Development and Challenges of Antimicrobial Peptides for Therapeutic Applications

1
Synthetic Biology Center, Massachusetts Institute of Technology (MIT), Cambridge, MA 02139, USA
2
Synthetic Biology Group, Research Laboratory of Electronics, Massachusetts Institute of Technology (MIT), Cambridge, MA 02139, USA
3
Broad Institute of MIT and Harvard, Cambridge, MA 02142, USA
4
Department of Electrical Engineering and Computer Science, Massachusetts Institute of Technology (MIT), Cambridge, MA 02142, USA
5
Harvard-MIT Division of Health Sciences and Technology, Cambridge, MA 02139, USA
6
Department of Biological Engineering, Massachusetts Institute of Technology (MIT), Cambridge, MA 02142, USA
*
Authors to whom correspondence should be addressed.
Antibiotics 2020, 9(1), 24; https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics9010024
Submission received: 13 December 2019
/
Revised: 27 December 2019
/
Accepted: 31 December 2019
/
Published: 13 January 2020
(This article belongs to the Special Issue Synthesis and Utility of Antimicrobial Peptides)
Abstract
:More than 3000 antimicrobial peptides (AMPs) have been discovered, seven of which have been approved by the U.S. Food and Drug Administration (FDA). Now commercialized, these seven peptides have mostly been utilized for topical medications, though some have been injected into the body to treat severe bacterial infections. To understand the translational potential for AMPs, we analyzed FDA-approved drugs in the FDA drug database. We examined their physicochemical properties, secondary structures, and mechanisms of action, and compared them with the peptides in the AMP database. All FDA-approved AMPs were discovered in Gram-positive soil bacteria, and 98% of known AMPs also come from natural sources (skin secretions of frogs and toxins from different species). However, AMPs can have undesirable properties as drugs, including instability and toxicity. Thus, the design and construction of effective AMPs require an understanding of the mechanisms of known peptides and their effects on the human body. This review provides an overview to guide the development of AMPs that can potentially be used as antimicrobial drugs.
1. Introduction
In the past several decades, multidrug-resistant bacteria have rapidly spread, causing increases in nosocomial infections and in-hospital mortality, and posing a threat to global health [1,2,3,4]. Moreover, the discovery of new classes of antibiotics has slowed down since 1987 [5,6]. The lack of new discoveries might be prompted by the conservative way we have searched for antibiotics, or this field may be saturated [5,6]; in other words, we may have already discovered many of the large natural structures that have antimicrobial activity. With the rise of antibiotic resistance, our last lines of effective antibiotics are failing [7,8,9]. Antimicrobial peptides (AMPs), a ubiquitous part of the innate immune defense in all classes of life, have been widely studied and show potential as small molecule antibiotics [10,11,12].
2. FDA Drug Approvals and Databases
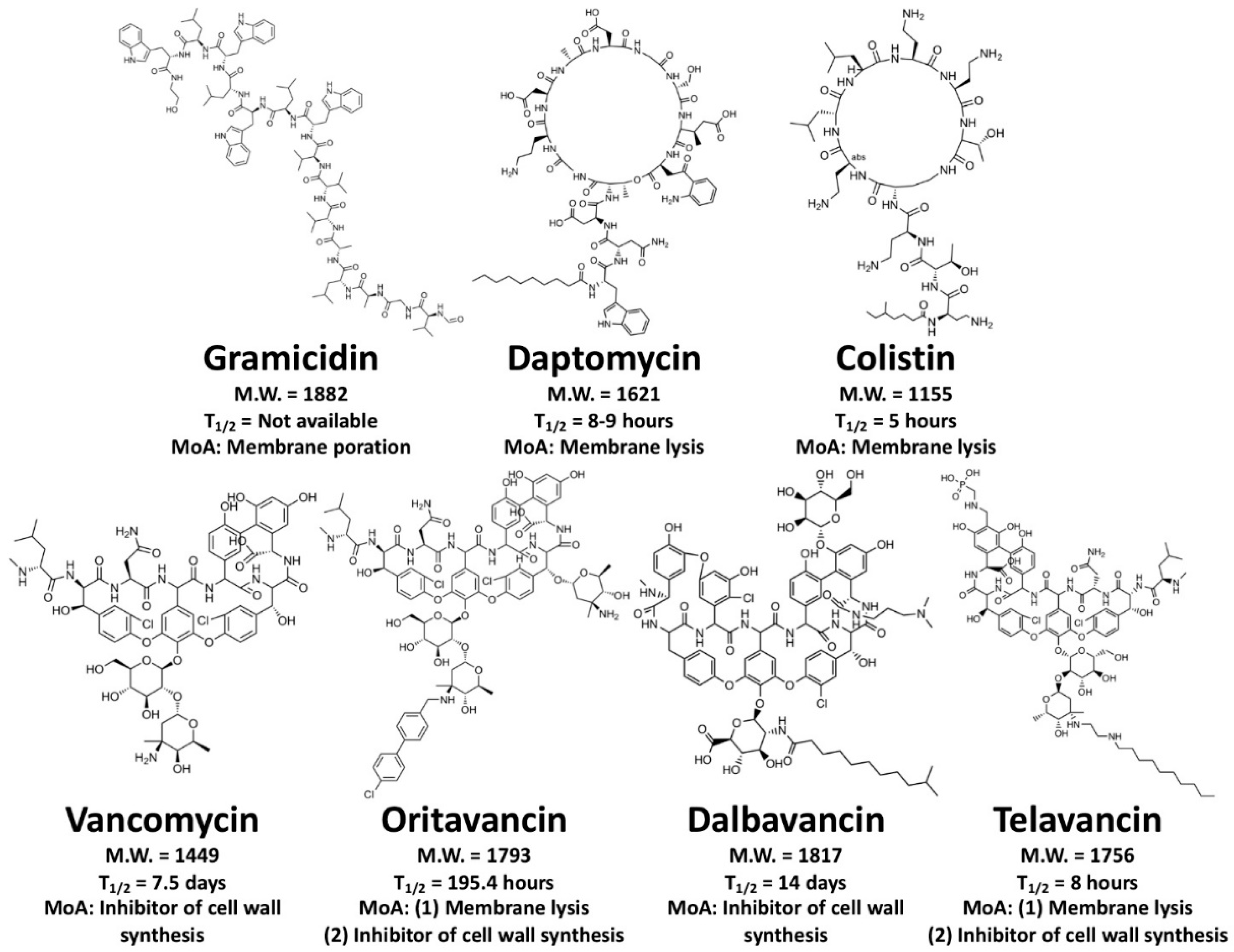
More than 3000 AMPs have been reported and characterized [13], but in their natural state, most are not suitable as drugs for human medicine. In fact, many of them failed prior to or during clinical trials [14]. To understand this problem, we looked into peptide drugs that have been approved by the U.S. Food and Drug Administration (FDA). Using the Therapeutic Proteins Database [THPdb, a subset of the FDA database (Drugs@FDA)], we analyzed all the peptide therapeutics so far approved by the FDA [15]. This database has a total of 852 peptide and protein therapeutics. Of these, 239 have been validated while the others are their derivatives and/or similar ingredients for therapeutics, and 27 of the 239 are small peptides (fewer than 50 amino acids). Six of the small peptides are AMPs: gramicidin D, daptomycin, vancomycin, oritavancin, dalbavancin, and telavancin (Figure 1).
Gramicidin D, first isolated from the soil bacterium Bacillus brevis and characterized in 1941, is a heterogeneous mixture of three pore-forming peptides: gramicidins A (80%), B (5%), and C (15%) [16,17,18,19,20,21]. Gramicidin D was approved by the FDA in 1955 as a constituent in Neosporin® [22], a triple antibiotic ointment for treating bacterial conjunctivitis. Daptomycin is a 13-residue cyclic lipopeptide antibiotic that binds onto the bacterial cell membrane, aggregates, and disrupts the membrane [23,24]. Daptomycin (also known as LY146032 [25]) and its derivative Cubicin (manufactured by Cubist Pharmaceuticals, now Merck & Co.) were approved in 2003 by the FDA to treat or prevent infectious diseases [26]. Cubicin and its new formulation Cubicin RF, which can be directly injected into the body, are antibiotics used for the treatment of complicated skin and skin structure infections (cSSSI) and Staphylococcus aureus bloodstream infections. Oritavancin, dalbavancin (formerly BI-397), and telavancin are small lipoglycopeptide antibiotics derived from vancomycin (approved by the FDA as an oral solution in 1983). These lipoglycopeptides are more potent and bactericidal than their prototype vancomycin, and they are effective against vancomycin-resistant bacteria. They inhibit bacterial cell wall formation [27,28], and telavancin and oritavancin also disrupt bacterial cell membranes and affect membrane permeability [29,30]. Similar to Cubicin, the therapeutic products Orbactiv (oritavancin), Dalvance (dalbavancin), and Vibativ (telavancin) are being used for injection against cSSSI caused by S. aureus, as well as other Gram-positive bacterial infections, and were approved by the FDA in 2014, 2014, and 2009, respectively.
One FDA-approved AMP is not mentioned in the THPdb. Colistin, also known as polymyxin E, is an antibacterial cyclic lipopeptide that was approved by the FDA in 1962 and is manufactured by Endo Pharmaceuticals, Inc, Malvern, Pennsylvania, U.S.A. as the product Coly-Mycins (colistin sulfate). Colistin is composed of 10 amino acids (sequence: KTKKKLLKKT; MW = 1145) and one fatty acid (6-methyl octanoic acid). Six of the constituent amino acids are positively charged (e.g., lysine), while the hydrophobic content is 20%. Colistin is active against several Gram-negative bacteria, e.g., Pseudomonas aeruginosa, Klebsiella pneumoniae, and Acinetobacter spp.
Peptide stability is a key requirement for the use of peptides as drugs [31,32,33,34]. Nevertheless, the hormone insulin and its analogs, which are among the most well-known peptides, have a short elimination half-life (4-6 min) in the bloodstream. Insulin was the first genetically engineered peptide hormone and was approved by the FDA in 1982 for the treatment of diabetes [35,36]. The elimination half-life of FDA-approved AMPs is much longer than that of insulin [13,31,32,33,34]. Daptomycin, oritavancin, dalbavancin, telavancin, and colistin have elimination half-lives of 8–9 h, 14 days, 8 h, 195.4 h, and 5 h, respectively (that of gramicidin has not been determined). More broadly, the average elimination half-life of FDA-approved new drugs is 50 h (median = 9 h), and of FDA-approved small peptides (less than 50 amino acids) for therapeutic use is 37 h (median = 3 h) (Figure 2). Thus, most of the FDA-approved peptides included in this analysis are stable in vivo, probably because those that are not biologically stable are unsuitable as drugs [31,32,33,34]. More rigid peptide structures may extend the elimination half-life [37,38,39]. For example, cyclic lipopeptides (e.g., daptomycin and colistin) and cyclic lipoglycopeptides (e.g., vancomycin, oritavancin, dalbavancin, and telavancin) are more stable than their linear counterparts [40]. In addition, introducing non-canonical amino acids into the peptide sequence can prevent biological degradation by proteases and extend the elimination half-life [41].
3. Antimicrobial Peptide Database
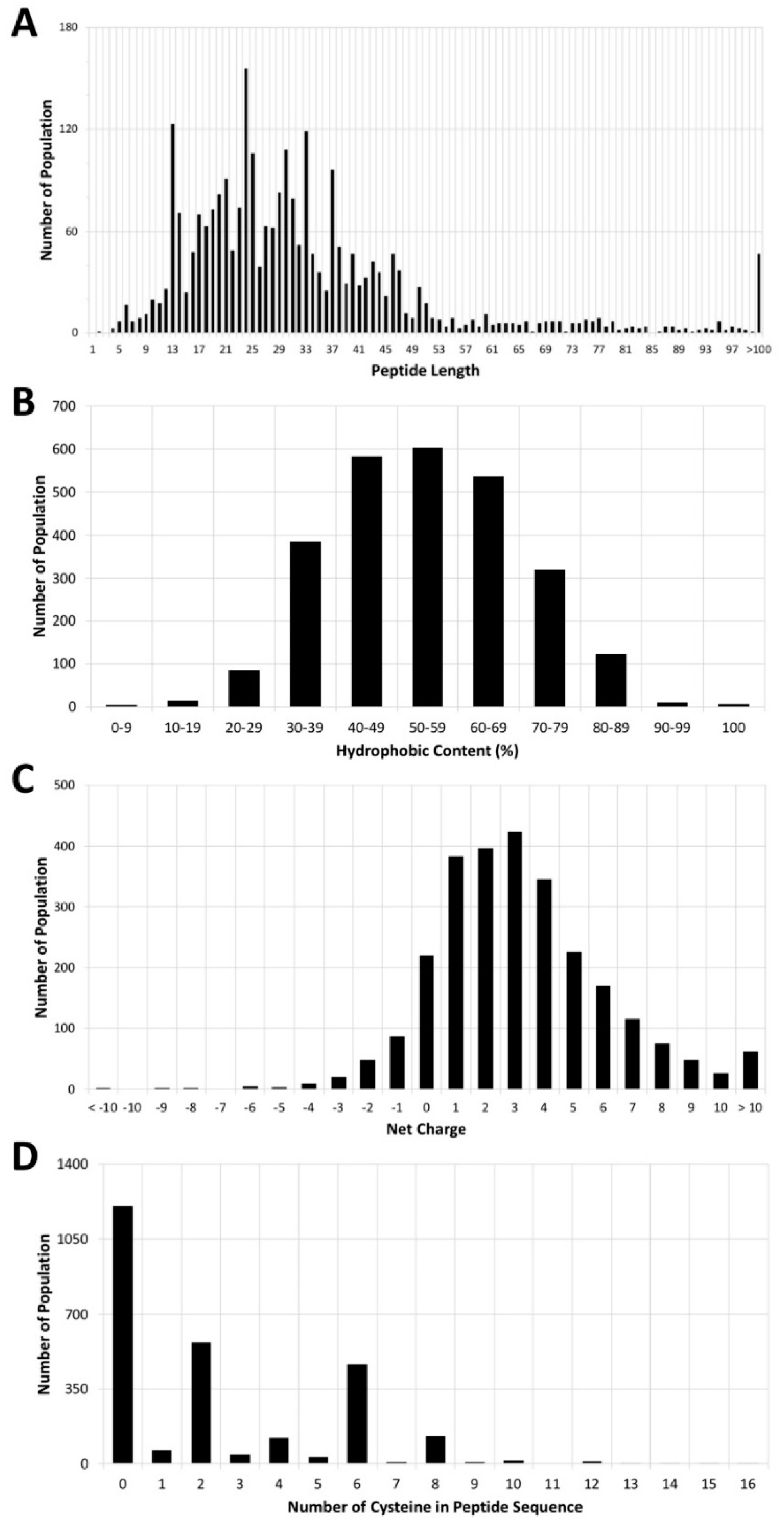
To date, only seven small AMPs have been approved by the FDA, so we extended our study to other AMPs that are under development and listed in the Antimicrobial Peptide Database (APD). A total of 3156 AMPs is listed in the APD, most of which were discovered in nature [13]. An analysis of 2700 of the 3156 AMPs in the APD showed that these peptides all have different structures and sequence motifs, and because they have a broad spectrum, they can kill a range of pathogens [42,43]. Interestingly, one-third of the AMPs are derived from frogs [44]. The average length of peptides in the APD is 33 amino acids, the median length is 28 amino acids, and more than 90% of the peptides, known as small peptides, have no more than 50 amino acids (Figure 3A). The average hydrophobic content of the peptides is 54% (Figure 3B), and the mean peptide net charge is +3 (Figure 3C). About 45% of the peptides do not contain cysteine; 21% and 17% of them have two cysteines and six cysteines, respectively (Figure 3D), which reveals the potential of a disulfide bond formation between two cysteines.
In the APD, 1869 of the 2700 peptides (~70% of the database) are small cationic amphipathic peptides. However, of the FDA-approved AMPs, only one, colistin, is in this category of small cationic amphipathic peptides. Gramicidin is small (10 amino acids) and has a net charge of +2, but it contains eight hydrophobic residues and two positively charged lysine residues, which makes it a small cationic hydrophobic peptide. Many studies have proposed that membrane-active AMPs selectively target and disrupt anionic bacterial cell membranes using electrostatic interactions [45,46,47,48,49]. However, daptomycin, a small amphipathic peptide with a neutral net charge, deviates from this pattern. Because vancomycin, oritavancin, dalbavancin, and telavancin are lipoglycopeptides, they are not included in the APD, which comprises only peptides and lipopeptides.
4. Current Development of Peptide Drugs
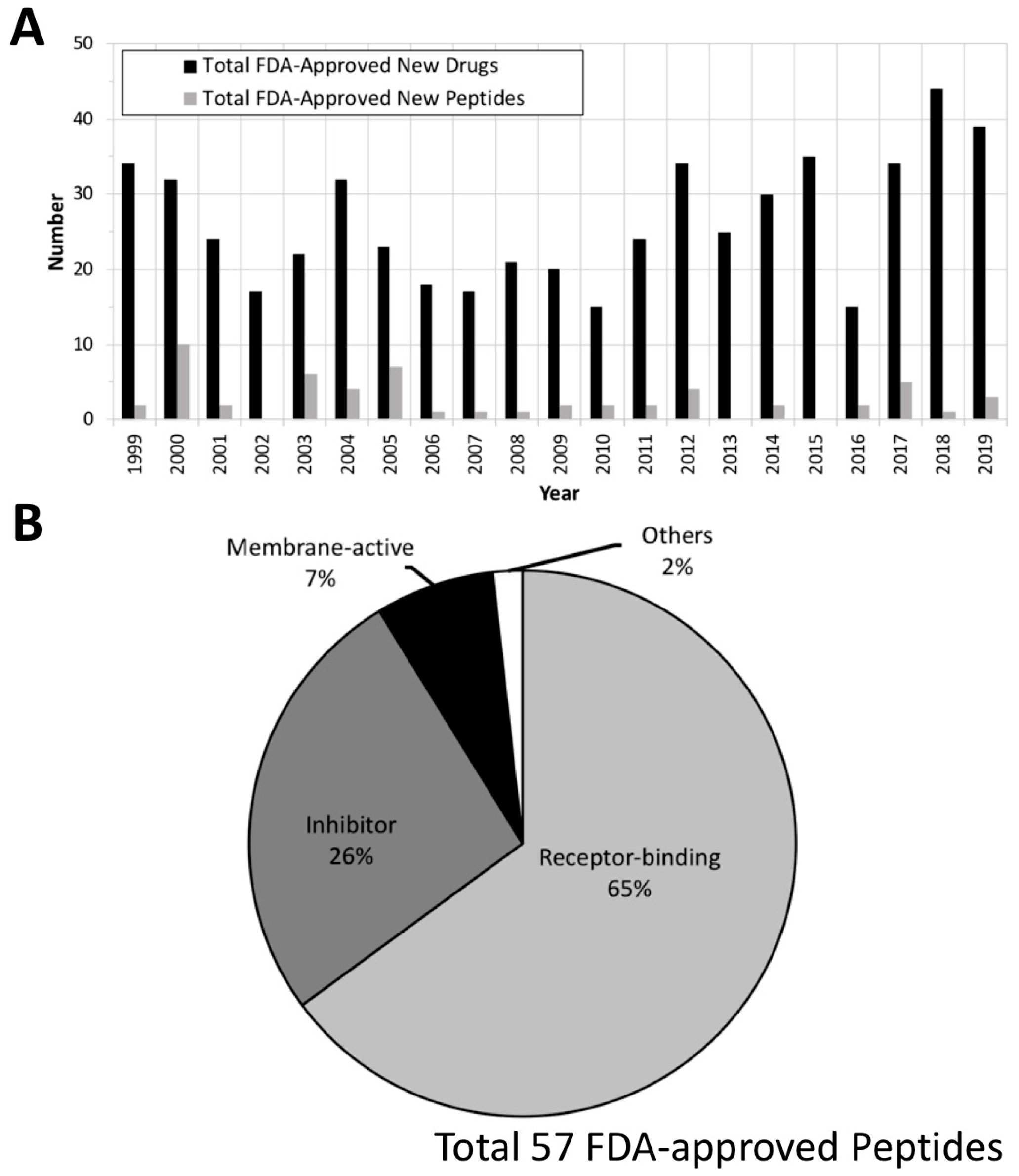
We further analyzed FDA-approved small peptide (less than 50 amino acids) therapeutics from the past 20 years (total 57 drugs; Table 1) using Drugs@FDA (http://www.fda.gov/drugsatfda) and DrugBank [50]. Details regarding newly approved compounds (submission classification: Type 1—New Molecular Entity) were extracted from Drugs@FDA, and the data were further confirmed by DrugBank. A total of 555 new molecules were approved and commercialized between January 1999 and December 2019 (Figure 4A). Many peptide therapeutics are not included in the THPdb, and the listed molecules are not limited to AMPs. Fifty-seven of the molecules are small peptide therapeutics, and most of these (37 drugs) are receptor-binding peptides that either activate or block the specific receptors to which they bind, causing a biological response. The rest of them are inhibitors of biological pathways (15 drugs), membrane-active peptides (MAPs; 4 drugs), or have other functions (1 drug) (Figure 4B). Below, we will discuss the various ways in which peptides can interact with cells to perform their therapeutic functions.
4.1. Receptor-Binding Peptides
Receptor-binding peptides, which include both agonists and antagonists, constitute the major category of therapeutic peptides that have been approved by the FDA. These peptides have been used as therapeutics and diagnostics for applications other than infectious diseases (see Table 1). Seventeen of the 37 FDA-approved receptor-binding peptides are insulin and its analogs, which are used for treating diabetes. Four of the 37 have been utilized as anti-cancer drugs: two for prostate cancer and two for neuroendocrine tumors. Some of the FDA-approved peptides have immune-modulating effects.
However, no receptor-binding peptide has yet received FDA-approval as an antimicrobial therapeutic [51]. Small-molecule drugs have been widely studied to modulate the immune system, e.g., drugs that interact with the toll-like receptor [52,53,54]. Given this capacity, it is possible that receptor-binding peptides could be used to treat infections by stimulating the immune system. Future directions of research may investigate the use of peptides to modulate the immune system instead of, or in addition to, killing bacteria directly.
In fact, several multifunctional AMPs have been used experimentally to modulate the immune response and kill pathogens [55,56,57]. For example, human cathelicidin LL-37 and human β defensins activate the toll-like receptor signal in the innate immune system [58,59]. Nevertheless, these peptides may be a double-edged sword in that a higher dose of AMPs (e.g., cathelicidin LL-37) or their proteolytic peptide fragments could result in off-target effects and trigger additional chronic inflammatory diseases, e.g., atopic dermatitis, rosacea, psoriasis, and hidradenitis suppurativa [60]. Clinical studies of LL-37 as a topical treatment for chronic leg ulcers has demonstrated safety [61]. LL-37 has entered phase II clinical studies for further investigation of its antimicrobial activity and its ability to modulate inflammation and the healing rate of diabetic foot ulcers (see more information on ClinicalTrials.gov website: https://clinicaltrials.gov; ClinicalTrials.gov Identifier: NCT04098562). More clinical studies of LL-37 are needed to explore the efficacy and potential side effects of this molecule [61]. In future work, synthetic peptides that have precise immunomodulatory effects together with direct antimicrobial activity may be designed as a promising route.
4.2. Membrane-Active Peptides (MAPs)
Five out of the seven FDA-approved AMPs are MAPs (Table 1): gramicidin, daptomycin, oritavancin, telavancin, and colistin. More specifically, gramicidin is a pore-forming peptide that forms ion channels as a transmembrane dimer. Daptomycin is a membrane lytic peptide that does not form pores but co-clusters with anionic lipids and lyses the membrane. These peptides aggregate and assemble in bacterial membranes, promoting membrane depolarization via different pathways [16,23,24,62,63,64]. Oritavancin and telavancin are dual-mechanism AMPs: they (i) inhibit bacterial cell wall synthesis and (ii) disrupt bacterial membranes. Although a few studies have suggested that these peptides have features similar to those of another pore-forming AMP, nisin [28,29,30,65], their actual mechanisms of membrane disruption remain unclear. Oritavancin and telavancin may either form membrane pores or channels, or lyse the membrane. Colistin forms pore-like aggregates in the bacterial cell membrane and disrupts the membrane; thus, it results in lytic cell death [66,67].
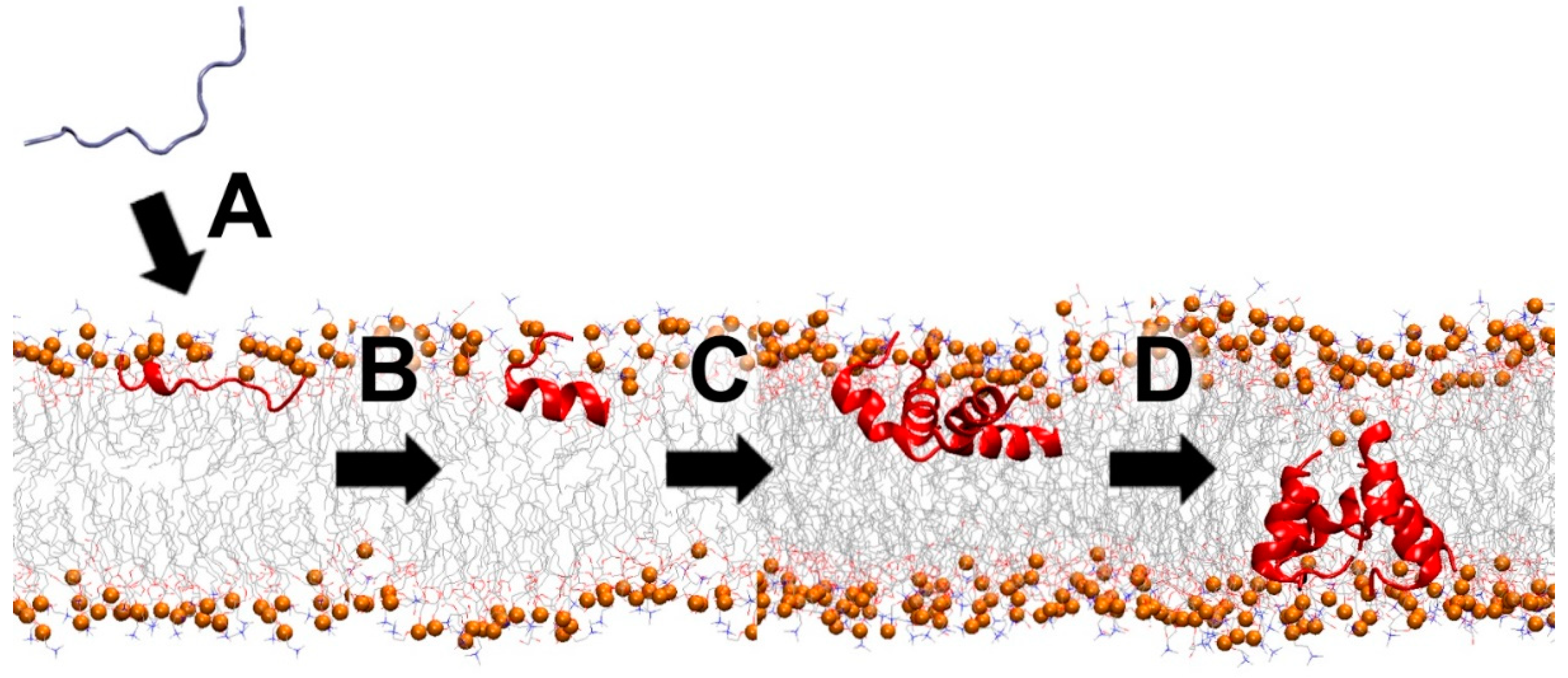
Membrane pore-forming AMPs constitute a large subgroup of MAPs. These peptides bind to cell membranes and spontaneously assemble in the lipid bilayer as a channel or pore-like structure, though not all are cytolytic (Figure 5). Well-known natural examples are gramicidin [68], colistin [67], melittin [69,70], maculatin [71], and alamethicin [72]. The two common models for the channel structures are barrel-stave and toroidal, depending on how the peptide interacts with the lipid headgroups [73]. These oligomeric structures can be a homogeneous population of oligomers or have diverse multimeric sizes that can conduct water and ions across the membrane. The pore size, which varies among different peptides, can be measured by several biophysical techniques and molecular dynamics simulations [44,71,74,75,76,77].
Membrane-lytic peptides, e.g., daptomycin [23,24,26,78,79], colistin [67], LL-37 [80], aurein 1.2 [81,82], and piscidin 1 [83,84], disrupt cell membranes, like detergents. These peptides accumulate on the membrane surface, carpet the membrane at a critical threshold concentration, and destabilize and permeabilize the membrane structure. Some membrane-lytic peptides, e.g., melittin and colistin, form pores at low peptide concentration and lyse the cell membrane above a threshold concentration or interact with specific membrane types [85].
Unlike receptor-binding peptides or peptide inhibitors that have specific binding targets, membrane-active peptides, whose activity is limited to specific cell membranes, are not well-defined. Their specificity is usually caused by their hydrophobic moment and electrostatic interactions, but exact mechanisms have not yet been determined [86,87,88]. These properties limit the ability to precisely tune the selectivity of membrane-active peptides toward a specific bacterial species [44,70,71,74,89,90]. Bacterial membranes are generally composed of more negative charges [91], whereas mammalian cell membranes contain abundant cholesterol and sphingomyelin, which make the membranes more rigid [92,93]. The advantage of utilizing membrane-active peptides for antibiotics is that bacteria may have less of a chance to develop drug resistance [94]. A deeper understanding of the molecular mechanisms underlying bacterial membrane disruption may enable further fine-tuning of the hydrophobic moment and charge distribution, and improvement of specificity.
4.3. Cell Wall-Inhibiting Peptides
Other FDA-approved AMPs are inhibitors of bacterial cell wall synthesis: vancomycin, oritavancin, dalbavancin, and telavancin. These glycopeptides bind to the D-alanyl-D-alanyl amino acids on peptidoglycan chains and prevent the incorporation of N-acetylmuramic acid and N-acetylglucosamine. Their binding blocks peptidoglycan elongation and cell wall formation, killing the bacteria [27,28,29,30,65,95].
4.4. Peptides Having Other Inhibitory Mechanisms
Other potential targets for inhibiting bacterial growth include DNA, RNA, and ribosomes (protein synthesis). Peptides with these mechanisms include edeine [96,97], tuberactinomycins [98], and dityromycin [99]. Edeine is an antimicrobial pentapeptide that binds to the binding site (P-site) of both 30S subunits and 70S ribosomes, thus inhibiting the binding of aminoacyl-tRNA and blocking translation initiation [96,97]. Tuberactinomycins, a group of cyclic peptides, inhibit prokaryotic protein synthesis and group I self-splicing via binding to the G-binding site and backbone of the intron RNA [98]. The antimicrobial cyclic decapeptide dityromycin has been shown to block elongation factor G (EF-G)-catalyzed translocation by disrupting the contact between EF-G and ribosomal protein S12, so that it deactivates the ribosome-EF-G complex and prevents translocation [99].
5. Conclusions and Future Perspectives
Peptide therapeutics have only made up a minority of all new molecular entities approved by the FDA (Figure 4A). Peptides have mostly been utilized to treat bacterial skin infections, pink eye, or wounds [100,101], e.g., Neosporin® (gramicidin; manufactured by Monarch Pharmaceuticals, Inc., Bristol, TN, USA), Cubicin® (daptomycin; manufactured by Merck & Co., Inc., Kenilworth, NJ, USA), Vancocin®HCl (vancomycin; manufactured by ANI Pharmaceuticals, Inc., Baudette, MN, USA), Orbactiv® (oritavancin; manufactured by Melinta Therapeutics, Inc., New Haven, CT, USA), DalvanceTM (dalbavancin; manufactured by Allergan Sales, LLC, Irvine, CA, USA), and Vibativ® (telavancin; manufactured by Theravance Biopharma, Inc., San Francisco, CA, USA). Several AMPs have been approved for direct injection into the human body, e.g., Cubicin, Vancocin, Orbactiv, Dalvance, Vibativ, and Coly-Mycins, because of their longer elimination half-life (ranging from hours to days) and better pharmacokinetics [31,32,33,34] compared with gramicidin or other AMPs. However, most of these lipopeptide antibiotics (except colistin) are used for treating Gram-positive bacterial infections, and only a few of them have been administered as oral solutions or tablets because of their poor penetration of the intestinal mucosa [102]. Oral vancomycin (Vancocin) is limited to the treatment of Gram-positive bacterial infections, such as Clostridium difficile diarrhea and staphylococcal enteritis, because of its poor absorption and ingestion in the body and the severity of these infections. AMPs to treat infections caused by Gram-negative bacteria are clearly needed.
Although vancomycin has been approved by the FDA, several clinical studies have shown that it may cause kidney damage in some patients or at high doses. Oritavancin and dalbavancin were, in fact, developed to improve the antibacterial activity of vancomycin, so that the dose could be reduced and toxicity lowered or prevented. Although the side effects of these compounds are mild, their effectiveness against drug-resistant Gram-positive organisms and for long-term treatment remains ambiguous [103,104,105]. Telavancin, another derivative of vancomycin, is more effective for treating a range of drug-resistant Gram-positive bacteria, but it has been reported that it may induce acute kidney injury and have a higher death rate than vancomycin [106,107]. Colistin may cause damage to the kidneys and the central nervous system in adult patients, and heavy use of colistin can result in the occurrence of colistin-resistant bacteria, making it problematic for regular use [108,109,110]. Other extensively studied pore-forming AMPs, such as alamethicin and melittin, are hemolytic and cytotoxic [69,111,112]; therefore, no clinical study has been conducted (see more information on ClinicalTrials.gov website: https://clinicaltrials.gov). This suggests that controlling the selectivity, reducing the toxicity, and lowering unexpected side effects are essential to the design of AMPs as human medicines [38,49].
The seven FDA-approved AMPs are small, with a molecular weight between 1145 and 1882. They are composed of several noncanonical amino acids and have chemical modifications or cyclic structures. These features optimize their pharmacokinetics and extend their elimination half-life so that they resist enzymatic degradation. The APD contains 3156 AMPs, 98% of which were discovered in nature [13]: many, in fact, were extracted from the skin secretions of frogs [44,113,114,115] or are toxins from other species, e.g., bees, snakes, and wasps [86,116,117]. In contrast, all the FDA-approved AMPs were discovered in or derived from Gram-positive bacteria commonly found in the soil: Brevibacillus brevis (gramicidin), Streptomyces roseosporus (daptomycin), Amycolatopsis orientalis (vancomycin, which is the prototype of oritavancin, dalbavancin, and telavancin), and Paenibacillus polymyxa (colistin) [118]. This coincidence is not surprising, as soil bacteria are also the source of many conventional antibiotics.
Numerous approaches to peptide design have been introduced to make AMPs less toxic for humans while maintaining or improving their potency to eliminate bacteria [11,42], e.g., rational design [119,120], combinatorial peptide libraries [75,121], high-throughput screening [122,123], database-guided approaches [124,125], structure-function-guided design [86,126,127], and molecular dynamics simulations [44]. Three major methods to improve AMP function have been described: (i) High-throughput screening can be used to identify potential AMPs [128,129,130]. The SPOT-synthesis technique, for instance, has been applied to medium- or high-throughput screens; with this technique, peptide arrays are synthesized on a cellulose membrane, and the peptides can be easily cleaved from the support for screening. In addition, combining computer algorithms, automated synthesis, and automated screening for drug design can rapidly accelerate and reduce the cost of labor for drug discovery [131]. (ii) Conjugation of peptides to other active molecules (for example, antibodies) or incorporation of peptides into nanoparticles or dendrimers allows the advantages of both types of biomolecules to be combined and overcomes their weaknesses [132]. Synthesizing AMP polymers using dendrimer or other AMP nanoparticles to increase the local concentration of the AMP can lower the required dose and combat multidrug-resistant bacteria [133,134,135,136]. (iii) The development of in vitro tests and computational predictions, for example, testing for similarities with allergens, can help to evaluate the immunogenicity and allergenic potential of newly developed AMPs [137], e.g., the basophil activation test [138], cytokine assays [139], lymphocyte activation analysis [140], and Structural Database of Allergenic Proteins (SDAP) [141,142,143]. Although these tools cannot fully predict the allergenic effects of a new AMP in the clinic, they provide a promising preclinical tools for evaluating peptide-based drugs.
We compared the AMPs from the APD [13] with the seven FDA-approved AMPs and found that the peptide sequences and physicochemical properties (e.g., hydrophobic content and net charge) vary widely among AMPs (Figure 3). These features merit additional study to determine what contribution they make to antimicrobial activity and ultimate clinical utility. It is unclear why the development of many natural peptides has stopped during preclinical stages or why the peptides failed to show sufficient antimicrobial activity and drug-like properties in clinical studies [5,10,134,144]. Their poor performance may derive from differences between the clinical setting and their native conditions. We speculate that in nature, peptides may participate in cooperative pathways with other chemical compounds or enzymes, and these conditions may increase their potency against bacteria. Synthetic AMPs used in isolation, on the other hand, may not have equally strong antimicrobial activity.
Based on these observations, it may be important not only to design synthetic AMPs that have antimicrobial activity, but to optimize them for desirable clinical properties, such as: (i) stabilizing peptide structures and introducing non-canonical amino acids into the peptide sequences to extend their elimination half-lives; (ii) mining antimicrobial agents from Gram-positive bacteria in soil and understanding which biochemical properties make these suitable for human use; (iii) discovering AMPs that can modulate the immune system; (iv) evaluating the potential synergy of AMPs with other chemical compounds or enzymes to enhance antimicrobial activity; (v) optimizing the design of computational tools for peptide therapeutics and high-throughput screening; and (vi) developing appropriate in vitro models that mimic in vivo conditions to evaluate the allergenic effects. Overall, AMPs can be a tool against drug-resistance bacteria and a source of promising therapeutics to treat infectious diseases. More investigation in the clinical setting is suggested.
Author Contributions
Conceptualization, C.H.C. and T.K.L.; writing—original draft preparation, C.H.C.; writing—review and editing, T.K.L.; formal analysis, C.H.C.; visualization, C.H.C.; funding acquisition, T.K.L. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded through a Defense Threat Reduction Agency grant (HDTRA11810041) to TKL. DTRA did not have any role in the design of the study, data analysis, or manuscript writing. TKL and CHC were supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under Award Number U19AI142780. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Acknowledgments
We thank Karen Pepper, Yong Lai, and Maria Eugenia Inda at MIT for reading over the document and providing input. We thank Bryan Lenneman, Tristan Bepler, and John Albin at MIT for valuable discussions.
Conflicts of Interest
TKL is a co-founder of Senti Biosciences, Synlogic, Engine Biosciences, Tango Therapeutics, Corvium, BiomX, and Eligo Biosciences. TKL also holds financial interests in nest.bio, Ampliphi, IndieBio, MedicusTek, Quark Biosciences, and Personal Genomics.
References
- Gold, H.S.; Moellering, R.C. Antimicrobial-drug resistance. N. Engl. J. Med. 1996, 335, 1445–1453. [Google Scholar] [CrossRef] [PubMed]
- Willyard, C. The drug-resistant bacteria that pose the greatest health threats. Nature 2017, 543, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alghoribi, M.F.; Gibreel, T.M.; Farnham, G.; Al Johani, S.M.; Balkhy, H.H.; Upton, M. Antibiotic-resistant ST38, ST131 and ST405 strains are the leading uropathogenic Escherichia coli clones in Riyadh, Saudi Arabia. J. Antimicrob. Chemother. 2015, 70, 2757–2762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petty, N.K.; Ben Zakour, N.L.; Stanton-Cook, M.; Skippington, E.; Totsika, M.; Forde, B.M.; Phan, M.D.; Gomes Moriel, D.; Peters, K.M.; Davies, M.; et al. Global dissemination of a multidrug resistant Escherichia coli clone. Proc. Natl. Acad. Sci. USA 2014, 111, 5694–5699. [Google Scholar] [CrossRef] [Green Version]
- Silver, L.L. Challenges of antibacterial discovery. Clin. Microbiol. Rev. 2011, 24, 71–109. [Google Scholar] [CrossRef] [Green Version]
- Conly, J.; Johnston, B. Where are all the new antibiotics? The new antibiotic paradox. Can. J. Infect. Dis. Med. Microbiol. 2005, 16, 159–160. [Google Scholar] [CrossRef] [Green Version]
- Hofer, U. The cost of antimicrobial resistance. Nat. Rev. Microbiol. 2019, 17, 3. [Google Scholar] [CrossRef]
- Collignon, P.; Beggs, J.J.; Walsh, T.R.; Gandra, S.; Laxminarayan, R. Anthropological and socioeconomic factors contributing to global antimicrobial resistance: A univariate and multivariable analysis. Lancet Planet Health 2018, 2, e398–e405. [Google Scholar] [CrossRef]
- Cassini, A.; Högberg, L.D.; Plachouras, D.; Quattrocchi, A.; Hoxha, A.; Simonsen, G.S.; Colomb-Cotinat, M.; Kretzschmar, M.E.; Devleesschauwer, B.; Cecchini, M.; et al. Attributable deaths and disability-adjusted life-years caused by infections with antibiotic-resistant bacteria in the EU and the European Economic Area in 2015: A population-level modelling analysis. Lancet Infect. Dis. 2019, 19, 56–66. [Google Scholar] [CrossRef] [Green Version]
- Wimley, W.C.; Hristova, K. Antimicrobial peptides: Successes, challenges and unanswered questions. J. Membr. Biol. 2011, 239, 27–34. [Google Scholar] [CrossRef] [Green Version]
- Torres, M.D.T.; Sothiselvam, S.; Lu, T.K.; de la Fuente-Nunez, C. Peptide design principles for antimicrobial applications. J. Mol. Biol. 2019, 431, 3547–3567. [Google Scholar] [CrossRef]
- Upton, M.; Cotter, P.; Tagg, J. Antimicrobial peptides as therapeutic agents. Int. J. Microbiol. 2012, 2012, 326503. [Google Scholar] [CrossRef]
- Wang, G.; Li, X.; Wang, Z. APD3: The antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016, 44, D1087–D1093. [Google Scholar] [CrossRef] [Green Version]
- Gordon, Y.J.; Romanowski, E.G.; McDermott, A.M. A review of antimicrobial peptides and their therapeutic potential as anti-infective drugs. Curr. Eye Res. 2005, 30, 505–515. [Google Scholar] [CrossRef]
- Usmani, S.S.; Bedi, G.; Samuel, J.S.; Singh, S.; Kalra, S.; Kumar, P.; Ahuja, A.A.; Sharma, M.; Gautam, A.; Raghava, G.P.S. THPdb: Database of FDA-approved peptide and protein therapeutics. PLoS ONE 2017, 12, e0181748. [Google Scholar] [CrossRef] [Green Version]
- Wallace, B.A. Gramicidin channels and pores. Annu. Rev. Biophys. Biophys. Chem. 1990, 19, 127–157. [Google Scholar] [CrossRef]
- Hotchkiss, R.D.; Dubos, R.J. The isolation of bactericidal substances from cultures of bacillus brevis. J. Biol. Chem. 1941, 141, 155–162. [Google Scholar]
- Lipmann, F.; Hotchkiss, R.D.; Dubos, R.J. The occurrence of d-amino acids in gramicidin and tyrocidine. J. Biol. Chem. 1941, 141, 163–170. [Google Scholar]
- Hotchkiss, R.D. The chemical nature of gramicidin and tyrocidine. J. Biol. Chem. 1941, 141, 171–186. [Google Scholar]
- Christensen, H.N.; Edwards, R.R.; Piersma, H.D. The composition of gramicidin and tyrocidine. J. Biol. Chem. 1941, 141, 187–196. [Google Scholar]
- Tishler, M.; Stokes, J.L.; Trenner, N.R.; Conn, J.B. Some properties of gramicidin. J. Biol. Chem. 1941, 141, 197–206. [Google Scholar]
- Hallett, J.W.; Wolkowicz, M.I.; Leopold, I.H. Ophthalmic use of neosporin. Am. J. Ophthalmol. 1956, 41, 850–853. [Google Scholar] [CrossRef]
- Lee, M.T.; Yang, P.Y.; Charron, N.E.; Hsieh, M.H.; Chang, Y.Y.; Huang, H.W. Comparison of the effects of daptomycin on bacterial and model membranes. Biochemistry 2018, 57, 5629–5639. [Google Scholar] [CrossRef]
- Kreutzberger, M.A.; Pokorny, A.; Almeida, P.F. Daptomycin-phosphatidylglycerol domains in lipid membranes. Langmuir 2017, 33, 13669–13679. [Google Scholar] [CrossRef]
- Eliopoulos, G.M.; Willey, S.; Reiszner, E.; Spitzer, P.G.; Caputo, G.; Moellering, R.C. In vitro and in vivo activity of LY 146032, a new cyclic lipopeptide antibiotic. Antimicrob. Agents Chemother. 1986, 30, 532–535. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, C.F.; Chambers, H.F. Daptomycin: Another novel agent for treating infections due to drug-resistant gram-positive pathogens. Clin. Infect. Dis. 2004, 38, 994–1000. [Google Scholar] [CrossRef] [Green Version]
- Chen, A.Y.; Zervos, M.J.; Vazquez, J.A. Dalbavancin: A novel antimicrobial. Int. J. Clin. Pract. 2007, 61, 853–863. [Google Scholar] [CrossRef] [Green Version]
- Zhanel, G.G.; Calic, D.; Schweizer, F.; Zelenitsky, S.; Adam, H.; Lagacé-Wiens, P.R.; Rubinstein, E.; Gin, A.S.; Hoban, D.J.; Karlowsky, J.A. New lipoglycopeptides: A comparative review of dalbavancin, oritavancin and telavancin. Drugs 2010, 70, 859–886. [Google Scholar] [CrossRef]
- Saravolatz, L.D.; Stein, G.E.; Johnson, L.B. Telavancin: A novel lipoglycopeptide. Clin. Infect. Dis. 2009, 49, 1908–1914. [Google Scholar] [CrossRef] [Green Version]
- Higgins, D.L.; Chang, R.; Debabov, D.V.; Leung, J.; Wu, T.; Krause, K.M.; Sandvik, E.; Hubbard, J.M.; Kaniga, K.; Schmidt, D.E.; et al. Telavancin, a multifunctional lipoglycopeptide, disrupts both cell wall synthesis and cell membrane integrity in methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2005, 49, 1127–1134. [Google Scholar] [CrossRef] [Green Version]
- Hamamoto, K.; Kida, Y.; Zhang, Y.; Shimizu, T.; Kuwano, K. Antimicrobial activity and stability to proteolysis of small linear cationic peptides with D-amino acid substitutions. Microbiol. Immunol. 2002, 46, 741–749. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Chau, J.K.; Perry, N.A.; de Boer, L.; Zaat, S.A.; Vogel, H.J. Serum stabilities of short tryptophan- and arginine-rich antimicrobial peptide analogs. PLoS ONE 2010, 5, e12684. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Yadav, V.; Smart, A.L.; Tajiri, S.; Basit, A.W. Toward oral delivery of biopharmaceuticals: An assessment of the gastrointestinal stability of 17 peptide drugs. Mol. Pharm. 2015, 12, 966–973. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Yadav, V.; Smart, A.L.; Tajiri, S.; Basit, A.W. Stability of peptide drugs in the colon. Eur. J. Pharm. Sci. 2015, 78, 31–36. [Google Scholar] [CrossRef]
- Johnson, I.S. Human insulin from recombinant DNA technology. Science 1983, 219, 632–637. [Google Scholar] [CrossRef]
- Duckworth, W.C.; Bennett, R.G.; Hamel, F.G. Insulin degradation: Progress and potential. Endocr. Rev. 1998, 19, 608–624. [Google Scholar]
- Zorzi, A.; Deyle, K.; Heinis, C. Cyclic peptide therapeutics: Past, present and future. Curr. Opin. Chem. Biol. 2017, 38, 24–29. [Google Scholar] [CrossRef] [Green Version]
- Lau, J.L.; Dunn, M.K. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorg. Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef]
- Lee, A.C.; Harris, J.L.; Khanna, K.K.; Hong, J.H. A comprehensive review on current advances in peptide drug development and design. Int. J. Mol. Sci. 2019, 20, 2383. [Google Scholar] [CrossRef] [Green Version]
- Bogdanowich-Knipp, S.J.; Chakrabarti, S.; Williams, T.D.; Dillman, R.K.; Siahaan, T.J. Solution stability of linear vs. cyclic RGD peptides. J. Pept. Res. 1999, 53, 530–541. [Google Scholar] [CrossRef]
- Rink, R.; Arkema-Meter, A.; Baudoin, I.; Post, E.; Kuipers, A.; Nelemans, S.A.; Akanbi, M.H.; Moll, G.N. To protect peptide pharmaceuticals against peptidases. J. Pharmacol. Toxicol. Methods 2010, 61, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Guha, S.; Ghimire, J.; Wu, E.; Wimley, W.C. Mechanistic landscape of membrane-permeabilizing peptides. Chem Rev. 2019, 119, 6040–6085. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.H.; Hofferek, V.; Separovic, F.; Reid, G.E.; Aguilar, M.I. The role of bacterial lipid diversity and membrane properties in modulating antimicrobial peptide activity and drug resistance. Curr. Opin. Chem. Biol. 2019, 52, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Starr, C.G.; Troendle, E.; Wiedman, G.; Wimley, W.C.; Ulmschneider, J.P.; Ulmschneider, M.B. Simulation-guided rational de novo design of a small pore-forming antimicrobial peptide. J. Am. Chem. Soc. 2019, 141, 4839–4848. [Google Scholar] [CrossRef] [PubMed]
- Hollmann, A.; Martinez, M.; Maturana, P.; Semorile, L.C.; Maffia, P.C. Antimicrobial peptides: Interaction with model and biological membranes and synergism with chemical antibiotics. Front. Chem. 2018, 6, 204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuzaki, K. Control of cell selectivity of antimicrobial peptides. Biochim. Biophys. Acta 2009, 1788, 1687–1692. [Google Scholar] [CrossRef] [Green Version]
- Matsuzaki, K. Why and how are peptide-lipid interactions utilized for self-defense? Magainins and tachyplesins as archetypes. Biochim. Biophys. Acta 1999, 1462, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Matsuzaki, K.; Sugishita, K.; Harada, M.; Fujii, N.; Miyajima, K. Interactions of an antimicrobial peptide, magainin 2, with outer and inner membranes of Gram-negative bacteria. Biochim. Biophys. Acta 1997, 1327, 119–130. [Google Scholar] [CrossRef] [Green Version]
- Henderson, J.M.; Iyengar, N.S.; Lam, K.L.H.; Maldonado, E.; Suwatthee, T.; Roy, I.; Waring, A.J.; Lee, K.Y.C. Beyond electrostatics: Antimicrobial peptide selectivity and the influence of cholesterol-mediated fluidity and lipid chain length on protegrin-1 activity. Biochim. Biophys. Acta Biomembr. 2019, 1861, 182977. [Google Scholar] [CrossRef] [PubMed]
- Law, V.; Knox, C.; Djoumbou, Y.; Jewison, T.; Guo, A.C.; Liu, Y.; Maciejewski, A.; Arndt, D.; Wilson, M.; Neveu, V.; et al. DrugBank 4.0: Shedding new light on drug metabolism. Nucleic Acids Res. 2014, 42, D1091–D1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahlapuu, M.; Håkansson, J.; Ringstad, L.; Björn, C. Antimicrobial peptides: An emerging category of therapeutic agents. Front. Cell. Infect. Microbiol. 2016, 6, 194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, T.; Henn, C.; de Jong, J.C.; Zimmer, C.; Kirsch, B.; Maurer, C.K.; Pistorius, D.; Müller, R.; Steinbach, A.; Hartmann, R.W. Identification of small-molecule antagonists of the Pseudomonas aeruginosa transcriptional regulator PqsR: Biophysically guided hit discovery and optimization. ACS Chem. Biol. 2012, 7, 1496–1501. [Google Scholar] [CrossRef]
- Worthington, R.J.; Blackledge, M.S.; Melander, C. Small-molecule inhibition of bacterial two-component systems to combat antibiotic resistance and virulence. Future Med. Chem. 2013, 5, 1265–1284. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Smith, C.; Yin, H. Targeting Toll-like receptors with small molecule agents. Chem. Soc. Rev. 2013, 42, 4859–4866. [Google Scholar] [CrossRef] [PubMed]
- Easton, D.M.; Nijnik, A.; Mayer, M.L.; Hancock, R.E. Potential of immunomodulatory host defense peptides as novel anti-infectives. Trends Biotechnol. 2009, 27, 582–590. [Google Scholar] [CrossRef]
- Haney, E.F.; Hancock, R.E. Peptide design for antimicrobial and immunomodulatory applications. Biopolymers 2013, 100, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Nijnik, A.; Hancock, R. Host defence peptides: Antimicrobial and immunomodulatory activity and potential applications for tackling antibiotic-resistant infections. Emerg. Health Threats J. 2009, 2, e1. [Google Scholar]
- Lee, E.Y.; Lee, M.W.; Wong, G.C.L. Modulation of toll-like receptor signaling by antimicrobial peptides. Semin. Cell Dev. Biol. 2019, 88, 173–184. [Google Scholar] [CrossRef]
- Van Harten, R.M.; van Woudenbergh, E.; van Dijk, A.; Haagsman, H.P. Cathelicidins: Immunomodulatory antimicrobials. Vaccines 2018, 6, 63. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, T.; Kulkarni, N.N.; Lee, E.Y.; Zhang, L.J.; Wong, G.C.L.; Gallo, R.L. Cathelicidin promotes inflammation by enabling binding of self-RNA to cell surface scavenger receptors. Sci. Rep. 2018, 8, 4032. [Google Scholar] [CrossRef]
- Grönberg, A.; Mahlapuu, M.; Ståhle, M.; Whately-Smith, C.; Rollman, O. Treatment with LL-37 is safe and effective in enhancing healing of hard-to-hear venous leg ulcers: A randomized, placebo-controlled clinical trial. Wound Repair Regen. 2014, 22, 613–621. [Google Scholar]
- Kelkar, D.A.; Chattopadhyay, A. The gramicidin ion channel: A model membrane protein. Biochim. Biophys. Acta 2007, 1768, 2011–2025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seydlová, G.; Sokol, A.; Lišková, P.; Konopásek, I.; Fišer, R. Daptomycin pore formation and stoichiometry depend on membrane potential of target membrane. Antimicrob. Agents Chemother. 2019, 63, e01589-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barboiu, M.; Le Duc, Y.; Gilles, A.; Cazade, P.A.; Michau, M.; Marie Legrand, Y.; van der Lee, A.; Coasne, B.; Parvizi, P.; Post, J.; et al. An artificial primitive mimic of the Gramicidin-A channel. Nat. Commun. 2014, 5, 4142. [Google Scholar] [CrossRef]
- Zhanel, G.G.; Trapp, S.; Gin, A.S.; DeCorby, M.; Lagacé-Wiens, P.R.; Rubinstein, E.; Hoban, D.J.; Karlowsky, J.A. Dalbavancin and telavancin: Novel lipoglycopeptides for the treatment of Gram-positive infections. Expert Rev. Anti-Infect. Ther. 2008, 6, 67–81. [Google Scholar] [CrossRef]
- Velkov, T.; Thompson, P.E.; Nation, R.L.; Li, J. Structure—Activity relationships of polymyxin antibiotics. J. Med. Chem. 2010, 53, 1898–1916. [Google Scholar] [CrossRef] [Green Version]
- Mohamed, Y.F.; Abou-Shleib, H.M.; Khalil, A.M.; El-Guink, N.M.; El-Nakeeb, M.A. Membrane permeabilization of colistin toward pan-drug resistant Gram-negative isolates. Braz. J. Microbiol. 2016, 47, 381–388. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.W. Deformation free energy of bilayer membrane and its effect on gramicidin channel lifetime. Biophys. J. 1986, 50, 1061–1070. [Google Scholar] [CrossRef] [Green Version]
- Raghuraman, H.; Chattopadhyay, A. Melittin: A membrane-active peptide with diverse functions. Biosci. Rep. 2007, 27, 189–223. [Google Scholar] [CrossRef]
- Leveritt, J.M.; Pino-Angeles, A.; Lazaridis, T. The structure of a melittin-stabilized pore. Biophys. J. 2015, 108, 2424–2426. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Chen, C.H.; Hu, D.; Ulmschneider, M.B.; Ulmschneider, J.P. Spontaneous formation of structurally diverse membrane channel architectures from a single antimicrobial peptide. Nat. Commun. 2016, 7, 13535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, R.O.; Richards, F.M. A voltage-gated ion channel model inferred from the crystal structure of alamethicin at 1.5-A resolution. Nature 1982, 300, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Harroun, T.A.; Weiss, T.M.; Ding, L.; Huang, H.W. Barrel-stave model or toroidal model? A case study on melittin pores. Biophys. J. 2001, 81, 1475–1485. [Google Scholar] [CrossRef] [Green Version]
- Wiedman, G.; Fuselier, T.; He, J.; Searson, P.C.; Hristova, K.; Wimley, W.C. Highly efficient macromolecule-sized poration of lipid bilayers by a synthetically evolved peptide. J. Am. Chem. Soc. 2014, 136, 4724–4731. [Google Scholar] [CrossRef]
- Li, S.; Kim, S.Y.; Pittman, A.E.; King, G.M.; Wimley, W.C.; Hristova, K. Potent macromolecule-sized poration of lipid bilayers by the macrolittins, a synthetically evolved family of pore-forming peptides. J. Am. Chem. Soc. 2018, 140, 6441–6447. [Google Scholar] [CrossRef]
- Wiedman, G.; Herman, K.; Searson, P.; Wimley, W.C.; Hristova, K. The electrical response of bilayers to the bee venom toxin melittin: Evidence for transient bilayer permeabilization. Biochim. Biophys. Acta 2013, 1828, 1357–1364. [Google Scholar] [CrossRef] [Green Version]
- Harriss, L.M.; Cronin, B.; Thompson, J.R.; Wallace, M.I. Imaging multiple conductance states in an alamethicin pore. J. Am. Chem. Soc. 2011, 133, 14507–14509. [Google Scholar] [CrossRef]
- LaPlante, K.L.; Rybak, M.J. Daptomycin—A novel antibiotic against Gram-positive pathogens. Expert Opin. Pharmacother. 2004, 5, 2321–2331. [Google Scholar] [CrossRef]
- Alder, J.D. Daptomycin: A new drug class for the treatment of Gram-positive infections. Drugs Today 2015, 41, 81–90. [Google Scholar] [CrossRef]
- Neville, F.; Cahuzac, M.; Konovalov, O.; Ishitsuka, Y.; Lee, K.Y.; Kuzmenko, I.; Kale, G.M.; Gidalevitz, D. Lipid headgroup discrimination by antimicrobial peptide LL-37: Insight into mechanism of action. Biophys. J. 2006, 90, 1275–1287. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, D.I.; Le Brun, A.P.; Whitwell, T.C.; Sani, M.A.; James, M.; Separovic, F. The antimicrobial peptide aurein 1.2 disrupts model membranes via the carpet mechanism. Phys. Chem. Chem. Phys. 2012, 14, 15739–15751. [Google Scholar] [CrossRef] [PubMed]
- Ambroggio, E.E.; Separovic, F.; Bowie, J.H.; Fidelio, G.D.; Bagatolli, L.A. Direct visualization of membrane leakage induced by the antibiotic peptides: Maculatin, citropin, and aurein. Biophys. J. 2005, 89, 1874–1881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrin, B.S.; Fu, R.; Cotton, M.L.; Pastor, R.W. Simulations of membrane-disrupting peptides II: AMP piscidin 1 favors surface defects over pores. Biophys. J. 2016, 111, 1258–1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, K.C.; Lee, S.H.; Hour, A.L.; Pan, C.Y.; Lee, L.H.; Chen, J.Y. Five different piscidins from Nile tilapia, Oreochromis niloticus: Analysis of their expressions and biological functions. PLoS ONE 2012, 7, e50263. [Google Scholar] [CrossRef]
- Ladokhin, A.S.; White, S.H. ‘Detergent-like’ permeabilization of anionic lipid vesicles by melittin. Biochim. Biophys. Acta 2001, 1514, 253–260. [Google Scholar] [CrossRef] [Green Version]
- Torres, M.D.T.; Pedron, C.N.; Higashikuni, Y.; Kramer, R.M.; Cardoso, M.H.; Oshiro, K.G.N.; Franco, O.L.; Silva, P.I., Jr.; Silva, F.D.; Oliveira, V.X., Jr.; et al. Structure-function-guided exploration of the antimicrobial peptide polybia-CP identifies activity determinants and generates synthetic therapeutic candidates. Commun. Biol. 2018, 1, 221. [Google Scholar] [CrossRef]
- Walther, T.H.; Ulrich, A.S. Transmembrane helix assembly and the role of salt bridges. Curr. Opin. Struct. Biol. 2014, 27, 63–68. [Google Scholar] [CrossRef] [Green Version]
- Ulmschneider, M.B.; Ulmschneider, J.P.; Schiller, N.; Wallace, B.A.; von Heijne, G.; White, S.H. Spontaneous transmembrane helix insertion thermodynamically mimics translocon-guided insertion. Nat. Commun. 2014, 5, 4863. [Google Scholar] [CrossRef]
- Pino-Angeles, A.; Lazaridis, T. Effects of peptide charge, orientation, and concentration on melittin transmembrane pores. Biophys. J. 2018, 114, 2865–2874. [Google Scholar] [CrossRef] [Green Version]
- Walker, L.R.; Marzluff, E.M.; Townsend, J.A.; Resager, W.C.; Marty, M.T. Native mass spectrometry of antimicrobial peptides in lipid nanodiscs elucidates complex assembly. Anal. Chem. 2019, 91, 9284–9291. [Google Scholar] [CrossRef]
- Epand, R.F.; Savage, P.B.; Epand, R.M. Bacterial lipid composition and the antimicrobial efficacy of cationic steroid compounds (Ceragenins). Biochim. Biophys. Acta 2007, 1768, 2500–2509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maxfield, F.R.; van Meer, G. Cholesterol, the central lipid of mammalian cells. Curr. Opin. Cell Biol. 2010, 22, 422–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goluszko, P.; Nowicki, B. Membrane cholesterol: A crucial molecule affecting interactions of microbial pathogens with mammalian cells. Infect. Immun. 2005, 73, 7791–7796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Rojas, A.; Moreno-Morales, J.; Mason, A.J.; Rolff, J. Cationic antimicrobial peptides do not change recombination frequency in Escherichia coli. Biol. Lett. 2018, 14, 20180006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Candiani, G.; Abbondi, M.; Borgonovi, M.; Romanò, G.; Parenti, F. In-vitro and in-vivo antibacterial activity of BI 397, a new semi-synthetic glycopeptide antibiotic. J. Antimicrob. Chemother. 1999, 44, 179–192. [Google Scholar] [CrossRef] [Green Version]
- Dinos, G.; Wilson, D.N.; Teraoka, Y.; Szaflarski, W.; Fucini, P.; Kalpaxis, D.; Nierhaus, K.H. Dissecting the ribosomal inhibition mechanisms of edeine and pactamycin: The universally conserved residues G693 and C795 regulate P-site RNA binding. Mol. Cell 2004, 13, 113–124. [Google Scholar] [CrossRef]
- Szer, W.; Kurylo-Borowska, Z. Interactions of edeine with bacterial ribosomal subunits. Selective inhibition of aminoacyl-tRNA binding sites. Biochim. Biophys. Acta 1972, 259, 357–368. [Google Scholar] [CrossRef]
- Wank, H.; Rogers, J.; Davies, J.; Schroeder, R. Peptide antibiotics of the tuberactinomycin family as inhibitors of group I intron RNA splicing. J. Mol. Biol. 1994, 236, 1001–1010. [Google Scholar] [CrossRef]
- Bulkley, D.; Brandi, L.; Polikanov, Y.S.; Fabbretti, A.; O’Connor, M.; Gualerzi, C.O.; Steitz, T.A. The antibiotics dityromycin and GE82832 bind protein S12 and block EF-G-catalyzed translocation. Cell Rep. 2014, 6, 357–365. [Google Scholar] [CrossRef]
- Nijnik, A.; Hancock, R.E. The roles of cathelicidin LL-37 in immune defences and novel clinical applications. Curr. Opin. Hematol. 2009, 16, 41–47. [Google Scholar] [CrossRef]
- Mangoni, M.L.; McDermott, A.M.; Zasloff, M. Antimicrobial peptides and wound healing: Biological and therapeutic considerations. Exp. Dermatol. 2016, 25, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Vlieghe, P.; Lisowski, V.; Martinez, J.; Khrestchatisky, M. Synthetic therapeutic peptides: Science and market. Drug Discov. Today 2010, 15, 40–56. [Google Scholar] [CrossRef] [PubMed]
- Scheinfeld, N. Dalbavancin: A review. Drugs Today (Barc) 2007, 43, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Rosenthal, S.; Decano, A.G.; Bandali, A.; Lai, D.; Malat, G.E.; Bias, T.E. Oritavancin (Orbactiv): A new-generation lipoglycopeptide for the treatment of acute bacterial skin and skin structure infections. Pharm. Ther. 2018, 43, 143–179. [Google Scholar]
- Smith, J.R.; Roberts, K.D.; Rybak, M.J. Dalbavancin: A novel lipoglycopeptide antibiotic with extended activity against gram-positive infections. Infect. Dis. Ther. 2015, 4, 245–258. [Google Scholar] [CrossRef] [Green Version]
- Cavanaugh, C.; Moeckel, G.W.; Perazella, M.A. Telavancin-associated acute kidney injury. Clin. Nephrol. 2019, 91, 187–191. [Google Scholar] [CrossRef]
- Nnedu, O.N.; Pankey, G.A. Update on the emerging role of telavancin in hospital-acquired infections. Ther. Clin. Risk Manag. 2015, 11, 605–610. [Google Scholar] [CrossRef] [Green Version]
- Wolinsky, E.; Hines, J.D. Neurotoxic and nephrotoxic effects of colistin in patients with renal disease. N. Engl. J. Med. 1962, 266, 759–762. [Google Scholar] [CrossRef]
- Lim, L.M.; Ly, N.; Anderson, D.; Yang, J.C.; Macander, L.; Jarkowski, A.; Forrest, A.; Bulitta, J.B.; Tsuji, B.T. Resurgence of colistin: A review of resistance, toxicity, pharmacodynamics, and dosing. Pharmacotherapy 2010, 30, 1279–1291. [Google Scholar] [CrossRef]
- Poirel, L.; Jayol, A.; Nordmann, P. Polymyxins: Antibacterial activity, susceptibility testing, and resistance mechanisms encoded by plasmids or chromosomes. Clin. Microbiol. Rev. 2017, 30, 557–596. [Google Scholar] [CrossRef] [Green Version]
- Bechinger, B. Structure and functions of channel-forming peptides: Magainins, cecropins, melittin and alamethicin. J. Membr. Biol. 1997, 156, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Leitgeb, B.; Szekeres, A.; Manczinger, L.; Vágvölgyi, C.; Kredics, L. The history of alamethicin: A review of the most extensively studied peptaibol. Chem. Biodivers. 2007, 4, 1027–1051. [Google Scholar] [CrossRef] [PubMed]
- Ladram, A.; Nicolas, P. Antimicrobial peptides from frog skin: Biodiversity and therapeutic promises. Front. Biosci. 2016, 21, 1341–1371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conlon, J.M.; Mechkarska, M. Host-defense peptides with therapeutic potential from skin secretions of frogs from the family pipidae. Pharmaceuticals 2014, 7, 58–77. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Liu, H.; Yang, H.; Yu, H.; You, D.; Ma, Y.; Ye, H.; Lai, R. Proteomic analysis of skin defensive factors of tree frog Hyla simplex. J. Proteome Res. 2011, 10, 4230–4240. [Google Scholar] [CrossRef]
- Wehbe, R.; Frangieh, J.; Rima, M.; El Obeid, D.; Sabatier, J.M.; Fajloun, Z. Bee venom: Overview of main compounds and bioactivities for therapeutic interests. Molecules 2019, 24, 2997. [Google Scholar] [CrossRef] [Green Version]
- Munawar, A.; Ali, S.A.; Akrem, A.; Betzel, C. Snake venom peptides: Tools of biodiscovery. Toxins (Basel) 2018, 10, 474. [Google Scholar] [CrossRef] [Green Version]
- Cochrane, S.A.; Vederas, J.C. Lipopeptides from bacillus and paenibacillus spp.: A gold mine of antibiotic candidates. Med. Res. Rev. 2016, 36, 4–31. [Google Scholar] [CrossRef]
- Ballantine, R.D.; Li, Y.X.; Qian, P.Y.; Cochrane, S.A. Rational design of new cyclic analogues of the antimicrobial lipopeptide tridecaptin A1. Chem. Commun. 2018, 54, 10634–10637. [Google Scholar] [CrossRef]
- Ballantine, R.D.; McCallion, C.E.; Nassour, E.; Tokajian, S.; Cochrane, S.A. Tridecaptin-inspired antimicrobial peptides with activity against multidrug-resistant Gram-negative bacteria. MedChemComm 2019, 10, 484–487. [Google Scholar] [CrossRef]
- Krauson, A.J.; He, J.; Wimley, W.C. Gain-of-function analogues of the pore-forming peptide melittin selected by orthogonal high-throughput screening. J. Am. Chem. Soc. 2012, 134, 12732–12741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Touti, F.; Gates, Z.P.; Bandyopadhyay, A.; Lautrette, G.; Pentelute, B.L. In-solution enrichment identifies peptide inhibitors of protein-protein interactions. Nat. Chem. Biol. 2019, 15, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Gates, Z.P.; Vinogradov, A.A.; Quartararo, A.J.; Bandyopadhyay, A.; Choo, Z.N.; Evans, E.D.; Halloran, K.H.; Mijalis, A.J.; Mong, S.K.; Simon, M.D.; et al. Xenoprotein engineering via synthetic libraries. Proc. Natl. Acad. Sci. USA 2018, 115, E5298–E5306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, B.; Wang, G. Ab initio design of potent anti-MRSA peptides based on database filtering technology. J. Am. Chem. Soc. 2012, 134, 12426–12429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porto, W.F.; Irazazabal, L.; Alves, E.S.F.; Ribeiro, S.M.; Matos, C.O.; Pires, Á.; Fensterseifer, I.C.M.; Miranda, V.J.; Haney, E.F.; Humblot, V.; et al. In silico optimization of a guava antimicrobial peptide enables combinatorial exploration for peptide design. Nat. Commun. 2018, 9, 1490. [Google Scholar] [CrossRef] [PubMed]
- Sani, M.A.; Lee, T.H.; Aguilar, M.I.; Separovic, F. Proline-15 creates an amphipathic wedge in maculatin 1.1 peptides that drives lipid membrane disruption. Biochim. Biophys. Acta 2015, 1848, 2277–2289. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, D.I.; Lee, T.H.; Sani, M.A.; Aguilar, M.I.; Separovic, F. Proline facilitates membrane insertion of the antimicrobial peptide maculatin 1.1 via surface indentation and subsequent lipid disordering. Biophys. J. 2013, 104, 1495–1507. [Google Scholar] [CrossRef] [Green Version]
- Hilpert, K. High-throughput screening for antimicrobial peptides using the SPOT technique. Methods Mol. Biol. 2010, 618, 125–133. [Google Scholar]
- López-Pérez, P.M.; Grimsey, E.; Bourne, L.; Mikut, R.; Hilpert, K. Screening and optimizing antimicrobial peptides by using SPOT-synthesis. Front. Chem. 2017, 5, 25. [Google Scholar] [CrossRef]
- Hilpert, K.; Winkler, D.F.; Hancock, R.E. Peptide arrays on cellulose support: SPOT synthesis, a time and cost efficient method for synthesis of large numbers of peptides in a parallel and addressable fashion. Nat. Protoc. 2007, 2, 1333–1349. [Google Scholar] [CrossRef]
- Coley, C.W.; Thomas, D.A.; Lummiss, J.A.M.; Jaworski, J.N.; Breen, C.P.; Schultz, V.; Hart, T.; Fishman, J.S.; Rogers, L.; Gao, H.; et al. A robotic platform for flow synthesis of organic compounds informed by AI planning. Science 2019, 365, eaax1566. [Google Scholar] [CrossRef] [PubMed]
- Touti, F.; Lautrette, G.; Johnson, K.D.; Delaney, J.C.; Wollacott, A.; Tissire, H.; Viswanathan, K.; Shriver, Z.; Mong, S.K.; Mijalis, A.J.; et al. Antibody-bactericidal macrocyclic peptide conjugates to target gram-negative bacteria. ChemBioChem 2018, 19, 2039–2044. [Google Scholar] [CrossRef] [PubMed]
- Lam, S.J.; O’Brien-Simpson, N.M.; Pantarat, N.; Sulistio, A.; Wong, E.H.; Chen, Y.Y.; Lenzo, J.C.; Holden, J.A.; Blencowe, A.; Reynolds, E.C.; et al. Combating multidrug-resistant Gram-negative bacteria with structurally nanoengineered antimicrobial peptide polymers. Nat. Microbiol. 2016, 1, 16162. [Google Scholar] [CrossRef] [PubMed]
- Biswaro, L.S.; da Costa Sousa, M.G.; Rezende, T.M.B.; Dias, S.C.; Franco, O.L. Antimicrobial peptides and nanotechnology, recent advances and challenges. Front. Microbiol. 2018, 9, 855. [Google Scholar] [CrossRef] [Green Version]
- Rajchakit, U.; Sarojini, V. Recent developments in antimicrobial-peptide-conjugated gold nanoparticles. Bioconjug. Chem. 2017, 28, 2673–2686. [Google Scholar] [CrossRef]
- Casciaro, B.; Moros, M.; Rivera-Fernández, S.; Bellelli, A.; de la Fuente, J.M.; Mangoni, M.L. Gold-nanoparticles coated with the antimicrobial peptide esculentin-1a(1-21)NH. Acta Biomater. 2017, 47, 170–181. [Google Scholar] [CrossRef] [Green Version]
- Deptuła, M.; Wardowska, A.; Dzierżyńska, M.; Rodziewicz-Motowidło, S.; Pikuła, M. Antibacterial peptides in dermatology-strategies for evaluation of allergic potential. Molecules 2018, 23, 414. [Google Scholar] [CrossRef] [Green Version]
- Pikuła, M.; Zieliński, M.; Specjalski, K.; Barańska-Rybak, W.; Dawgul, M.; Langa, P.; Jassem, E.; Kamysz, W.; Trzonkowski, P. In vitro evaluation of the allergic potential of antibacterial peptides: Camel and citropin. Chem. Biol. Drug Des. 2016, 87, 562–568. [Google Scholar] [CrossRef]
- Brunetti, J.; Roscia, G.; Lampronti, I.; Gambari, R.; Quercini, L.; Falciani, C.; Bracci, L.; Pini, A. Immunomodulatory and anti-inflammatory activity in vitro and in vivo of a novel antimicrobial candidate. J. Biol. Chem. 2016, 291, 25742–25748. [Google Scholar] [CrossRef] [Green Version]
- Pacor, S.; Giangaspero, A.; Bacac, M.; Sava, G.; Tossi, A. Analysis of the cytotoxicity of synthetic antimicrobial peptides on mouse leucocytes: Implications for systemic use. J. Antimicrob. Chemother. 2002, 50, 339–348. [Google Scholar] [CrossRef] [Green Version]
- Schein, C.H.; Ivanciuc, O.; Braun, W. Bioinformatics approaches to classifying allergens and predicting cross-reactivity. Immunol. Allergy Clin. N. Am. 2007, 27, 1–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanciuc, O.; Schein, C.H.; Braun, W. SDAP: Database and computational tools for allergenic proteins. Nucleic Acids Res. 2003, 31, 359–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanciuc, O.; Gendel, S.M.; Power, T.D.; Schein, C.H.; Braun, W. AllerML: Markup language for allergens. Regul. Toxicol. Pharmacol. 2011, 60, 151–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, J.; Sun, L.; Huang, S.; Zhu, C.; Li, P.; He, J.; Mackey, V.; Coy, D.H.; He, Q. The antimicrobial peptides and their potential clinical applications. Am. J. Transl. Res. 2019, 11, 3919–3931. [Google Scholar] [PubMed]
Figure 1.
Chemical structures of seven FDA-approved AMPs. From upper left to bottom right: gramicidin (linear peptide; pore-forming peptide), daptomycin (cyclic lipopeptide; membrane-lytic peptide), colistin (cyclic lipopeptide; membrane-lytic peptide), vancomycin (lipoglycopeptide; inhibitor of cell wall synthesis), oritavancin (lipoglycopeptide; dual-mechanism: membrane-lytic peptide and inhibitor of cell wall synthesis), dalbavancin (lipoglycopeptide; inhibitor of cell wall synthesis), and telavancin (lipoglycopeptide; dual-mechanism: membrane-lytic peptide and inhibitor of cell wall synthesis). MoA indicates “mechanism of action”. T1/2 indicates the elimination half-life.
Figure 1.
Chemical structures of seven FDA-approved AMPs. From upper left to bottom right: gramicidin (linear peptide; pore-forming peptide), daptomycin (cyclic lipopeptide; membrane-lytic peptide), colistin (cyclic lipopeptide; membrane-lytic peptide), vancomycin (lipoglycopeptide; inhibitor of cell wall synthesis), oritavancin (lipoglycopeptide; dual-mechanism: membrane-lytic peptide and inhibitor of cell wall synthesis), dalbavancin (lipoglycopeptide; inhibitor of cell wall synthesis), and telavancin (lipoglycopeptide; dual-mechanism: membrane-lytic peptide and inhibitor of cell wall synthesis). MoA indicates “mechanism of action”. T1/2 indicates the elimination half-life.
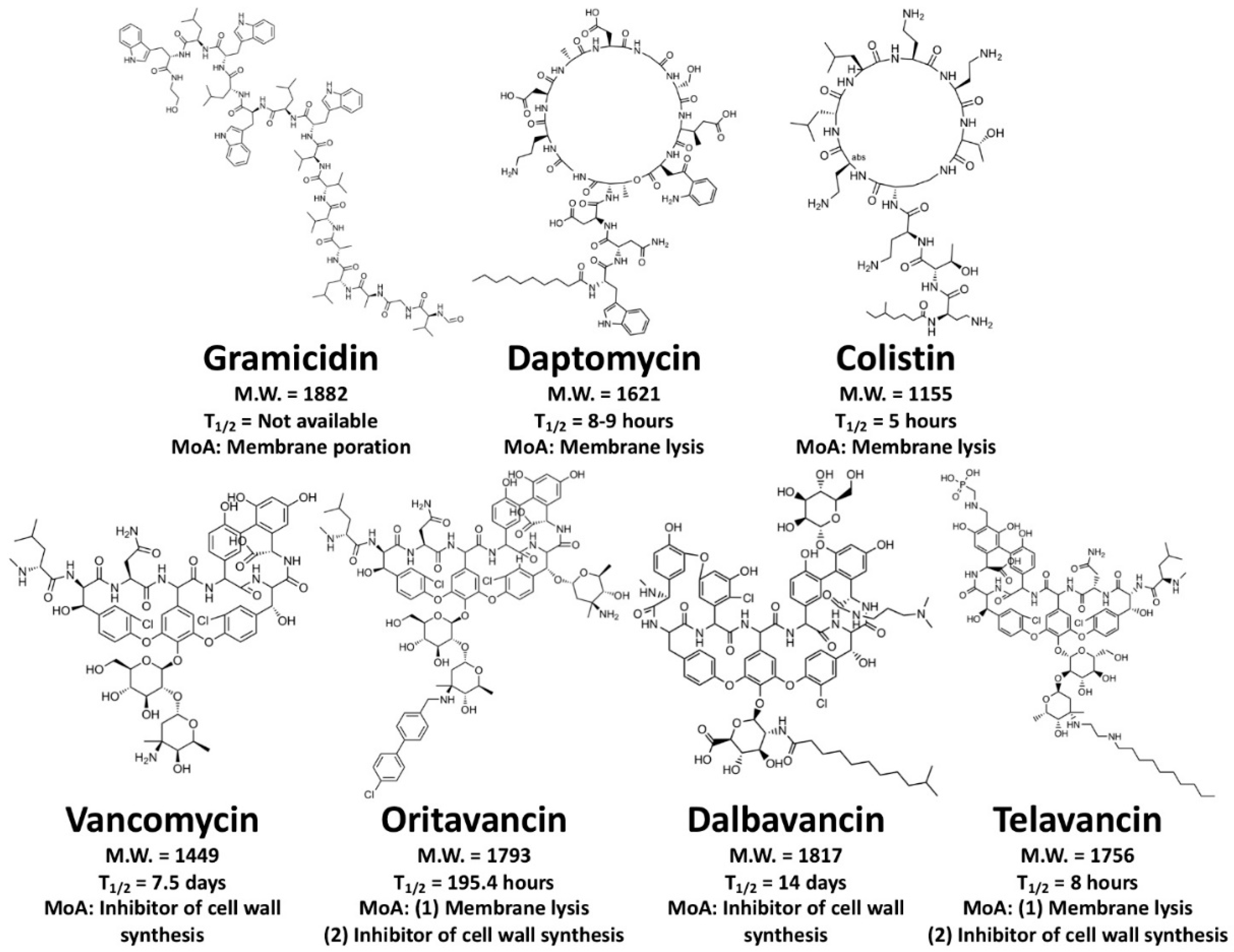
Figure 2.
The elimination half-life (hours) of all FDA-approved drugs (509 validated molecules out of 555 molecules; black) and FDA-approved small peptides (54 validated molecules out of 57 molecules; gray). The FDA-approved drugs also include the FDA-approved small peptides. The FDA-approved small peptides are summarized in Table 1. The raw data (submission classification: Type 1—New Molecular Entity between January 1999 and December 2019) were collected from Drugs@FDA (http://www.fda.gov/drugsatfda).
Figure 2.
The elimination half-life (hours) of all FDA-approved drugs (509 validated molecules out of 555 molecules; black) and FDA-approved small peptides (54 validated molecules out of 57 molecules; gray). The FDA-approved drugs also include the FDA-approved small peptides. The FDA-approved small peptides are summarized in Table 1. The raw data (submission classification: Type 1—New Molecular Entity between January 1999 and December 2019) were collected from Drugs@FDA (http://www.fda.gov/drugsatfda).

Figure 3.
Analysis of Antimicrobial Peptide Database (APD). This analysis took into account 2700 of the AMPs listed in the APD. Distributions of (A) peptide length, (B) hydrophobic content, (C) net charge, and (D) number of cysteine residues in the peptide sequence. The raw data were collected from the APD [13].
Figure 3.
Analysis of Antimicrobial Peptide Database (APD). This analysis took into account 2700 of the AMPs listed in the APD. Distributions of (A) peptide length, (B) hydrophobic content, (C) net charge, and (D) number of cysteine residues in the peptide sequence. The raw data were collected from the APD [13].
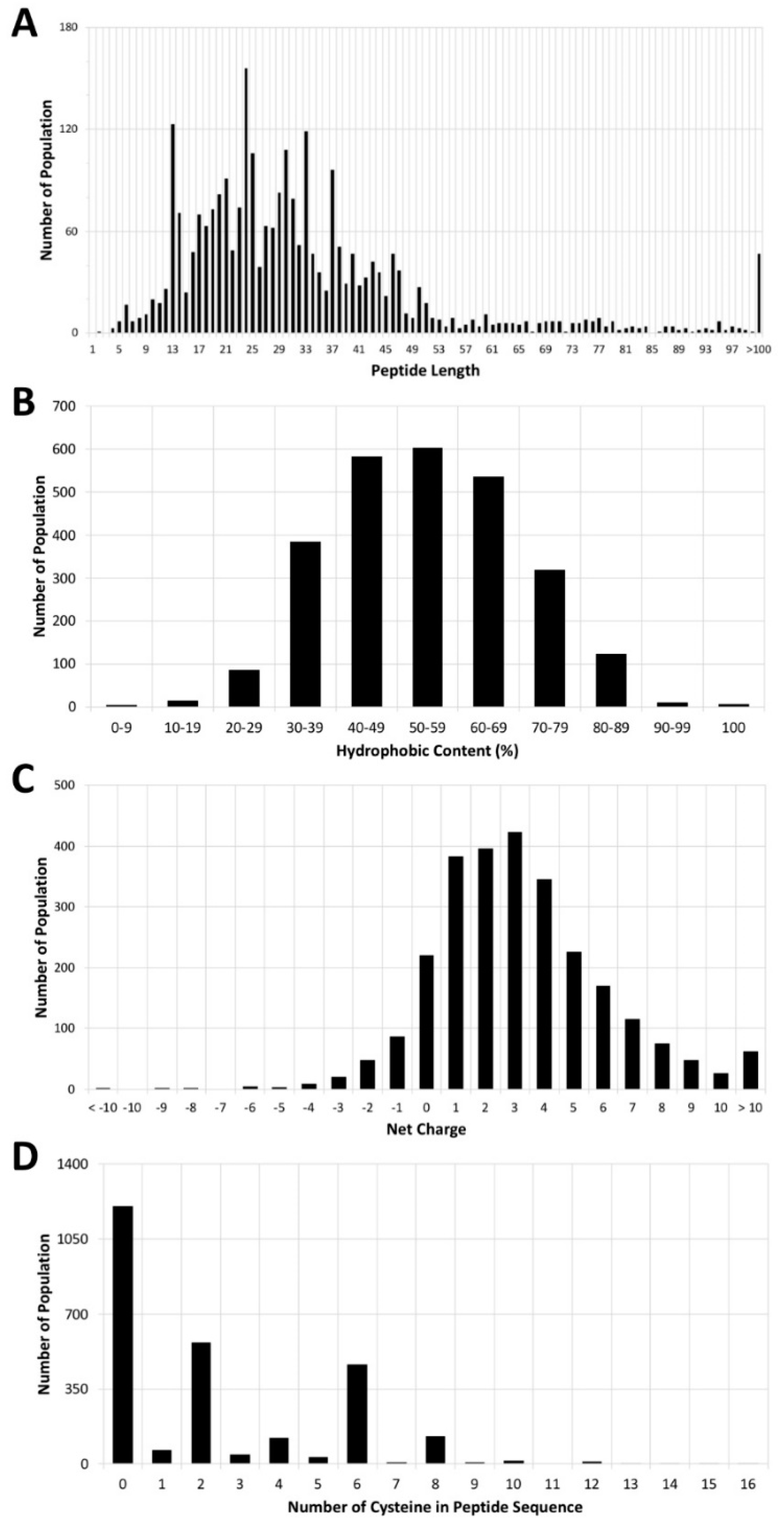
Figure 4.
Analysis of the FDA-approved new drugs between January 1999 and December 2019. (A) Annual number of the total FDA-approved new drugs (black) and peptide/protein therapeutics (gray). (B) Known mechanisms of the FDA-approved peptide/protein therapeutics. “Inhibitor” includes membrane inhibition as well as other mechanisms of action. The raw data were collected from Drugs@FDA (http://www.fda.gov/drugsatfda).
Figure 4.
Analysis of the FDA-approved new drugs between January 1999 and December 2019. (A) Annual number of the total FDA-approved new drugs (black) and peptide/protein therapeutics (gray). (B) Known mechanisms of the FDA-approved peptide/protein therapeutics. “Inhibitor” includes membrane inhibition as well as other mechanisms of action. The raw data were collected from Drugs@FDA (http://www.fda.gov/drugsatfda).

Figure 5.
Schematic partitioning for membrane-active peptides (MAPs) that interact with the lipid bilayer. Water-soluble peptides spontaneously (A) bind onto the interface of the lipid bilayer, (B) fold and (C) aggregate at the membrane interface, and (D) form a channel and/or pore in the lipid membrane. Orange spheres represent the phosphorus atoms of the lipid headgroups.
Figure 5.
Schematic partitioning for membrane-active peptides (MAPs) that interact with the lipid bilayer. Water-soluble peptides spontaneously (A) bind onto the interface of the lipid bilayer, (B) fold and (C) aggregate at the membrane interface, and (D) form a channel and/or pore in the lipid membrane. Orange spheres represent the phosphorus atoms of the lipid headgroups.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Summary of the small peptide (less than 50 amino acids) therapeutics approved by the FDA between January 1999 and December 2019. Raw data (submission classification: Type 1—New Molecular Entity) were collected from Drugs@FDA (http://www.fda.gov/drugsatfda) and the data were further confirmed by DrugBank [50]. “MAP” is defined as “membrane-active peptide”.
Table 1.
Summary of the small peptide (less than 50 amino acids) therapeutics approved by the FDA between January 1999 and December 2019. Raw data (submission classification: Type 1—New Molecular Entity) were collected from Drugs@FDA (http://www.fda.gov/drugsatfda) and the data were further confirmed by DrugBank [50]. “MAP” is defined as “membrane-active peptide”.
| DRUG NAME | APPROVAL DATE | NDA NUMBER | ACTIVE INGREDIENTS | COMPANY | MW | PEPTIDE | APPLICATIONS | CATEGORY | ELIMINATION HALF-LIFE |
|---|---|---|---|---|---|---|---|---|---|
| SCENESSE | 10/8/2019 | 210797 | AFAMELANOTIDE | CLINUVEL INC | 1646.85 | Synthetic peptide | Prevents skin damage from the sun in patients with erythropoietic protoporphyria | Receptor binding | 30 min |
| GALLIUM DOTATOC GA68 | 8/21/2019 | 210828 | GALLIUM DOTATOC GA-68 | UNIV IOWA HOSPS AND CLINICS PET IMAGING CENTER | 1489.65 | Cyclic octapeptide | Neuroendocrine tumors (NETs) diagnosis | Receptor binding | 2–4 h |
| VYLEESI (AUTOINJECTOR) | 6/21/2019 | 210557 | BREMELANOTIDE ACETATE | AMAG PHARMS INC | 1025.2 | Cyclic heptapeptide | Hypoactive sexual desire disorder (HSDD) treatment | Receptor binding | 1.9–4 h |
| LUTATHERA | 1/26/2018 | 208700 | LUTETIUM DOTATATE LU-177 | AAA USA INC | 1609.6 | Cyclic peptide-radionuclide conjugate | Gastroenteropan-creatic neuroendocrine tumors (GEP-NETs) treatment | Receptor binding | 3.5–71 h |
| GIAPREZA | 12/21/2017 | 209360 | ANGIOTENSIN II ACETATE | LA JOLLA PHARMA | 1046.2 | Synthetic peptide | Treatment of sepsis, septic shock, diabetes mellitus, and acute renal failure | Receptor binding | <1 min |
| OZEMPIC | 12/5/2017 | 209637 | SEMAGLUTIDE | NOVO NORDISK INC | 4113.58 | Chemically modified peptide | Improving glycemic control in patients with type 2 diabetes mellitus | Receptor binding | 7 days |
| TYMLOS | 4/28/2017 | 208743 | ABALOPARATIDE | RADIUS HEALTH INC | 3961 | Synthetic peptide | Osteoporosis treatment | Receptor binding | 1.7 h |
| PARSABIV | 2/7/2017 | 208325 | ETELCALCETIDE | KAI PHARMS INC | 1047.5 | Synthetic peptide | Treatment of secondary hyperparathyroidism | Receptor binding | 3–4 days |
| TRULANCE | 1/19/2017 | 208745 | PLECANATIDE | SALIX | 1682 | Cyclic peptide | Chronic idiopathic constipation (CIC) treatment | Receptor binding | N/A |
| ADLYXIN | 7/27/2016 | 208471 | LIXISENATIDE | SANOFI-AVENTIS US | 4858.5 | Synthetic peptide | Type 2 diabetes mellitus (T2DM) treatment | Receptor binding | 1–3.5 h |
| NETSPOT | 6/1/2016 | 208547 | GALLIUM DOTATATE GA-68 | AAA USA INC | 1435.6 | Cyclic peptide-radionuclide conjugate | Neuroendocrine tumors (NETs) diagnosis | Receptor binding | 1 h |
| ORBACTIV | 8/6/2014 | 206334 | ORITAVANCIN DIPHOSPHATE | MELINTA THERAP | 1989.09 | Lipoglycopeptide | Treatment of complicated skin and skin structure infections (cSSSI) caused by gram-positive bacteria | MAP | 195.4 h |
| DALVANCE | 5/23/2014 | 021883 | DALBAVANCIN HYDROCHLORIDE | ALLERGAN SALES LLC | ~1800 | Lipoglycopeptide | Treatment of complicated skin and skin structure infections (cSSSI) caused by gram-positive bacteria | Inhibitor | 346 h |
| GATTEX KIT | 12/21/2012 | 203441 | TEDUGLUTIDE RECOMBINANT | NPS PHARMS INC | 3752 | Glucagon-like peptide-2 | Short bowel syndrome (SBS) treatment | Receptor binding | 1.3–2 h |
| SIGNIFOR | 12/14/2012 | 200677 | PASIREOTIDE DIASPARTATE | NOVARTIS | 1313.41 | Cyclohexapeptide | Treatment of Cushing’s disease | Receptor binding | 10–13 h |
| LINZESS | 8/30/2012 | 202811 | LINACLOTIDE | ALLERGAN SALES LLC | 1526.8 | Cyclic peptide | Treatment of irritable bowel syndrome | Receptor binding | N/A |
| KYPROLIS | 7/20/2012 | 202714 | CARFILZOMIB | ONYX THERAP | 719.9 | Modified tetrapeptidyl epoxide | Multiple myeloma treatment | Inhibitor | ≤1 h |
| FIRAZYR | 8/25/2011 | 022150 | ICATIBANT ACETATE | SHIRE ORPHAN THERAP | 1304.5 | Synthetic peptide | Treatment of angioedema, liver disease, burns, and burn infections | Receptor binding | 1.4 h |
| INCIVEK | 5/23/2011 | 201917 | TELAPREVIR | VERTEX PHARMS | 679.85 | Chemically modified peptide | Treatment of chronic Hepatitis C | Inhibitor | 4–11 h |
| EGRIFTA | 11/10/2010 | 022505 | TESAMORELIN ACETATE | THERATECHNOLOGIES | 5135.9 | Synthetic peptide | Human immunodeficiency virus (HIV) treatment | Receptor binding | 26–38 min |
| VICTOZA | 1/25/2010 | 022341 | LIRAGLUTIDE RECOMBINANT | NOVO NORDISK INC | 3751.2 | Synthetic peptide | Improving glycemic control in patients with type 2 diabetes mellitus | Receptor binding | 13 h |
| ISTODAX | 11/5/2009 | 022393 | ROMIDEPSIN | CELGENE | 540.71 | Bicyclic peptide | Treatment of cutaneous T-cell lymphoma (CTCL) and/or peripheral T-cell lymphoma (PTCL) | Inhibitor | 3 h |
| VIBATIV | 9/11/2009 | 022110 | TELAVANCIN HYDROCHLORIDE | CUMBERLAND PHARMS | 1755.6 | Lipoglycopeptide | Treatment of complicated skin and skin structure infections (cSSSI) caused by Gram-positive bacteria | MAP | 8 h |
| FIRMAGON | 12/24/2008 | 022201 | DEGARELIX ACETATE | FERRING | 1632.3 | Synthetic peptide | Prostate cancer treatment | Receptor binding | 53 h |
| SOMATULINE DEPOT | 30/08/2007 | 022074 | LANREOTIDE ACETATE | IPSEN PHARMA | 1096.34 | Cyclical octapeptide | Treatment of neuroendocrine tumors (NETs) and acromegaly | Receptor binding | 22 days |
| ERAXIS | 2/17/2006 | 021632 | ANIDULAFUNGIN | VICURON | 1140.3 | Lipopeptide | Anti-fungal drug | Inhibitor | 40–50 h |
| LEVEMIR | 6/16/2005 | 021536 | INSULIN DETEMIR RECOMBINANT | NOVO NORDISK INC | 5916.9 | A long-acting basal insulin analog | Treatment of hyperglycemia caused by type 1 and type 2 diabetes | Receptor binding | 57 h |
| LEVEMIR FLEXPEN | 6/16/2005 | 021536 | INSULIN DETEMIR RECOMBINANT | NOVO NORDISK INC | 5916.9 | A long-acting basal insulin analog | Treatment of hyperglycemia caused by type 1 and type 2 diabetes | Receptor binding | 5–7 h |
| LEVEMIR FLEXTOUCH | 6/16/2005 | 021536 | INSULIN DETEMIR RECOMBINANT | NOVO NORDISK INC | 5916.9 | A long-acting basal insulin analog | Treatment of hyperglycemia caused by type 1 and type 2 diabetes | Receptor binding | 5–7 h |
| LEVEMIR INNOLET | 6/16/2005 | 021536 | INSULIN DETEMIR RECOMBINANT | NOVO NORDISK INC | 5916.9 | A long-acting basal insulin analog | Treatment of hyperglycemia caused by type 1 and type 2 diabetes | Receptor binding | 5–7 h |
| LEVEMIR PENFILL | 6/16/2005 | 021536 | INSULIN DETEMIR RECOMBINANT | NOVO NORDISK INC | 5916.9 | A long-acting basal insulin analog | Treatment of hyperglycemia caused by type 1 and type 2 diabetes | Receptor binding | 5–7 h |
| BYETTA | 4/28/2005 | 021773 | EXENATIDE SYNTHETIC | ASTRAZENECA AB | 4186.6 | Synthetic peptide | Improving glycemic control in patients with type 2 diabetes mellitus | Receptor binding | 2.4 h |
| SYMLIN | 3/16/2005 | 021332 | PRAMLINTIDE ACETATE | ASTRAZENECA AB | 3949.4 | Peptide hormone | Treatment of type 1 and type 2 diabetes mellitus | Receptor binding | 48 min |
| PRIALT | 12/28/2004 | 021060 | ZICONOTIDE ACETATE | TERSERA THERAPS LLC | 2639 | Synthetic peptide | Chronic pain treatment | Inhibitor | 2.9–6.5 h |
| APIDRA | 4/16/2004 | 021629 | INSULIN GLULISINE RECOMBINANT | SANOFI AVENTIS US | 5823 | Human insulin analog | Treatment of hyperglycemia caused by type 1 and type 2 diabetes | Inhibitor | 13–86 min |
| APIDRA SOLOSTAR | 4/16/2004 | 021629 | INSULIN GLULISINE RECOMBINANT | SANOFI AVENTIS US | 5823 | Human insulin analog | Treatment of hyperglycemia caused by type 1 and type 2 diabetes | Inhibitor | 13–86 min |
| CHIRHOSTIM | 4/9/2004 | 021256 | SECRETIN SYNTHETIC HUMAN | CHIRHOCLIN | 3039.44 | Gastrointestinal peptide hormone | (1) Pancreatic secretions to aid in the diagnosis of pancreatic exocrine dysfunction(2) Gastrin secretion to aid in the diagnosis of gastrinoma(3) Pancreatic secretions to facilitate the identification of the ampulla of Vater and accessory papilla during ERCP | Inhibitor | 45 min |
| PLENAXIS | 11/25/2003 | 021320 | ABARELIX | SPECIALTY EUROPEAN | 1416.06 | Synthetic peptide | Prostate cancer treatment | Inhibitor | 13.2 days |
| CUBICIN | 9/12/2003 | 021572 | DAPTOMYCIN | CUBIST PHARMS LLC | 1620.67 | Cyclic lipopeptide | Treatment of complicated skin and skin structure infections (cSSSI) caused by Gram-positive bacteria | MAP | 8.1–9 h |
| CUBICIN RF | 9/12/2003 | 021572 | DAPTOMYCIN | CUBIST PHARMS LLC | 1620.67 | Cyclic lipopeptide | Treatment of complicated skin and skin structure infections (cSSSI) caused by Gram-positive bacteria | MAP | 8.1–9 h |
| REYATAZ | 6/20/2003 | 021567 | ATAZANAVIR SULFATE | BRISTOL MYERS | 704.9 | Azapeptide | Human immunodeficiency virus (HIV) treatment | Inhibitor | 6.5–7.9 h |
| VELCADE | 5/13/2003 | 021602 | BORTEZOMIB | MILLENNIUM PHARMS | 384.24 | Chemically modified peptide | Multiple myeloma treatment | Inhibitor | 9–15 h |
| FUZEON | 3/13/2003 | 021481 | ENFUVIRTIDE | ROCHE | 4492 | Synthetic peptide | Human immunodeficiency virus (HIV) treatment | Inhibitor | 3.8 h |
| NATRECOR | 8/10/2001 | 020920 | NESIRITIDE RECOMBINANT | SCIOS LLC | 3464 | Cyclic peptide | Acute decompensated heart failure (ADHF) treatment | Receptor binding | 18 min |
| CANCIDAS | 1/26/2001 | 021227 | CASPOFUNGIN ACETATE | MERCK | 1213.42 | Cyclic lipopeptide | Anti-fungal drug | Inhibitor | 9–11 h |
| ANGIOMAX | 12/15/2000 | 020873 | BIVALIRUDIN | SANDOZ INC | 2180 | Synthetic peptide | Treatment of heparin-induced thrombocytopenia and for the prevention of thrombosis | Inhibitor | 22 min–3.5 h |
| CETROTIDE | 8/11/2000 | 021197 | CETRORELIX | EMD SERONO INC | 1431.06 | Synthetic peptide | For prevention of premature ovulation in women undergoing fertility treatments with controlled ovulation | Receptor binding | ~62.8 h |
| TRELSTAR | 6/15/2000 | 020715 | TRIPTORELIN PAMOATE | ALLERGAN SALES LLC | 1699.9 | Synthetic peptide | Prostate cancer treatment | Receptor binding | 6 min–3 h |
| NOVOLOG | 6/7/2000 | 020986 | INSULIN ASPART RECOMBINANT | NOVO NORDISK INC | 5825.8 | Peptide hormone | Treatment of hyperglycemia caused by type 1 and type 2 diabetes | Receptor binding | 81 min |
| NOVOLOG FLEXPEN | 6/7/2000 | 020986 | INSULIN ASPART RECOMBINANT | NOVO NORDISK INC | 5825.8 | Peptide hormone | Treatment of hyperglycemia caused by type 1 and type 2 diabetes | Receptor binding | 81 min |
| NOVOLOG FLEXTOUCH | 6/7/2000 | 020986 | INSULIN ASPART RECOMBINANT | NOVO NORDISK INC | 5825.8 | Peptide hormone | Treatment of hyperglycemia caused by type 1 and type 2 diabetes | Receptor binding | 81 min |
| NOVOLOG INNOLET | 6/7/2000 | 020986 | INSULIN ASPART RECOMBINANT | NOVO NORDISK INC | 5825.8 | Peptide hormone | Treatment of hyperglycemia caused by type 1 and type 2 diabetes | Receptor binding | 81 min |
| NOVOLOG PENFILL | 6/7/2000 | 020986 | INSULIN ASPART RECOMBINANT | NOVO NORDISK INC | 5825.8 | Peptide hormone | Treatment of hyperglycemia caused by type 1 and type 2 diabetes | Receptor binding | 81 min |
| LANTUS | 4/20/2000 | 021081 | INSULIN GLARGINE RECOMBINANT | SANOFI AVENTIS US | 6063 | Peptide hormone | Treatment of hyperglycemia caused by type 1 and type 2 diabetes | Receptor binding | N/A |
| LANTUS SOLOSTAR | 4/20/2000 | 021081 | INSULIN GLARGINE RECOMBINANT | SANOFI AVENTIS US | 6063 | Peptide hormone | Treatment of hyperglycemia caused by type 1 and type 2 diabetes | Receptor binding | N/A |
| NEO TECT KIT | 8/3/1999 | 021012 | TECHNETIUM TC-99M DEPREOTIDE | CIS BIO INTL SA | 486.14 | Cyclic peptide | (1) Detecting coronary artery disease(2) Evaluating myocardial function | Others | 6.02 h |
| GANIRELIX ACETATE | 7/29/1999 | 021057 | GANIRELIX ACETATE | ORGANON USA INC | 1570.35 | Peptide hormone | For inhibition of premature LH surges in women undergoing controlled ovarian hyperstimulation | Receptor binding | 16.2 h |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Chen, C.H.; Lu, T.K. Development and Challenges of Antimicrobial Peptides for Therapeutic Applications. Antibiotics 2020, 9, 24. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics9010024
AMA Style
Chen CH, Lu TK. Development and Challenges of Antimicrobial Peptides for Therapeutic Applications. Antibiotics. 2020; 9(1):24. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics9010024
Chicago/Turabian StyleChen, Charles H., and Timothy K. Lu. 2020. "Development and Challenges of Antimicrobial Peptides for Therapeutic Applications" Antibiotics 9, no. 1: 24. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics9010024
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.