3.2. Synthetic Procedures
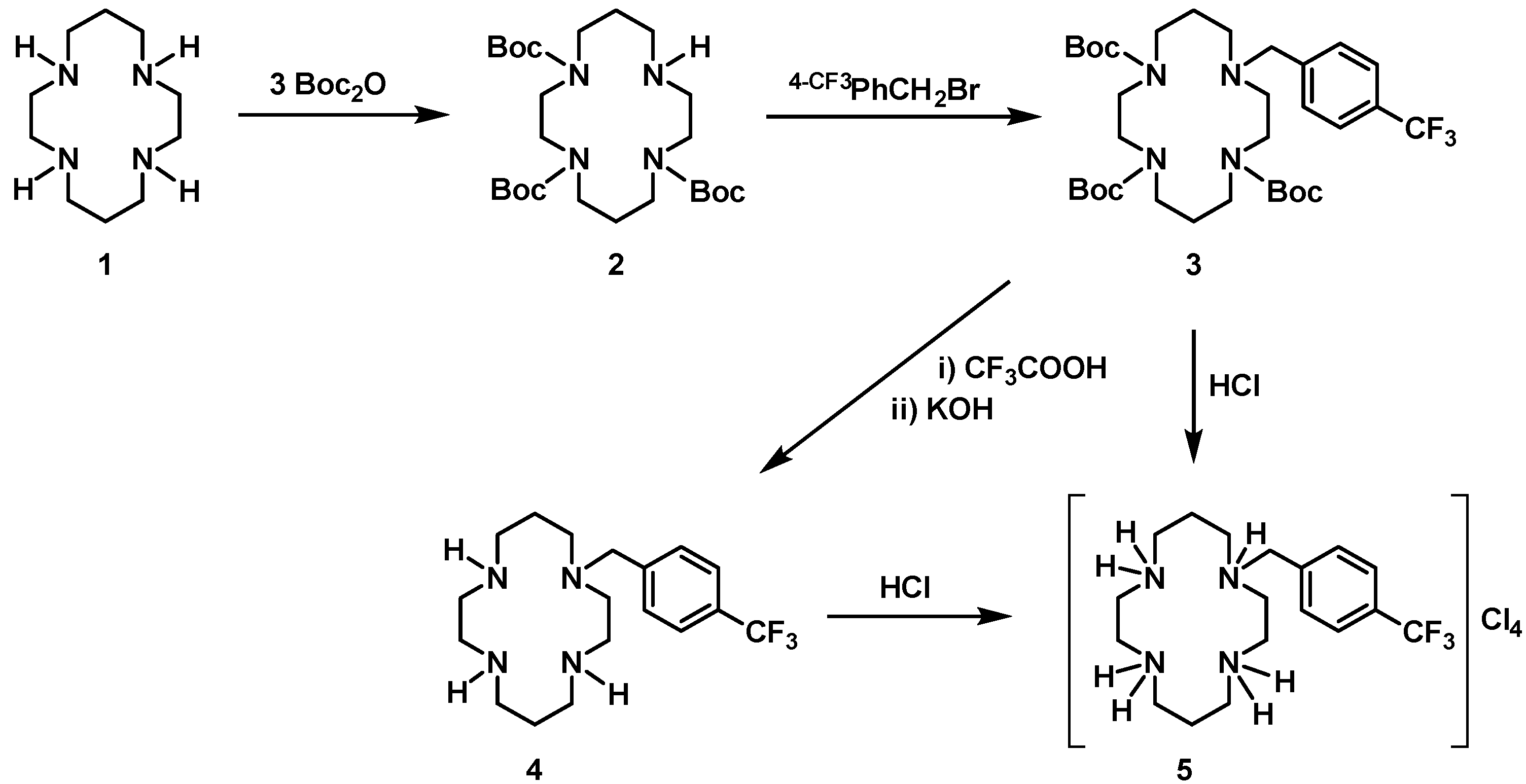
Boc3(4-CF3PhCH2)Cyclam (3): Compound 2 (1.02 g, 2.04 mmol) was dissolved in a minimum volume of dimethylformamide. K2CO3 (0.70 g, 5.06 mmol) and 4-(trifluoromethyl)benzyl bromide (0.49 g, 2.05 mmol) were added. The reaction mixture was left stirring overnight. A saturated solution of KHCO3 and brine were added, and the product extracted with small portions of chloroform. The organic phases were combined and dried with anhydrous MgSO4. After filtration, the solvent was evaporated under reduced pressure and the product was obtained in a 98% yield (1.32 g, 2.00 mmol). 1H NMR (CDCl3, 300.1 MHz, 296 K): δ (ppm) 7.50 (d, 3JH-H = 8 Hz, 2H, o-Ph or m-Ph), 7.34 (d, 3JH-H = 8 Hz, 2H, o-Ph or m-Ph), 3.53 (s, 2H, PhCH2N), 3.30 (overlapping, 12H total, 6H, [C3]CH2N and 6H, [C2]CH2N), 2.56 (m, 2H [C2]CH2N), 2.34 (m, 2H [C3]CH2N), 1.84 (m, 2H, CH2CH2CH2), 1.65 (m, 2H, CH2CH2CH2), 1.41-1.21 (br, 27H, CH3). 13C{1H} NMR (CDCl3, 75.5 MHz, 296 K): δ (ppm) 155.8 (CO), 155.6 (overlapping, CO), 143.4 (i-Ph), 129.2 (o-Ph or m-Ph), 127.4 (q, 2JC-F = 97 Hz, p-Ph), 125.2 (o-Ph or m-Ph), 124.3 (q, 1JC-F = 272 Hz, CF3), 79.7 (C(CH3)3), 79.6 (C(CH3)3), 79.5 (C(CH3)3), 59.5 (PhCH2N), 53.5 ([C2]CH2N), 51.9 ([C3]CH2N), 47.9-46.3 (overlapping, [C2]CH2N and [C3]CH2N), 28.5-28.4 (overlapping, CH2CH2CH2 and CH3). 19F NMR (CDCl3, 282.4 MHz, 296K): δ (ppm) −62.4 (s, CF3). MS (CH3CN, ESI): m/z 659.21 [M+H]+, 501.20 [M-CF3PHCH2+H]+.
H3(4-CF3PhCH2)Cyclam (4): Compound 3 (0.66 g, 1.00 mmol) was dissolved in 20 mL of dichloromethane and 10 mL of trifluoracetic acid (0.13 mol) were added. The reaction mixture was refluxed overnight. The solvent was evaporated to dryness to give a brow oil that was dissolved in water. KOH was added until the reaction mixture reached pH ≈ 13. The product was extracted with dichloromethane, the organic phase was washed with brine and dried with anhydrous MgSO4. After filtration, the solvent was evaporated to dryness producing the product as a white solid in a 31% yield (0.11 g, 0.31 mmol). 1H NMR (CDCl3, 400.1 MHz, 296 K): δ (ppm) 7.57 (d, 3JH-H = 8 Hz, 2H, m-Ph), 7.48 (d, 3JH-H = 8 Hz, 2H, o-Ph), 4.39 (br, 3H, NH) 3.65 (s, 2H, PhCH2N), 2.90 (m, 2H, [C2]CH2N), 2.85 (m, 2H [C3]CH2N), 2.79 (overlapping, 4H total, 2H, [C3]CH2N and 2H, [C2]CH2N), 2.71 (overlapping, 4H total, 2H, [C3]CH2N and 2H, [C2]CH2N), 2.60 (overlapping, 4H total, 2H, [C3]CH2N and 2H, [C2]CH2N), 1.89 (m, 2H, CH2CH2CH2), 1.81 (m, 2H, CH2CH2CH2). 13C{1H} NMR (CDCl3, 100.6 MHz, 296 K): δ (ppm) 142.8 (i-Ph), 129.7 (q, 2JC-F = 30 Hz, p-Ph), 129.6 (o-Ph), 127.0 (q, 1JC-F = 264 Hz, CF3), 125.5 (q, 3JC-F = 4 Hz, m-Ph), 57.9 (PhCH2N), 53.5 ([C2]CH2N or [C3]CH2N), 53.1 [C2]CH2N or [C3]CH2N), 50.5 ([C2]CH2N or [C3]CH2N), 49.3 ([C3]CH2N), 48.8 ([C2]CH2N or [C3]CH2N), 48.6 ([C2]CH2N or [C3]CH2N), 47.2 ([C2]CH2N or [C3]CH2N), 47.1 ([C2]CH2N), 27.0 (CH2CH2CH2), 25.6 (CH2CH2CH2). 19F NMR (CDCl3, 376.5 MHz, 296K): δ (ppm) −62.4 (s, CF3). Anal. calcd for C18H29F3N4: C, 60.31; H, 8.16; N, 15.63. Found: C, 60.28; H, 8.19; N, 15.57.
[H4{H3(4-CF3PhCH2)Cyclam}]Cl4 (5): Method A: Compound 3 (0.66 g, 1.00 mmol) was dissolved in dichloromethane and a concentrated aqueous solution of HCl (37%) was added until pH ≈ 1. The reaction mixture was left stirring overnight at room temperature. The solvent was evaporated to dryness producing the product as white solid in 56% yield (0.29 g, 0.56 mmol). Method B: Compound 4 (0.11 g, 0.31 mmol) was dissolved in ethanol and a concentrated aqueous solution of HCl (37%) was added until pH ≈ 1. The product was precipitated out of the solution. After filtration, the product was dried producing the compound as a white solid in a 48% yield (0.08 g, 0.15 mmol). 1H NMR (D2O/(CD3)2CO, 300.1 MHz, 296 K): δ (ppm) 7.87 (d, 3JH-H = 9 Hz, 2H, m-Ph), 7.78 (d, 3JH-H = 9 Hz, 2H, o-Ph), 4.48 (s, 2H, PhCH2N), 3.70 (m, 6H, [C2]CH2N), 3.59 (m, 2H [C2]CH2N), 3.50 (m, 6H, [C3]CH2N), 3.36 (m, 2H [C3]CH2N), 2.33 (m, 4H, CH2CH2CH2). 13C {1H} NMR (D2O/(CD3)2CO, 75.5 MHz, 296 K): δ (ppm) 135.1 (i-Ph), 131.8 (o-Ph), 131.2 (q, 2JC-F = 32 Hz, p-Ph), 126.4 (d, 3JC-F = 3 Hz, m-Ph), 124.2 (q, 1JC-F = 272 Hz, CF3), 57.4 (PhCH2N), 48.7 ([C3]CH2N), 46.3 ([C2]CH2N), 43.0 ([C3]CH2N), 42.5 (overlapping, [C3]CH2N), 39.5 (overlapping, [C2]CH2N), 39.1 ([C2]CH2N), 20.0 (CH2CH2CH2), 19.7 (CH2CH2CH2). 19F NMR (D2O/(CD3)2CO, 282.4 MHz, 296K): δ (ppm) −62.6 (s, CF3). Anal. calcd for C18H33Cl4F3N4.H2O: C, 41.39; H, 6.75; N, 10.73. Found: C, 41.29; H, 6.60; N, 10.68.
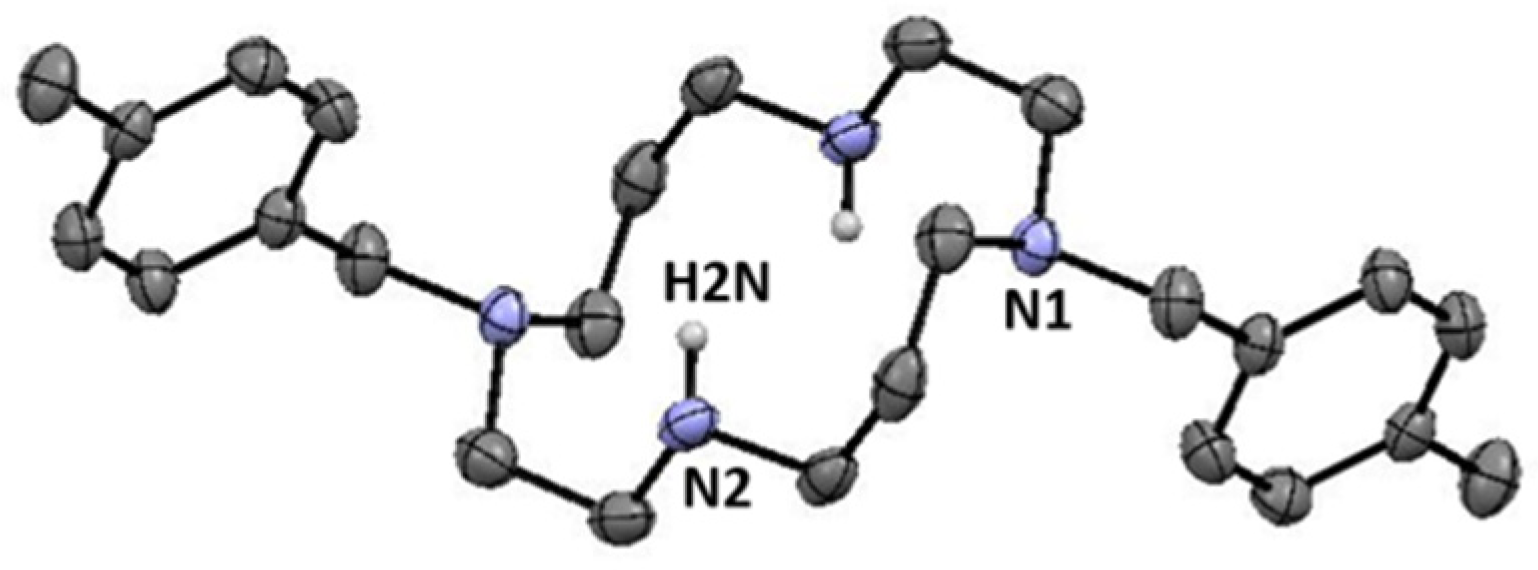
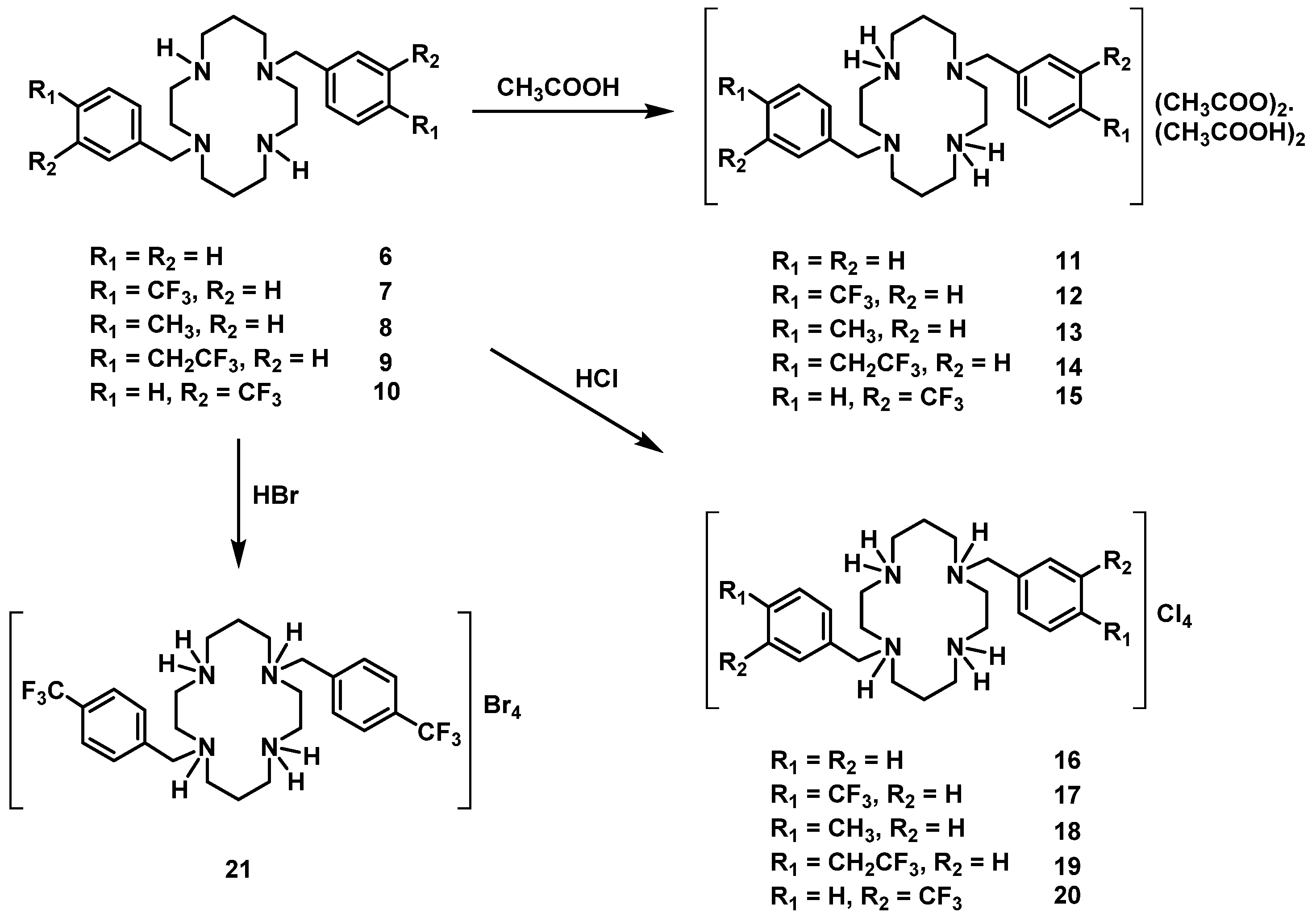
H2(4-CH3PhCH2)2Cyclam (8): 1,4,8,11-tetraazatriciclo[9.3.1.14,8]hexadecane (5.00 g, 22.3 mmol) was dissolved in a minimum volume of acetonitrile and 2.2 equiv. of 4-(methyl)benzyl bromide (9.10 g, 49.2 mmol) were added. The solution was stirred overnight at room temperature resulting in a white precipitate that was filtered, washed with acetonitrile, and dried under reduced pressure. A portion of the product obtained was hydrolyzed in an aqueous NaOH solution (3M) for 4 h under stirring at room temperature. The product was extracted with small portions of chloroform that were combined and dried with anhydrous MgSO4. After filtration, the solvent was evaporated to dryness giving an oil that was converted into a white solid after successive freeze–trituration–pump–thaw cycles. Compound 8 was obtained in a 92% yield (3.80 g, 9.30 mmol). Suitable crystals for single crystal X-ray diffraction were obtained from a concentrated chloroform solution. 1H NMR (CDCl3, 400.1 MHz, 296 K): δ (ppm) 7.19 (d, 3JH-H = 8 Hz, 4H, o-Ph or m-Ph), 7.10 (d, 3JH-H = 8 Hz, 4H, o-Ph or m-Ph), 3.69 (s, 4H, PhCH2N), 2.79 (s, 2H, NH), 2.73 (m, 4H, [C3]CH2N), 2.69 (m, 4H, [C2]CH2N), 2.55 (m, 4H, [C2]CH2N), 2.51 (m, 4H, [C3]CH2N), 2.29 (s, 6H, CH3), 1.83 (m, 4H, CH2CH2CH2). 13C {1H} NMR (CDCl3, 100.6 MHz, 296 K): δ (ppm) 136.6 (i-Ph or p-Ph), 134.2 (i-Ph or p-Ph), 129.7 (o-Ph or m-Ph), 128.9 (o-Ph or m-Ph), 57.5 (PhCH2N), 54.2 ([C2]CH2N), 51.8 ([C3]CH2N), 50.5 ([C3]CH2N), 47.9 ([C2]CH2N), 26.1 (CH2CH2CH2), 21.8 (CH3). Anal. calcd for C26H40N4: C, 76.42; H, 9,87; N, 13.71. Found: C, 76.06; H, 9.91; N, 13.69.
H2(4-CF3CH2PhCH2)2Cyclam (9): Compound 9 was prepared by the same procedure described for 8 by reaction of 1,4,8,11-tetraazatriciclo[9.3.1.14,8]hexadecane with 1-(bromomethyl)-4-(2,2,2-trifluoroethyl)benzene being obtained as a white solid in a 52% yield (0.28 g, 0.51 mmol). 1H NMR (CDCl3, 400.1 MHz, 296 K): δ (ppm) 7.29 (d, 3JH-H = 8 Hz, 4H, o-Ph or m-Ph), 7.21 (d, 3JH-H = 8 Hz, 4H, o-Ph or m-Ph), 3.71 (s, 4H, PhCH2N), 3.31 (q, 4H, 3JH-F = 11 Hz, CH2CF3), 2.72-2.53 (overlapping, 18H total, 2H, NH, 8H, [C3]CH2N and 8H, [C2]CH2N), 1.83 (m, 4H, CH2CH2CH2). 13C{1H} NMR (CDCl3, 100.6 MHz, 296 K): δ (ppm) 137.6 (i-Ph), 130.1 (o-Ph or m-Ph), 129.9 (o-Ph or m-Ph), 129.0 (p-Ph), 125.9 (q, 1JC-F = 279 Hz, CH2CF3), 57.6 (PhCH2N), 54.3 ([C2]CH2N), 51.7 ([C3]CH2N), 50.1 ([C2]CH2N or [C3]CH2N), 47.9 ([C2]CH2N or [C3]CH2N), 40.0 (q, 2JH-F = 29 Hz, CH2CF3), 26.1 (CH2CH2CH2). 19F NMR (CDCl3), 376.5 MHz, 296K): δ (ppm) −65.9 (s, CF3). Anal. calcd for C28H38F6N4: C, 61.75; H, 7.03; N, 10.29. Found: C, 60.82; H, 6.75; N, 10.00.
H2(3-CF3PhCH2)2Cyclam (10): Compound 10 was prepared by the same procedure described for 8 by reaction of 1,4,8,11-tetraazatriciclo[9.3.1.14,8]hexadecane with 1-(bromomethyl)-3-(trifluoromethyl)benzene being obtained as a white solid in a 61% yield (2.55 g, 4.94 mmol). 1H NMR (CDCl3, 400.1 MHz, 296 K): δ (ppm) 7.55 (s, 2H, o-Ph), 7.49-7.44 (overlapping, 4H total, o-Ph and m-Ph), 7.37 (d, 3JH-H = 8 Hz, 2H, p-Ph), 3.68 (s, 4H, PhCH2N), 2.74-2.69 (overlapping, 10H total, 2H, NH, 4H, [C3]CH2N and 4H, [C2]CH2N), 2.59 (m, 4H, [C2]CH2N), 2.51 (m, 4H, [C3]CH2N), 1.83 (m, 4H, CH2CH2CH2). 13C{1H} NMR (CDCl3, 100.6 MHz, 296 K): δ (ppm) 139.3 (i-Ph), 132.6 (o-Ph or m-Ph), 130.6 (q, 2JC-F = 32 Hz, m-Ph), 128.7 (o-Ph or m-Ph), 125.8 (q, 3JC-F = 3 Hz, o-Ph), 124.3, (q, 1JC-F = 273 Hz, CF3), 124.0 (q, 3JC-F = 4 Hz, p-Ph), 57.6 (PhCH2N), 53.9 ([C2]CH2N), 51.3 ([C3]CH2N), 49.1 ([C2]CH2N or [C3]CH2N), 47.5 ([C2]CH2N or [C3]CH2N), 25.9 (CH2CH2CH2). 19F NMR (CDCl3), 376.5 MHz, 296K): δ (ppm) −62.5 (s, CF3). Anal. calcd for C26H34F6N4: C, 60.45; H, 6.63; N, 10.85. Found: C, 60.21; H, 6.63; N, 10.82.
[H2{H2(PhCH2)2Cyclam}](CH3COO)2.(CH3COOH)2 (11): Compound 6 (0.35 g, 0.92 mmol) was dissolved in a small volume of acetonitrile and 1 mL of glacial acetic acid was added to the solution. This mixture was refluxed for 1 h and the solvent was evaporated under reduced pressure producing a white solid that was washed with diethyl ether and dried in a vacuum. The product was obtained as a white solid in an 84% yield (0.48 g, 0.77 mmol). 1H NMR (CDCl3, 400.1 MHz, 296 K): δ (ppm) 10.81 (overlapping, 6H total, 4H, NH2+ and 2H, CH3COOH), 7.31-7.30 (overlapping, 6H total, 2H, p-Ph and 4H, m-Ph), 7.21 (d, 3JH-H = 8 Hz, 4H, o-Ph), 3.97 (s, 4H, PhCH2N), 3.25 (m, 4H, [C3]CH2N), 3.20 (m, 4H, [C2]CH2N), 2.78 (m, 4H, [C2]CH2N), 2.74 (m, 4H, [C3]CH2N), 2.00-1.97 (overlapping, 16H total, 4H, CH2CH2CH2, 6H, CH3COO− and 6H, CH3COOH). 13C{1H} NMR (CDCl3,100.6 MHz, 296 K): δ (ppm) 176.5 (COO), 134.0 (i-Ph), 130.8 (o-Ph), 128.4 (m-Ph), 127.7 (p-Ph), 53.9 (PhCH2N), 51.9 ([C3]CH2N), 48.5 (overlapping, [C2]CH2N and [C3]CH2N), 45.4 ([C2]CH2N), 22.8 (CH2CH2CH2), 22.5 (CH3COO). Anal. calcd for C28H44N4O4.(CH3COOH)2: C, 61.91; N, 9.03; H, 8.44. Found: C, 61.85; N, 9.81; H, 8.40.
[H2{H2(4-CH3PhCH2)2Cyclam}](CH3COO)2.(CH3COOH)2 (13): Compound 13 was prepared by the same procedure described for 11 using compound 8 (1.00 g, 2.45 mmol) as the starting material. The product was obtained as a white solid in an 80% yield (1.27 g, 1.96 mmol). 1H NMR (D2O, 300.1 MHz, 296 K): δ (ppm) 7.31 (d, 3JH-H = 6 Hz, 4H, o-Ph or m-Ph), 7.23 (d, 3JH-H = 6 Hz, 4H, o-Ph or m-Ph), 3.48 (s, 4H, PhCH2N), 3.31 (m, 4H, [C3]CH2N or [C2]CH2N), 3.23 (m, 4H, [C3]CH2N or [C2]CH2N), 2.80 (m, 4H, [C3]CH2N or [C2]CH2N), 2.76 (m, 4H, [C3]CH2N or [C2]CH2N), 2.25 (s, 6H, CH3), 1.99 (overlapping, 16H total, 4H, CH2CH2CH2, 6H, CH3COO- and 6H, CH3COOH). 1H NMR (CDCl3, 300.1 MHz, 296 K): δ (ppm) 10.73 (overlapping, 6H total, 4H, NH2+ and 2H, CH3COOH), 7.12 (d, 3JH-H = 6 Hz, 4H, o-Ph or m-Ph), 7.02 (d, 3JH-H = 9 Hz, 4H, o-Ph or m-Ph), 3.79 (s, 4H, PhCH2N), 3.16-3.14 (overlapping, 8H total, 4H, [C3]CH2N and 4H, [C2]CH2N), 2.75 (m, 4H, [C2]CH2N), 2.66 (m, 4H, [C3]CH2N), 2.32 (s, 6H, CH3), 1.99 (overlapping, 16H total, 4H, CH2CH2CH2, 6H, CH3COO- and 6H, CH3COOH). 13C{1H} NMR (CDCl3, 75.5 MHz, 296 K): δ (ppm) 176.6 (COO), 137.3 (i-Ph), 130.9 (p-Ph), 130.8 (o-Ph or m-Ph), 129.1 (o-Ph or m-Ph), 52.7 (PhCH2N), 51.3 ([C3]CH2N), 48.7 ([C2]CH2N), 48.3 ([C3]CH2N), 45.6 ([C2]CH2N), 22.8 (CH2CH2CH2), 22.6 (CH3COO), 21.2 (CH3). Anal. calcd for C30H48N4O4.(CH3COOH)2: C, 62.94; H,8.70; N,8.64. Found: C, 62.26; H, 9.17; N, 8.46.
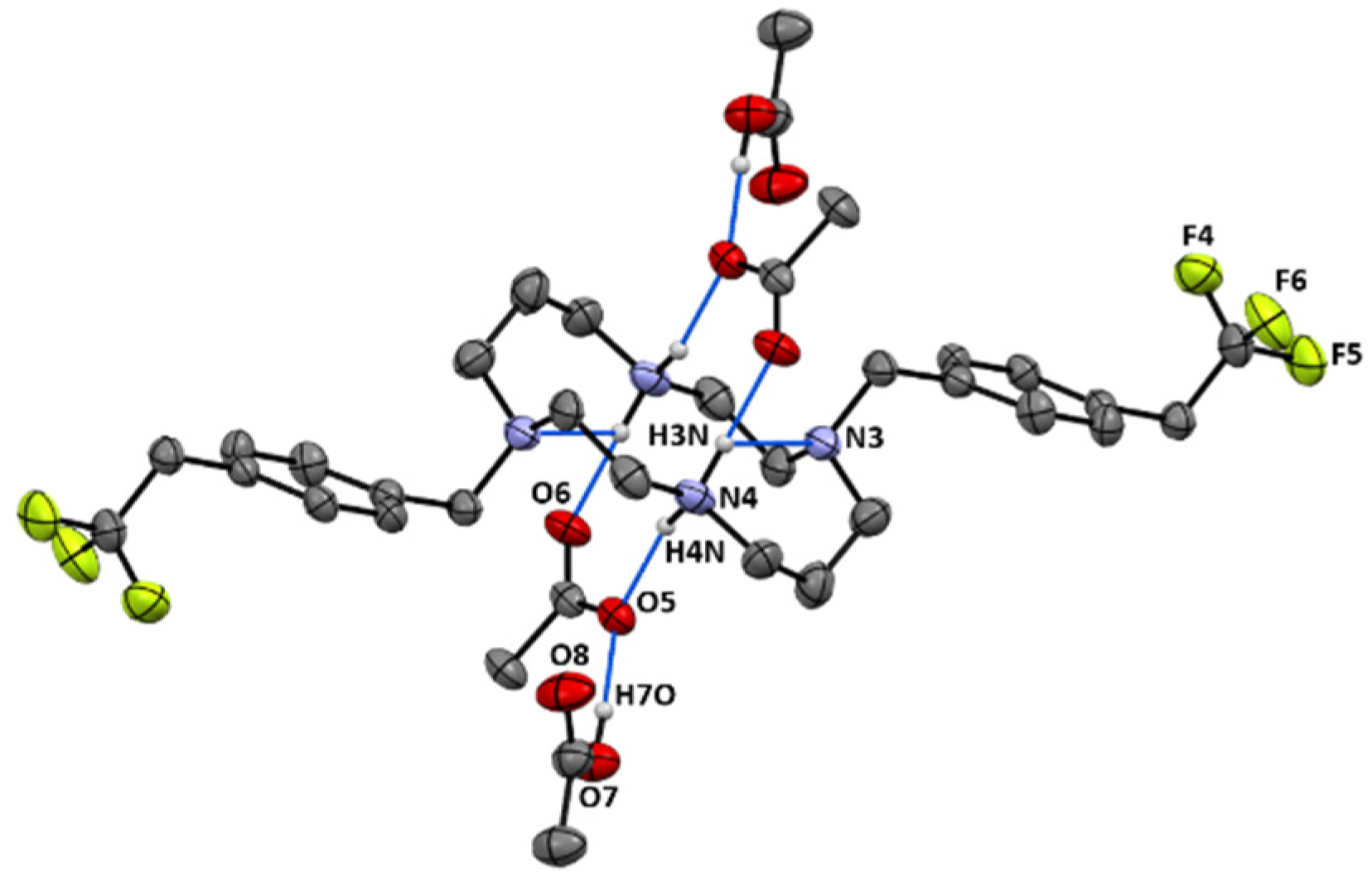
[H2{H2(4-CF3CH2PhCH2)2Cyclam}](CH3COO)2.(CH3COOH)2 (14): Compound 14 was prepared by the same procedure described for 11 using compound 9 (0.24 g, 0.44 mmol) as the starting material. The product was obtained as a white solid in an 84% yield (0.29 g, 0.37 mmol). Suitable crystals for single crystal X-ray diffraction were obtained from an aqueous acetic acid solution. 1H NMR (D2O, 400.1 MHz, 296 K): δ (ppm) 7.46 (d, 3JH-H = 8 Hz, 4H, o-Ph or m-Ph), 7.35 (d, 3JH-H = 8 Hz, 4H, o-Ph or m-Ph), 3.60 (s, 4H, PhCH2N), 3.47 (q, 4H, 3JH-F = 11 Hz, CH2CF3), 3.33 ([C3]CH2N or [C2]CH2N), 3.28 (m, 4H, [C3]CH2N or [C2]CH2N), 2.83 (m, 4H, [C3]CH2N or [C2]CH2N), 2.73 (m, 4H, [C3]CH2N or [C2]CH2N), 1.99 (overlapping, 16H total, 4H, CH2CH2CH2, 6H, CH3COO- and 6H, CH3COOH). 1H NMR (CDCl3, 400.1 MHz, 296 K): δ (ppm) 10.28 (overlapping, 6H total, 4H, NH2+ and 2H, COOH), 7.25 (d, 3JH-H = 8 Hz, 4H, o-Ph or m-Ph), 7.16 (d, 3JH-H = 8 Hz, 4H, o-Ph or m-Ph), 3.83 (s, 4H, PhCH2N), 3.34 (m, 3JH-F = 11 Hz, 4H, CH2CF3), 3.10 (overlapping, 8H total, 4H, [C2]CH2N and 4H, [C3]CH2N), 2.76 (m, 4H, [C2]CH2N), 2.68 (m, 4H, [C3]CH2N), 2.00-1.96 (overlapping, 16H total, 4H, CH2CH2CH2, 6H, CH3COO- and 6H, CH3COOH). 13C{1H} NMR (CDCl3,100.6 MHz, 296 K): δ (ppm) 176.7 (COO), 134.6 (i-Ph), 131.0 (o-Ph or m-Ph), 130.2 (o-Ph or m-Ph), 129.6 (m, 3JC-F = 3 Hz, p-Ph), 125.8 (q, 1JC-F = 277 Hz, CH2CF3), 53.3 (PhCH2N), 51.1 ([C3]CH2N), 49.1 ([C2]CH2N), 48.2 ([C3]CH2N or [C2]CH2N), 45.9 ([C3]CH2N or [C2]CH2N), 40.0 (q, 2JH-F = 29 Hz, CH2CF3), 23.1 (CH2CH2CH2), 22.6 (CH3COO). 19F NMR (D2O, 376.5 MHz, 296 K): δ (ppm) −65.8 (s, CH2CF3). 19F NMR (CDCl3, 376.5 MHz, 296 K): δ (ppm) −65.9 (s, CH2CF3). Anal. calcd for C32H46F6N4O4.(CH3COOH)2: C, 55.09; H, 6.94; N, 7.14. Found: C, 55.00; H, 6.86; N, 7.01.
[H2{H2(3-CF3PhCH2)2Cyclam}](CH3COO)2.(CH3COOH)2 (15): Compound 15 was prepared by the same procedure described for 11 using compound 10 (0.55 g, 1.06 mmol) as the starting material. The product was obtained as a white solid in a 28% yield (0.23 g, 0.30 mmol). 1H NMR (D2O, 400.1 MHz, 296 K): δ (ppm) 7.70 (d, 3JH-H = 8 Hz, 2H, p-Ph), 7.65 (s, 2H, o-Ph), 7.62 (t, 3JH-H = 8 Hz, 2H, m-Ph), 7.56 (d, 3JH-H = 8 Hz, 2H, o-Ph), 3.70 (s, 4H, PhCH2N), 3.33-3.32 (overlapping, 8H total, 4H, [C3]CH2N and 4H, [C2]CH2N), 2.82 (m, 4H, [C2]CH2N), 2.69 (m, 4H, [C3]CH2N), 2.02 (m, 4H, CH2CH2CH2), 1.98 (overlapping, 12H total, 6H, CH3COO− and 6H, CH3COOH). 1H NMR (CDCl3, 300.1 MHz, 296 K): δ (ppm) 10.61 (overlapping, 6H total, 4H, NH2+ and 2H, COOH), 7.54 (d, 3JH-H = 8 Hz, 2H, p-Ph), 7.44 (t, 3JH-H = 8 Hz, 2H, m-Ph), 7.39 (s, 2H, o-Ph), 7.36 (d, 3JH-H = 8 Hz, 2H, o-Ph), 3.90 (s, 4H, PhCH2N), 3.13 (overlapping, 8H total, 4H, [C2]CH2N and 4H, [C3]CH2N), 2.77 (m, 4H, [C2]CH2N), 2.67 (m, 4H, [C3]CH2N), 2.00 (overlapping, 16H total, 4H, CH2CH2CH2, 6H, CH3COO− and 6H, CH3COOH). 13C{1H} NMR (CDCl3, 75.5 MHz, 296 K): δ (ppm) 176.7 (COO), 135.5 (o-Ph), 134.1 (i-Ph), 130.7 (q, 2JC-F = 32 Hz, m-Ph), 128.9 (m-Ph), 127.0 (q, 3JC-F = 4 Hz, o-Ph), 124.5 (q, 3JC-F = 4 Hz, p-Ph), 124.2 (q, 1JC-F = 272 Hz, CF3), 52.8 (PhCH2N), 51.3 ([C3]CH2N), 48.8 ([C2]CH2N), 48.5 ([C3]CH2N or [C2]CH2N), 46.0 ([C3]CH2N or [C2]CH2N), 23.0 (CH2CH2CH2), 22.5 (CH3COO). 19F NMR (D2O, 376.5 MHz, 296 K): δ (ppm) −62.4 (s, CF3). 19F NMR (CDCl3, 282.4 MHz, 296 K): δ (ppm) −62.6 (s, CF3). Anal. calcd for C30H42F6N4O4.(CH3COOH)2: C, 53.96; H, 6.66; N, 7.40. Found: C, 53.85; H, 6.74; N, 7.46.
[H4{H2(PhCH2)2Cyclam}]Cl4 (16): Compound 6 (1.00 g, 2.63 mmol) was dissolved in a minimum volume of ethanol and a concentrated aqueous solution of HCl (37%) was added dropwise until the solution reached pH ≈ 1. The white precipitate which formed was filtered, washed with ethanol, and dried under reduced pressure giving the product in a 51% yield (0.73 g, 1.34 mmol). 1H NMR (D2O/(CD3)2CO, 300.1 MHz, 296 K): δ (ppm) 7.57 (overlapping, 10H total, Ph), 4.50 (s, 4H, PhCH2N), 3.68-3.44 (overlapping, 16H total, 8H, [C3]CH2N and 8H, [C2]CH2N), 1.19 (m, 4H, CH2CH2CH2). 13C{1H} NMR (D2O/(CD3)2CO, 75.5 MHz, 296 K): δ (ppm) 131.2 (o-Ph or m-Ph), 129.7 (p-Ph), 129.5 (o-Ph or m-Ph), 129.2 (i-Ph), 57.3 (PhCH2N), 47.2 ([C2]CH2N), 44.7 ([C2]CH2N), 42.1 ([C3]CH2N), 37.5 ([C3]CH2N), 17.5 (CH2CH2CH2). Anal. calcd for C24H40Cl4N4.H2O: C, 52.95; H, 7.78; N, 10.29. Found: C, 52.78; H, 7.64; N, 10.25.
[H4{H2(4-CF3PhCH2)2Cyclam}]Cl4 (17): Compound 17 was prepared by the same procedure described for 16 using compound 7 (1.00 g, 1.94 mmol) as the starting material. The product was obtained as a white solid in a 66% yield (0.88 g, 1.29 mmol). 1H NMR (D2O/(CD3)2CO, 400.1 MHz, 296 K): δ (ppm) 7.85 (d, 3JH-H = 8 Hz, 4H, m-Ph), 7.75 (d, 3JH-H = 8 Hz, 4H, o-Ph), 4.24 (s, 4H, PhCH2N), 3.65 (m, 4H, [C2]CH2N), 3.51 (m, 4H, [C3]CH2N), 3.42 (m, 4H, [C2]CH2N), 3.20 (m, 4H, [C3]CH2N), 2.28 (m, 4H, CH2CH2CH2). 13C{1H} NMR (D2O/(CD3)2CO, 100.6 MHz, 296 K): δ (ppm) 136.2 (i-Ph), 131.7 (o-Ph), 130.8 (q, 2JC-F = 24 Hz, p-Ph), 126.3 (d, 3JC-F = 3 Hz, m-Ph), 124.3 (q, 1JC-F = 272 Hz, CF3), 57.1 (PhCH2N), 49.8 ([C3]CH2N), 47.7 ([C2]CH2N), 44.9 ([C3]CH2N), 41.2 ([C2]CH2N), 20.4 (CH2CH2CH2). 19F NMR (CDCl3, 376.5 MHz, 296 K): δ (ppm) −62.6 (s, CF3). Anal. calcd for C26H38Cl4F6N4.H2O: C, 45.90; H, 5.93; N, 8.23. Found: C, 46.01; H, 5.80; N, 8.23.
[H4{H2(4-CH3PhCH2)2Cyclam}]Cl4 (18): Compound 18 was prepared by the same procedure described for 16 using compound 8 (0.80 g, 1.96 mmol) as the starting material. The product was obtained as a white solid in an 88% yield (0.99 g, 1.73 mmol). 1H NMR (D2O/C6D5N, 300.1 MHz, 296 K): δ (ppm) 6.51(d, 3JH-H = 6 Hz, 4H, o-Ph or m-Ph), 6.51 (d, 3JH-H = 6 Hz, 4H, o-Ph or m-Ph), 2.92 (m, 4H, [C3]CH2N), 2.83 (overlapping, 8H total, 4H, [C2]CH2N and 4H, PhCH2N), 2.09 (m, 4H, [C2]CH2N), 1.96 (m, 4H, [C3]CH2N), 1.42 (m, 4H, CH2CH2CH2) 1.37 (s, 6H, CH3). 13C{1H} NMR (D2O/C6D5N, 75.5 MHz, 296 K): δ (ppm) 139.9 (i-Ph), 133.2 (p-Ph), 132.7 (o-Ph or m-Ph), 131.6 (o-Ph or m-Ph), 57.3 ([C3]CH2N), 53.9 ([C3]CH2N), 53.0 ([C2]CH2N), 50.9 (PhCH2N), 47.5 ([C2]CH2N), 24.2 (CH2CH2CH2), 22.7 (CH3). Anal. calcd for C26H44Cl4N4.H2O: C, 54.55; H, 8.10; N, 9.79. Found: C, 54.41; H, 8.30; N, 9.57.
[H4{H2(4-CF3CH2PhCH2)2Cyclam}]Cl4 (19): Compound 19 was prepared by the same procedure described for 16 using compound 9 (0.35 g, 0.64 mmol) as the starting material. The product was quantitatively obtained as a white solid. 1H NMR (D2O/(CD3)2CO, 300.1 MHz, 296 K): δ (ppm) 7.63 (d, 3JH-H = 8 Hz, 4H, o-Ph or m-Ph), 7.57 (d, 3JH-H = 8 Hz, 4H, o-Ph or m-Ph), 4.41 (s, 4H, PhCH2N), 3.73-3.60 (overlapping, 12H total, 8H, [C2]CH2N and 4H, 3JH-F = 11 Hz, CH2CF3), 3.52 (m, 4H, [C3]CH2N), 3.42 (m, 4H, [C3]CH2N), 2.33 (m, 4H, CH2CH2CH2). 13C{1H} NMR (D2O/(CD3)2CO, 75.5 MHz, 296 K): δ (ppm) 132.8 (i-Ph), 131.6 (o-Ph or m-Ph), 131.5 (p-Ph and o-Ph or m-Ph), 126.4 (q, 1JC-F = 277 Hz, CH2CF3), 58.2 (PhCH2N), 48.6 ([C3]CH2N), 45.2 ([C2]CH2N), 42.4 ([C3]CH2N), 39.0 (q, 2JC-F = 29 Hz, CH2CF3), 38.2 ([C2]CH2N), 19.2 (CH2CH2CH2). 19F NMR (D2O/(CD3)2CO, 282.4 MHz, 296 K): δ (ppm) −65.6 (s, CH2CF3). Anal. calcd for C28H42Cl4F6N4.(H2O)2: C, 46.29; N, 7.71; H, 6.38. Found: C, 46.29; N, 747; H, 6.24.
[H4{H2(3-CF3PhCH2)2Cyclam}]Cl4 (20): Compound 20 was prepared by the same procedure described for 16 using compound 10 (0.70 g, 1.36 mmol) as the starting material. The product was obtained as a white solid in a 68% yield (0.61 g, 0.92 mmol). 1H NMR (D2O/(CD3)2CO, 300.1 MHz, 296 K): δ (ppm) 7.93-7.92 (overlapping, 8H total, Ph), 4.25 (s, 4H, PhCH2N), 3.76 (m, 4H, [C2]CH2N), 3.70 (m, 4H, [C3]CH2N), 3.28 (m, 4H, [C2]CH2N), 3.09 (m, 4H, [C3]CH2N), 2.38 (m, 4H, CH2CH2CH2). 13C{1H} NMR (D2O/(CD3)2CO, 75.5 MHz, 296 K): δ (ppm) 128.4 (i-Ph), 128.1 (o-Ph or m-Ph), 123.9 (q, 2JC-F = 32 Hz, m-Ph), 123.3 (o-Ph or m-Ph), 121.4 (q, 1JC-F = 270 Hz, CF3), 120.4 (q, 3JC-F = 3 Hz, o-Ph or p-Ph), 118.6 (q, 3JC-F = 4 Hz, o-Ph or p-Ph), 48.8 (PhCH2N), 44.0 ([C3]CH2N), 42.1 ([C2]CH2N), 39.7 ([C3]CH2N), 36.9 ([C2]CH2N), 15.3 (CH2CH2CH2). 19F NMR (D2O/(CD3)2CO, 282.4 MHz, 296 K): δ (ppm) -59.8 (s, CF3). Anal. calcd for C26H38Cl4F6N4: C, 47.14; H, 5.78; N, 8.46. Found: C, 46.51; H, 5.78; N, 8.40.
[H4{H2(4-CF3PhCH2)2Cyclam}]Br4 (21): Compound 7 (0.70 g, 1.36 mmol) was dissolved in a minimum volume of ethanol and a concentrated aqueous solution of HBr (48%) was added dropwise until the solution reached pH ≈ 1. The white precipitate which formed was filtered, washed with ethanol, and dried under reduced pressure giving the product in a 71% yield (0.84 g, 0.96 mmol). 1H NMR (D2O/(CD3)2CO, 300.1 MHz, 296 K): δ (ppm) 7.83 (d, 3JH-H = 8 Hz, 4H, m-Ph), 7.72 (d, 3JH-H = 8 Hz, 4H, o-Ph), 4.19 (s, 4H, PhCH2N), 3.63 (m, 4H, [C3]CH2N), 3.50 (m, 4H, [C3]CH2N), 3.37 (m, 4H, [C2]CH2N), 3.16 (m, 4H, [C2]CH2N), 2.21 (m, 4H, CH2CH2CH2). 13C{1H} NMR (D2O/(CD3)2CO, 75.5 MHz, 296 K): δ (ppm) 135.4 (i-Ph), 131.4 (o-Ph), 131.0 (q, 2JC-F = 33 Hz, p-Ph), 126.2 (m-Ph), 124.2 (q, 1JC-F = 273 Hz, CF3), 57.4 (PhCH2N), 49.2 ([C2]CH2N), 46.7 ([C2]CH2N or [C3]CH2N), 43.9 ([C2]CH2N or [C3]CH2N), 40.1 ([C3]CH2N), 19.8 (CH2CH2CH2). 19F NMR (CDCl3, 282.4 MHz, 296 K): δ (ppm) −62.6 (s, CF3). Anal. calcd for C26H38Br4F6N4.(H2O)2: C, 35.64; H, 4.83; N, 6.39. Found: C, 35.73; H, 4.82; N, 6.13.
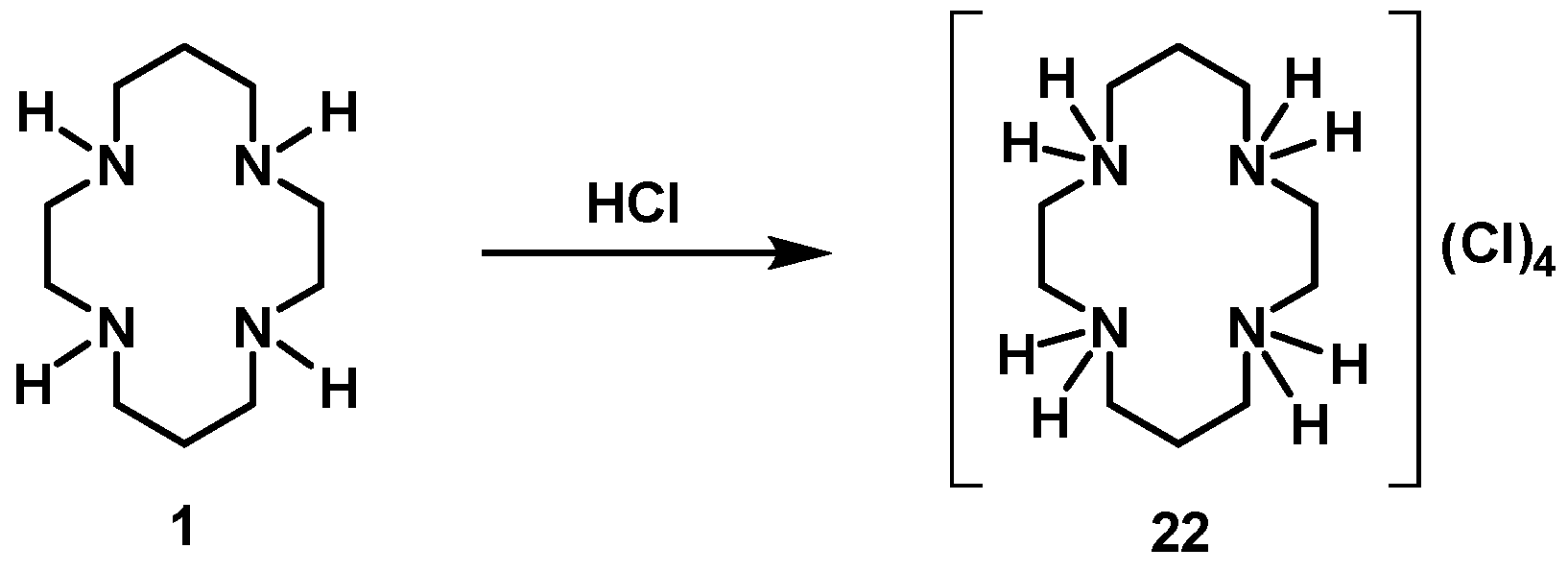
[H4(H4Cyclam)]Cl4 (22): Compound 22 was prepared by the same procedure described for 16 using cyclam (0.59 g, 2.94 mmol) as the starting material. The product was obtained as a white solid in an 89% yield (1.00 g, 2.62 mmol). 1H NMR (D2O/(CD3)2CO, 400.1 MHz, 296 K): δ (ppm) 3.59 (m, 8H, [C2]CH2N), 3.44 (m, 8H, [C3]CH2N), 2.25 (m, 4H, CH2CH2CH2). 13C{1H} NMR (D2O/(CD3)2CO, 100.6 MHz, 296 K): δ (ppm) 40.5 ([C3]CH2N), 37.3 ([C2]CH2N), 17.6 (CH2CH2CH2). Anal. calcd for C10H28Cl4N4.(H2O)2: C, 31.43; H, 8.44; N, 14.66. Found: C, 31.34; H, 7.59; N, 14.56.
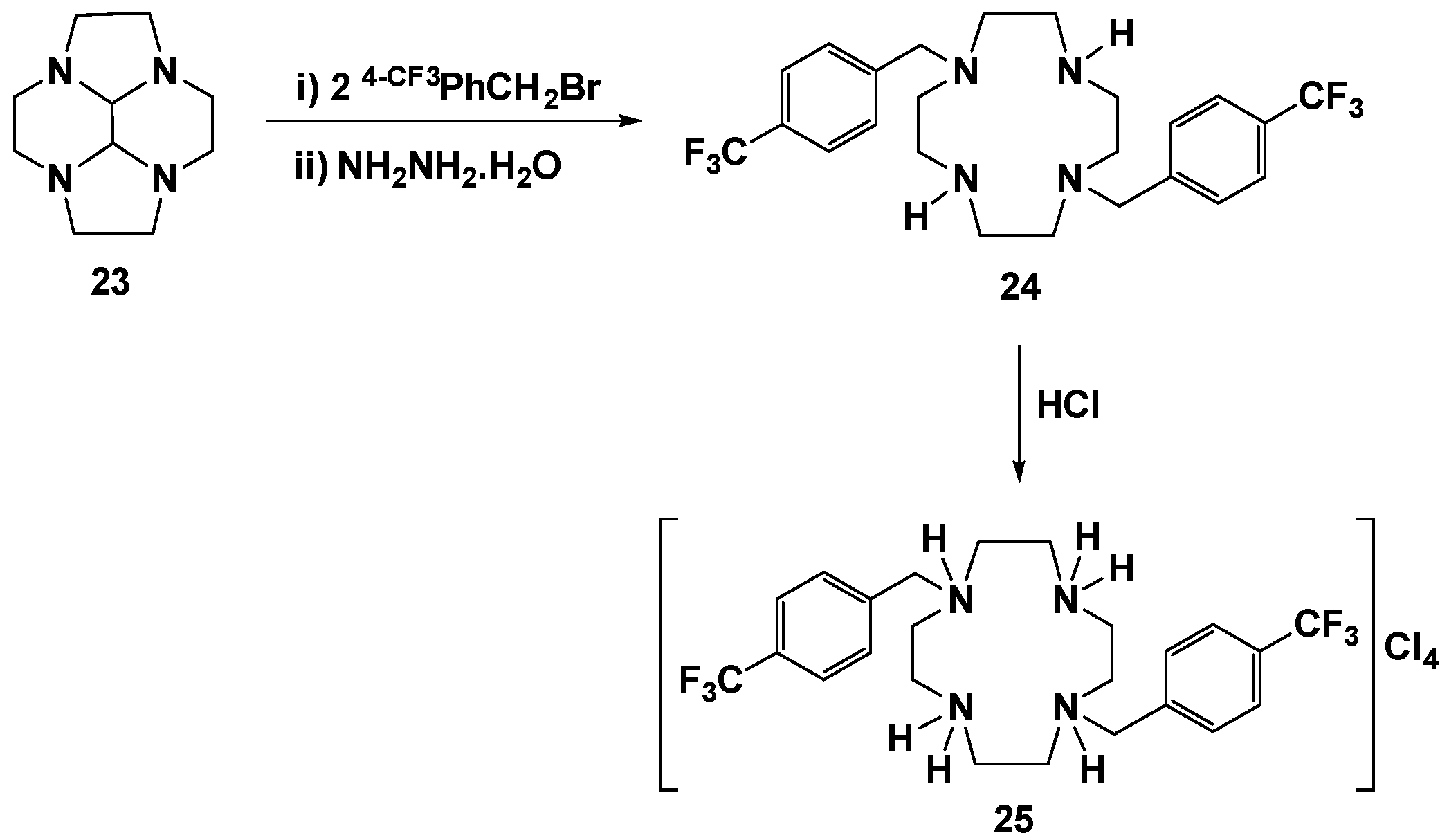
H2(4-CF3PhCH2)2Cyclen (24): Compound 23 (0.40 g, 2.06 mmol) was dissolved in the minimum volume of acetonitrile and two equiv. of 4-(trifluoromethyl)benzyl bromide (1.03 g, 4.31 mmol) were added. The solution was stirred overnight at room temperature. The white precipitate which formed was then separated by filtration, washed with acetonitrile, and dried under reduced pressure. The obtained product was heated overnight at 100 ºC in a 5 mL of hydrazine hydrate solution (50%–60%). The mixture was allowed to cool to room temperature and then placed in a cold bath to promote precipitation. The precipitate was filtered off, washed with ethanol, and dried in a vacuum giving compound 24 in a 45% yield (0.45 g, 0.92 mmol). 1H NMR (CDCl3, 300.1 MHz, 296 K): δ (ppm) 7.60 (d, 3JH-H = 8 Hz, 4H, m-Ph), 7.45 (d, 3JH-H = 8 Hz, 4H, o-Ph), 3.69 (s, 4H, PhCH2N), 2.69-2.65 (overlapping, 18H total, 16H, CH2N and 2H, NH). 13C{1H} NMR (CDCl3, 75.5 MHz, 296 K): δ (ppm) 143.4 (i-Ph), 129.8 (q, 2JC-F = 32 Hz, p-Ph), 129.3 (o-Ph), 125.6 (q, 3JC-F = 4 Hz, m-Ph), 124.2 (q, 1JC-F = 272 Hz, CF3), 60.1 (PhCH2N), 52.1 (CH2N), 45.7 (CH2N). 19F NMR (CDCl3, 282.4 MHz, 296 K): δ (ppm) −62.5 (s, CF3). Anal. Calcd. for C24H30F6N4.½H2O: C, 57.94; H, 6.28; N, 11.26. Found: C, 57.70; H, 6.32; N, 11.52.
H4[H2(4-CF3PhCH2)2Cyclen]Cl4 (25): Compound 24 (0.43 g, 0.88 mmol) was dissolved in a minimum volume of ethanol and a concentrated aqueous solution of HCl (37%) was added dropwise until the solution reached pH ≈ 1. The brown suspension was centrifugated giving a precipitate that was washed several times with small portions of acetonitrile and diethyl ether and further dried under reduced pressure. Compound 25 was obtained as a yellow solid in a 48% yield (0.27 g, 0.42 mmol). 1H NMR (D2O/(CD3)2CO, 300.1 MHz, 296 K): δ (ppm) 7.87 (d, 3JH-H = 8 Hz, 4H, m-Ph), 7.75 (d, 3JH-H = 8 Hz, 4H, o-Ph), 4.14 (s, 4H, PhCH2N), 3.50 (m, 8H CH2N), 3.12 (m, 8H CH2N). 13C{1H} NMR (D2O/(CD3)2CO, 75.5 MHz, 296 K): δ (ppm) 140.0 (i-Ph), 131.1 (o-Ph), 129.8 (q, 2JC-F = 32 Hz, p-Ph), 126.0 (q, 3JC-F = 4 Hz, m-Ph), 124.6 (q, 1JC-F = 271 Hz, CF3), 56.7 (PhCH2N), 47.8 (CH2N), 42.9 (CH2N). 19F NMR (D2O/(CD3)2CO, 282.4 MHz, 296 K): δ (ppm) −59.8 (s, CF3). Anal. Calcd. for C24H34Cl4F6N4: C, 45.44; H, 5.40; N, 8.83. Found: C, 46.04; H, 5.92; N, 10.70.

 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}













