of Pharmacy at Virginia Commonwealth University (VCU). This work was also supported by VCU’s CTSA (UL1TR000058 from the National Center for Advancing Translational Sciences) and the CCTR Endowment Fund of Virginia Commonwealth University.
Acknowledgments
We thank Julia A. Hotinger for helpful discussions.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Salmond, G.P.C.; Reeves, P.J. Membrane traffic wardens and protein secretion in Gram-negative bacteria. Trends Biochem. Sci. 1993, 18, 7–12. [Google Scholar] [CrossRef]
- Cornelis, G.R.; Gijsegem, F.V. Assembly and function of type III secretory systems. Annu. Rev. Microbiol. 2000, 54, 735–774. [Google Scholar] [CrossRef] [PubMed]
- Grado, M.A.; Abe, A.G.; Gauthier, A.; Steele-Mortimer, O.; DeVinny, R.; Finlay, B.B. Identification of the intimin-binding domain of Tir of enteropathogenic Escherichia coli. Cell Microbiol. 1999, 1, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, K.G.; Giron, J.A.; Jerse, A.N.N.E.; Mcdaniel, T.K.; Donnenberg, M.S.; Kaper, J.B. Enteropathogenic Escherichia coli contains a putative type III secretion system necessary for the export of proteins involved in attaching and effacing lesion formation. Proc. Natl. Acad. Sci. USA 1995, 92, 7996–8000. [Google Scholar] [CrossRef]
- Franzin, F.M.; Sircili, M.P. Locus of enterocyte effacement: A pathogenicity island involved in the virulence of enteropathogenic and enterohemorrhagic Escherichia coli subjected to a complex network of gene regulation. BioMed Res. Int. 2015, 2015, 534738. [Google Scholar] [CrossRef]
- Croxen, M.A.; Finlay, B.B. Molecular mechanisms of Escherichia coli pathogenicity. Nat. Rev. Microbiol. 2010, 8, 26–38. [Google Scholar] [CrossRef]
- Dai, W.; Li, Z. Conserved type III secretion system exerts important roles in Chlamydia trachomatis. Int. J. Exp. Pathol. 2014, 7, 5404–5414. [Google Scholar]
- Bartra, S.S.; Lorica, C.; Qian, L.; Gong, X.; Bahnan, W. Chromosomally-encoded Yersinia pestis type III secretion effector proteins promote infection in cells and in mice. Front. Cell. Infect. Microbiol. 2019, 9. [Google Scholar] [CrossRef]
- Marketon, M.M.; Depaolo, R.W.; Debord, K.L. Plague bacteria target immune cells during infection. Science 2005, 309, 1739–1742. [Google Scholar] [CrossRef]
- Crawford, J.A.; Kaper, J.B. The N-terminus of enteropathogenic Escherichia coli (EPEC) Tir mediates transport across bacterial and eukaryotic cell membranes. Mol. Microbiol. 2002, 46, 855–868. [Google Scholar] [CrossRef]
- Fasciano, A.C.; Shaban, L.; Mecsas, J. Promises and challenges of the type three secretion system injectisome as an antivirulence target. EcoSal Plus 2019. [Google Scholar] [CrossRef]
- Yang, J.; Hocking, D.M.; Cheng, C.; Dogovski, C.; Perugini, M.A.; Holien, J.K.; Parker, M.W.; Hartland, E.L.; Tauschek, M.; Robins-Browne, R.M. Disarming bacterial virulence through chemical inhibition of the DNA binding domain of an AraC-like transcriptional activator protein. J. Biol. Chem. 2013, 288, 31115–31126. [Google Scholar] [CrossRef]
- Marshall, N.C.; Finlay, B.B.; Marshall, N.C.; Finlay, B.B. Targeting the type III secretion system to treat bacterial infections. Expert Opin. Ther. Targets 2014, 18, 137–152. [Google Scholar] [CrossRef]
- Kimura, K.; Iwatsuki, M.; Nagai, T.; Matsumoto, A.; Takahashi, Y.; Shiomi, K.; Omura, S.; Abe, A. A small-molecule inhibitor of the bacterial type III secretion system protects against in vivo infection with Citrobacter rodentium. J. Antibiot. 2011, 64, 197–203. [Google Scholar] [CrossRef]
- Linington, R.G.; Robertson, M.; Gauthier, A.; Finlay, B.B.; Van Soest, R.; Andersen, R.J. Caminoside A, an antimicrobial glycolipid isolated from the marine sponge Caminus sphaeroconia. Org. Lett. 2002, 4, 4089–4092. [Google Scholar] [CrossRef]
- Kola, M.; Urba, K. Antibiotic selective pressure and development of bacterial resistance. Int. J. Antimicrob. Agents 2001, 17, 357–363. [Google Scholar] [CrossRef]
- Loquet, A.; Sgourakis, N.G.; Gupta, R.; Giller, K.; Riedel, D.; Goosmann, C.; Griesinger, C.; Kolbe, M.; Baker, D.; Becker, S.; et al. Atomic model of the type III secretion system needle. Nature 2012, 486, 276–279. [Google Scholar] [CrossRef] [Green Version]
- Cornelis, G.R. The type III secretion injectisome. Nat. Rev. Microbiol. 2006, 4, 811–825. [Google Scholar] [CrossRef]
- Galán, J.E.; Waksman, G. Protein-injection machines in bacteria. Cell 2018, 172, 1306–1318. [Google Scholar] [CrossRef]
- Sekiya, K.; Ohishi, M.; Ogino, T.; Tamano, K.; Sasakawa, C.; Abe, A. Supermolecular structure of the enteropathogenic Escherichia coli type III secretion system and its direct interaction with the EspA-sheath-like structure. Proc. Natl. Acad. Sci. USA 2001, 98, 11638–11643. [Google Scholar] [CrossRef]
- Bzdzion, L.; Krezel, H.; Wrzeszcz, K.; Grzegorek, I.; Nowinska, K.; Chodaczek, G.; Swietnicki, W. Design of small molecule inhibitors of type III secretion system ATPase EscN from enteropathogenic Escherichia coli. Acta Biochim. Pol. 2017, 64, 49–63. [Google Scholar] [CrossRef]
- Andrade, A.; Pardo, J.P.; Espinosa, N.; Pérez-Hernández, G.; González-Pedrajo, B. Enzymatic characterization of the enteropathogenic Escherichia coli type III secretion ATPase EscN. Arch. Biochem. Biophys. 2007, 468, 121–127. [Google Scholar] [CrossRef]
- Bergeron, J.R.C.; Fernández, L.; Wasney, G.A.; Vuckovic, M.; Reffuveille, F.; Hancock, R.E.W.; Strynadka, N.C.J. The structure of a type 3 secretion system (T3SS) ruler protein suggests a molecular mechanism for needle length sensing. J. Biol. Chem. 2016, 291, 1676–1691. [Google Scholar] [CrossRef]
- Munera, D.; Crepin, V.F.; Marches, O.; Frankel, G. N-terminal type III secretion signal of enteropathogenic Escherichia coli translocator proteins. J. Bacteriol. 2010, 192, 3534–3539. [Google Scholar] [CrossRef]
- Deng, W.; Yu, H.B.; Li, Y.; Brett, B. SepD/SepL-dependent secretion signals of the type III secretion system translocator proteins in enteropathogenic Escherichia coli. J. Bacteriol. 2015, 197, 1263–1275. [Google Scholar] [CrossRef]
- Shaw, R.K.; Cleary, J.; Murphy, M.S.; Frankel, G.; Knutton, S. Interaction of enteropathogenic Escherichia coli with human intestinal mucosa: Role of effector proteins in brush border remodeling and formation of attaching and effacing lesions. Infect. Immun. 2005, 73, 1243–1251. [Google Scholar] [CrossRef]
- Park, D.; Lara-Tejero, M.; Waxham, M.N.; Li, W.; Hu, B. Visualization of the type III secretion-mediated Salmonella–host cell interface using cryo-electron tomography. eLife Sci. 2018, 7, 1–15. [Google Scholar] [CrossRef]
- Creasey, E.A.; Delahay, R.M.; Bishop, A.A.; Shaw, R.K.; Kenny, B.; Knutton, S.; Frankel, G. CesT Is a bivalent enteropathogenic Escherichia coli chaperone required for translocation of both Tir and Map. Mol. Microbiol. 2003, 47, 209–221. [Google Scholar] [CrossRef]
- Haack, K.R.; Robinson, C.L.; Miller, K.J.; Fowlkes, J.W.; Mellies, J.L. Interaction of Ler at the LEE5 (Tir) operon of enteropathogenic Escherichia coli. Infect. Immun. 2003, 71, 384–392. [Google Scholar] [CrossRef]
- Abe, A.; De Grado, M.; Pfuetzner, R.A.; Sánchez-Sanmartín, C.; Puente, Â.L.; Devinney, R.; Strynadka, N.C.; Finlay, B.B. Enteropathogenic Escherichia coli translocated intimin receptor, Tir, requires a specific chaperone for stable secretion. Mol. Microbiol. 1999, 33, 1162–1175. [Google Scholar] [CrossRef]
- Coburn, B.; Sekirov, I.; Finlay, B.B. Type III secretion systems and disease. Clin. Microbiol. Rev. 2007, 20, 535–549. [Google Scholar] [CrossRef]
- Hauser, A.R. The type III secretion system of Pseudomonas aeruginosa: Infection by injection. Nat. Rev. Microbiol. 2009, 7, 654–666. [Google Scholar] [CrossRef]
- Galan, J.E. Common themes in the design and function of bacterial effectors. Cell Host Microbe 2009, 5, 571–579. [Google Scholar] [CrossRef]
- Golubeva, Y.A.; Sadik, A.Y.; Ellermeier, J.R.; Slauch, J.M. Integrating global regulatory input into the Salmonella pathogenicity island 1 type III secretion system. Genetics 2012, 190, 79–90. [Google Scholar] [CrossRef]
- Figueira, R.; Holden, D.W. Functions of the Salmonella pathogenicity island 2 (SPI-2) type III secretion system effectors. Microbiology 2012, 2, 1147–1161. [Google Scholar] [CrossRef]
- Winstanley, C.; Hart, C.A. Type III secretion systems and pathogenicity islands. J. Med. Microbiol. 2001, 50, 116–126. [Google Scholar] [CrossRef]
- Wang, H.; Avican, K.; Fahlgren, A.; Erttmann, S.F.; Nuss, A.M.; Dersch, P.; Fallman, M.; Edgren, T.; Wolf-Watz, H. Increased plasmid copy number is essential for Yersinia T3SS function and virulence. Science 2016, 353, 492–495. [Google Scholar] [CrossRef]
- Mcdaniel, T.K.; Jarvis, K.G.; Donnenberg, M.S.; Kaper, J.B. A genetic locus of enterocyte effacement conserved among diverse enterobacterial pathogens. Proc. Natl. Acad. Sci. USA 1995, 92, 1664–1668. [Google Scholar] [CrossRef]
- Elliott, S.J.; Sperandio, V.; Giron, J.A.; Wainwright, L.; Hutcheson, S.W.; McDaniel, T.K. The locus of enterocyte effacement (LEE)-encoded regulator controls expression of both LEE- and non-LEE-encoded virulence factors in enteropathogenic and enterohemorrhagic Escherichia coli. Infect. Immun. 2000, 68, 6115–6126. [Google Scholar] [CrossRef]
- Deng, W.; Li, Y.; Vallance, B.A.; Finlay, B.B. Locus of enterocyte effacement from Citrobacter rodentium: Sequence analysis and evidence for horizontal transfer among attaching and effacing pathogens. Am. Soc. Microbiol. 2001, 69, 6323–6335. [Google Scholar] [CrossRef]
- Vidal, J.E.; Navarro-García, F. EspC translocation into epithelial cells by enteropathogenic Escherichia coli requires a concerted participation of type V and III secretion systems. Cell. Microbiol. 2008, 10, 1975–1986. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Xiuwen, H.; Yu, B. First total synthesis of caminoside A, an antimicrobial glycolipid from sponge. Synlett 2005, 3, 437–440. [Google Scholar] [CrossRef]
- Zhang, Z.; Zong, C.; Song, G.; Lv, G.; Chun, Y.; Wang, P.; Ding, N.; Li, Y. Total synthesis of caminoside B, a novel antimicrobial glycolipid isolated from the marine sponge Caminus sphaeroconia. Carbohydr. Res. 2010, 345, 750–760. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.; Lehr, H.H.; Teitel, S.; Maehr, H.; Grunberg, E. A new antibiotic X-5108 of Streptomyces origin. I. Production, isolation and properties. J. Antibiot. 1973, 26, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Chinali, G. Synthetic Analogs of aurodox and kirromycin active on elongation factor Tu from Escherichia coli. J. Antibiot. 1981, 34, 1039–1045. [Google Scholar] [CrossRef] [PubMed]
- Warawa, J.; Finlay, B.B.; Kenny, B. Type III secretion-dependent hemolytic activity of enteropathogenic Escherichia coli. Infect. Immun. 1999, 67, 5538–5540. [Google Scholar]
- Swimm, A.I.; Kalman, D. Cytosolic extract induces Tir translocation and pedestals in EPEC-infected red blood cells. PLoS Pathog. 2008, 4, e4. [Google Scholar] [CrossRef]
- Simpson, N.; Shaw, R.; Crepin, V.F.; Mundy, R.; Fitzgerald, A.J.; Straatman-Iwanowska, A.; Knutton, S.; Frankel, G. The enteropathogenic Escherichia coli type III secretion system effector Map binds EBP50/NHERF1: Implication for cell signaling and diarrhea. Mol. Microbiol. 2006, 60, 349–363. [Google Scholar] [CrossRef]
- McHugh, R.E.; O’Boyle, N.; Connoly, J.P.R.; Hoskisson, P.A.; Roe, A.J. Characterization of the mode of action of aurodox, a type III secretion system inhibitor from Streptomyces goldiniensis. Infect. Immun. 2019, 87, 1–12. [Google Scholar] [CrossRef]
- Tamura, S.; Takahashi, N.; Miyamoto, S.; Mori, R.; Suzuki, S.; Nagatsu, J. Isolation and physiological activities of piericidin A, a natural insecticide produced by Streptomyces. Agric. Biol. Chem. 1963, 27, 576–582. [Google Scholar] [CrossRef]
- Hall, C.; Wu, M.; Crane, F.L. Piericidin A: A new inhibitor of mitochondrial electron transport. Biochem. Biophys. Res. Commun. 1966, 25, 373–377. [Google Scholar] [CrossRef]
- Kroiss, J.; Kaltenpoth, M.; Schneider, B.; Schwinger, M.; Hertweck, C.; Maddula, R.K.; Strohm, E.; Svatoš, A. Symbiotic Streptomycetes provide antibiotic combination prophylaxis for wasp offspring. Nat. Chem. Biol. 2010, 6, 261–263. [Google Scholar] [CrossRef] [PubMed]
- Duncan, M.C.; Wong, R.; Dupzyk, A.J.; Bray, W.M.; Linington, R.G. Inhibitors of the Yersinia pseudotuberculosis type III secretion system. Antimicrob. Agents Chemother. 2014, 58, 1118–1126. [Google Scholar] [CrossRef] [PubMed]
- Auerbuch, V.; Golenbock, D.T.; Isberg, R.R. Innate immune recognition of Yersinia pseudotuberculosis type III secretion. PLoS Pathog. 2009, 5, e1000686. [Google Scholar] [CrossRef] [PubMed]
- Morgan, J.M.; Duncan, M.C.; Johnson, K.S.; Diepold, A.; Lam, H.; Dupzyk, A.J.; Martin, L.R.; Wong, R.; Armitage, J.P.; Linington, R.G. Piericidin A1 blocks Yersinia Ysc type III secretion system needle assembly. Am. Soc. Microbiol. 2017, 2, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Y.; Du, X.; Chen, H.; Liu, J.; Zhao, B.; Huang, D.; Li, G.; Xu, Q.; Zhang, M.; Weimer, B.C.; et al. Cytosporone B is an agonist for nuclear orphan receptor Nur77. Nat. Chem. Biol. 2008, 4, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Cao, X.; Rico-bautista, E.; Yu, J.; Chen, L.; Chen, J.; Bobkov, A.; Wolf, D.A.; Zhang, X.; Dawson, M.I. Cytosporone B on cancer cell viability. Med. Chem. Commun. 2013, 4, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Liu, Q.; Li, L.; Wang, W.; Yao, L.; Yang, M.; Liu, B.; Chen, W.; Zhan, Y.; Zhang, M.; et al. The orphan receptor TR3 suppresses intestinal tumorigenesis in mice by downregulating wnt signaling. Gastrointrotestinal Neoplasia 2012, 61, 714–724. [Google Scholar]
- Lee, S.; Li, X.; Khan, S.; Safe, S. Targeting NR4A1 (TR3) in cancer cells and tumors. Expert Opin. Ther. Targets 2011, 15, 195–206. [Google Scholar] [CrossRef]
- Li, J.; Lv, C.; Sun, W.; Li, Z.; Han, X.; Li, Y.; Shen, Y. Cytosporone B, an inhibitor of the type III secretion system of Salmonella enterica serovar Typhimurium. Antimicrob. Agents Chemother. 2013, 57, 2191–2198. [Google Scholar] [CrossRef]
- Huang, H.; Zhang, L.; Zhang, X.; Ji, X.; Ding, X.; Shen, X.; Jiang, H.; Liu, H. Total synthesis of cytosporone B. Chin. J. Chem. 2010, 28, 1041–1043. [Google Scholar] [CrossRef]
- Iwatsuki, M.; Uchida, R.; Yoshijima, H.; Ui, H.; Shiomi, K.; Matsumoto, A.; Takahashi, Y.; Abe, A.; Tomoda, H.; Satoshi, O. Guadinomines, type III secretion system inhibitors, produced by Streptomyces sp. K01-0509 I. Taxonomy, fermentation, isolation and biological properties. J. Antibiot. 2008, 61, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Iwatsuki, M.; Uchida, R.; Yoshijima, H.; Ui, H.; Shiomi, K.; Kim, Y.; Hirose, T.; Sunazuka, T.; Abe, A.; Tomoda, H. Guadinomines, type III secretion system inhibitors, produced by Streptomyces sp. K01-0509. II. Physico-chemical properties and structure elucidation. J. Antibiot. 2008, 61, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Hirose, T.; Sunazuka, T.; Tsuchiya, S.; Tanaka, T.; Kojima, Y.; Mori, R.; Iwatsuki, M.; Ōmura, S. Total synthesis and determination of the absolute configuration of guadinomines B and C2. Chem. Eur. J. 2008, 14, 8220–8238. [Google Scholar] [CrossRef]
- Holmes, T.C.; May, A.E.; Zaleta-Rivera, K.; Ruby, J.G.; Skewes-Cox, P.; Fischbach, M.A.; Derisi, J.L.; Iwatsuki, M.; Satoshi, O.; Khosla, C. Molecular insights into the biosynthesis of guadinomine: A type III secretion system inhibitor. J. Am. Chem. Soc. 2012, 134, 17797–17806. [Google Scholar] [CrossRef]
- Bohnhoff, B.Y.M.; Miller, C.P.; Martin, W.R. Resistance of the mouse’s intestinal tract to experimental Salmonella infection I. Factors which interfere with the initiation of infection. J. Exp. Med. 1964, 120, 805–816. [Google Scholar] [CrossRef]
- Sun, Y.; O’Riordan, M.X. Regulation of bacterial pathogenesis by intestinal short-chain fatty acids. Adv. Appl. Microbiol. 2013, 85, 93–118. [Google Scholar]
- Nakanishi, N.; Tashiro, K.; Kuhara, S.; Hayashi, T.; Sugimoto, N.; Tobe, T. Regulation of virulence by butyrate sensing in enterohaemorrhagic Escherichia coli. Microbiology 2009, 155, 521–530. [Google Scholar] [CrossRef]
- Pomare, E.W.; Branch, H.W.J.; Naylor, C.P.E.; Macfarlane, T. Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut 1987, 28, 1221–1227. [Google Scholar]
- Takao, M.; Yen, H.; Tobe, T. LeuO Enhances butyrate-induced virulence expression through a positive regulatory loop in enterohaemorrhagic Escherichia coli. Mol. Microbiol. 2014, 93, 1302–1313. [Google Scholar]
- Baek, C.; Wang, S.; Roland, K.L.; Curtiss, R., III. Leucine-responsive regulatory protein (Lrp) acts as a virulence repressor in Salmonella enterica serovar Typhimurium. J. Bacteriol. 2009, 191, 1278–1292. [Google Scholar] [CrossRef] [PubMed]
- Cordone, A.; Lucchini, S.; De Felice, M.; Ricca, E. Direct and indirect control of Lrp on LEE pathogenicity genes of Citrobacter rodentium. Fed. Eur. Microbiol. Soc. 2011, 325, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Crutcher, F.K.; Puckhaber, L.S.; Stipanovic, R.D.; Bell, A.A.; Nichols, R.L.; Lawrence, K.S.; Liu, J. Microbial resistance mechanisms to the antibiotic and phytotoxin fusaric acid. J. Chem. Ecol. 2017, 43, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Gaumman, E. Fusaric acid as a wilt toxin. Phytopathology 1957, 47, 342–357. [Google Scholar]
- Dong, X.; Ling, N.; Wang, M.; Shen, Q.; Guo, S. Fusaric acid is a crucial factor in the disturbance of leaf water imbalance in Fusarium-infected banana plants. Plant. Physiol. Biochem. 2012, 60, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Reveglia, P.; Cinelli, T.; Cimmino, A.; Masi, M.; Evidente, A. The main phytotoxic metabolite produced by a strain of Fusarium oxysporum inducing grapevine plant declining in Italy. Nat. Prod. Res. 2017, 32, 2398–2407. [Google Scholar] [CrossRef]
- Li, J.; Sun, W.; Guo, Z.; Lu, C.; Shen, Y. Fusaric acid modulates type three secretion system of Salmonella enterica serovar Typhimurium. Biochem. Biophys. Res. Commun. 2014, 449, 455–459. [Google Scholar] [CrossRef]
- Zetterström, C.E.; Hasselgren, J.; Salin, O.; Davis, R.A.; Quinn, R.J. The resveratrol tetramer (-)-hopeaphenol inhibits type III secretion in the Gram-negative pathogens Yersinia pseudotuberculosis and Pseudomonas aeruginosa. PLoS ONE 2013, 8, e81969. [Google Scholar] [CrossRef]
- Subramanian, R.; Raj, V.; Manigandan, K.; Elangovan, N. Antioxidant activity of hopeaphenol isolated from Shorea roxburghii stem bark extract. J. Taibah Univ. Sci. 2015, 9, 237–244. [Google Scholar] [CrossRef]
- International Union for Conservation of Nature (IUCN) Red List. Available online: https://www.iucnredlist.org/search?query=anisoptera&searchType=species (accessed on 5 September 2019).
- Babich, H.; Zuckerbraun, H.L.; Barber, I.B.; Babich, S.B.; Borenfreund, E. Cytotoxicity of sanguinarine chloride to cultured human cells from oral tissue. Pharmacol. Toxicol. 1996, 78, 397–403. [Google Scholar] [CrossRef]
- Mondal, A.; Gandhi, A.; Fimognari, C.; Atanasov, A.G.; Bishayee, A. Alkaloids for cancer prevention and therapy: Current progress and future perspectives. Eur. J. Pharmacol. 2019, 858. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Y.; Wang, T.; Deng, X.; Chu, X. Natural compound sanguinarine chloride targets the type III secretion system of Salmonella enterica serovar Typhimurium. Biochem. Biophys. Rep. 2018, 14, 149–154. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Y.; Qiu, J.; Luo, Z.; Deng, X. The herbal compound thymol protects mice from lethal infection by Salmonella typhimurium. Front. Microbiol. 2018, 9, 1–7. [Google Scholar] [CrossRef]
- Ghannadi, A.; Ebrahim, S.; Kabouche, A. Thymus Fontanesii Boiss. & Reut: A potential source of thymol-rich essential oil in North Africa. Zeitschrift fur Naturforschung C 2004, 59, 187–189. [Google Scholar]
- Yang, S.; Peng, Q.; Francisco, M.S.; Wang, Y.; Zeng, Q.; Yang, C. Type III secretion system genes of Dickeya dadantii 3937 are induced by plant phenolic acids. PLoS ONE 2008, 3, e2973. [Google Scholar] [CrossRef]
- Khokhani, D.; Zhang, C.; Li, Y.; Wang, Q.; Zeng, Q.; Yamazaki, A.; Hutchins, W.; Zhou, S.; Chen, X.; Yang, C. Discovery of plant phenolic compounds that act as type III secretion system inhibitors or inducers of the fire blight pathogen, Erwinia amylovora. Appl. Environ. Microbiol. 2013, 79, 5424–5436. [Google Scholar] [CrossRef]
Figure 1.
Example of type III secretion system (T3SS) structure.

Figure 2.
Gene map for the locus of enterocyte effacement (LEE) in E. coli, indicating the five major promoter regions [40].
Figure 2.
Gene map for the locus of enterocyte effacement (LEE) in E. coli, indicating the five major promoter regions [40].

Figure 3.
Structures of caminosides, inhibitors isolated from marine sponge C. sphaeroconia.

Figure 4.
Structure of aurodox, a polyketide isolated from Streptomyces sp. K06-0806.

Figure 5.
Piericidin A, a natural product T3SS inhibitor isolated from Streptomyces sp. 16–22 [50].
Figure 5.
Piericidin A, a natural product T3SS inhibitor isolated from Streptomyces sp. 16–22 [50].

Figure 6.
Analogs of cytosporone B (Csn-B) synthesized and analyzed for T3SS inhibitory activity by the Shen group.
Figure 6.
Analogs of cytosporone B (Csn-B) synthesized and analyzed for T3SS inhibitory activity by the Shen group.

Figure 7.
Guadinomines isolated from Streptomyces sp. K01-0509, and their activity against enteropathogenic Escherichia coli (EPEC).
Figure 7.
Guadinomines isolated from Streptomyces sp. K01-0509, and their activity against enteropathogenic Escherichia coli (EPEC).

Figure 8.
Butyric acid, a product of Bacterioidetes and other intestinal microbes [67].
Figure 8.
Butyric acid, a product of Bacterioidetes and other intestinal microbes [67].

Figure 9.
Fusaric acid, a fungal toxin isolated from Fusarium oxysporum.

Figure 10.
(-)-Hopeaphenol, a tetramer of resveratrol isolated from rainforest plants.

Figure 11.
Sanguinarine chloride, a natural product from bloodroot Sanguinaria canadensis.

Figure 12.
Thymol, a major component of essential oils from Thymus plants.

Figure 13.
Structures of trans-cinnamic acid (TCA), trans-4-methoxycinnamic acid (TMCA), and benzoic acid (BA), regulators of the T3SS.
Figure 13.
Structures of trans-cinnamic acid (TCA), trans-4-methoxycinnamic acid (TMCA), and benzoic acid (BA), regulators of the T3SS.

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Pendergrass, H.A.; May, A.E. Natural Product Type III Secretion System Inhibitors. Antibiotics 2019, 8, 162. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics8040162
AMA Style
Pendergrass HA, May AE. Natural Product Type III Secretion System Inhibitors. Antibiotics. 2019; 8(4):162. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics8040162
Chicago/Turabian StylePendergrass, Heather A., and Aaron E. May. 2019. "Natural Product Type III Secretion System Inhibitors" Antibiotics 8, no. 4: 162. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics8040162
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.