Design, Screening, and Testing of Non-Rational Peptide Libraries with Antimicrobial Activity: In Silico and Experimental Approaches

 ,
,  , , ,
, , ,  , , and
, , and
Abstract
:1. Introduction
2. Antimicrobial Peptides
2.1. Antibacterial
2.2. Antivirals
2.3. Antifungal
2.4. Antiparasitic
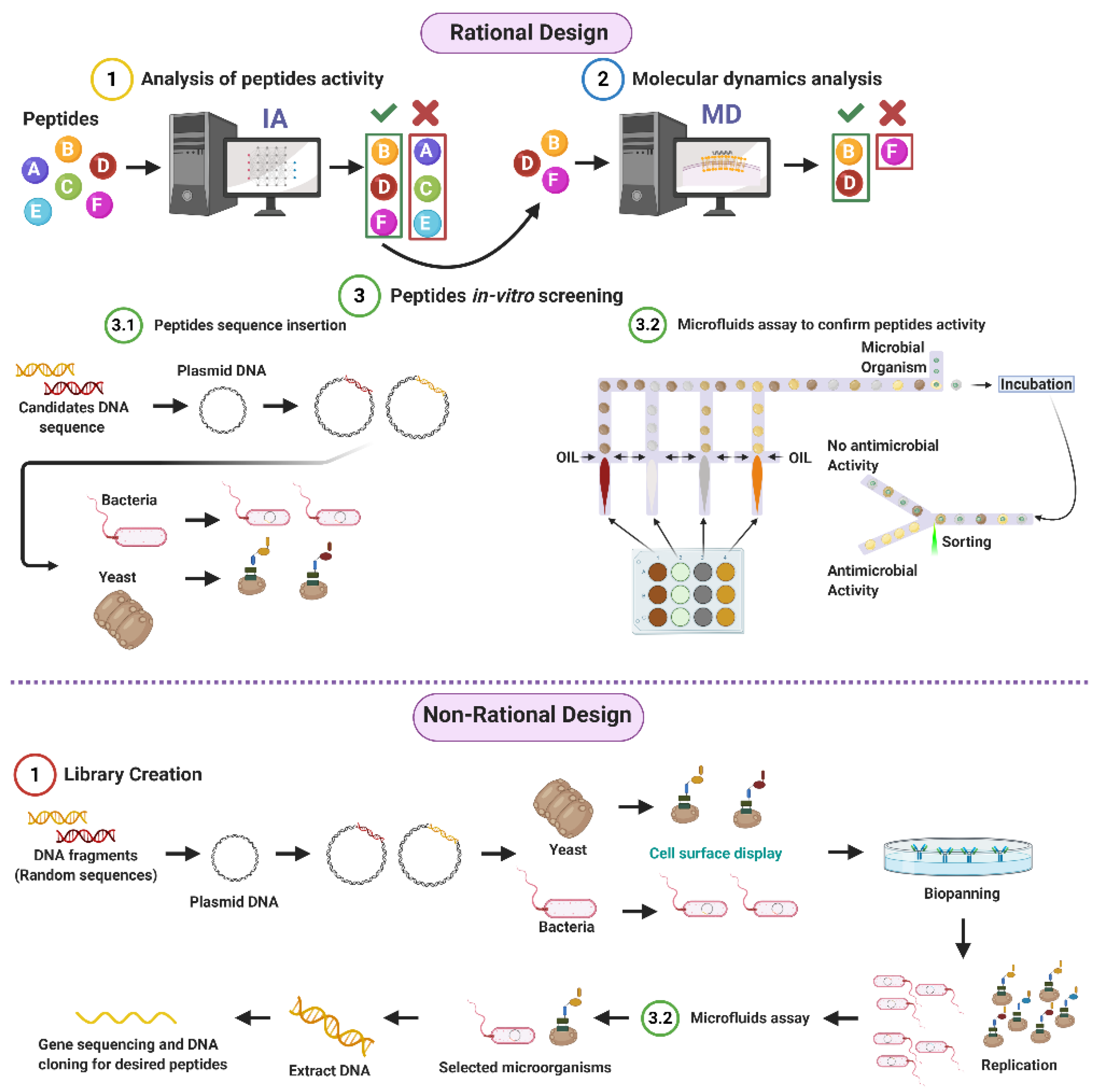
3. Peptide Library Design
3.1. Rational
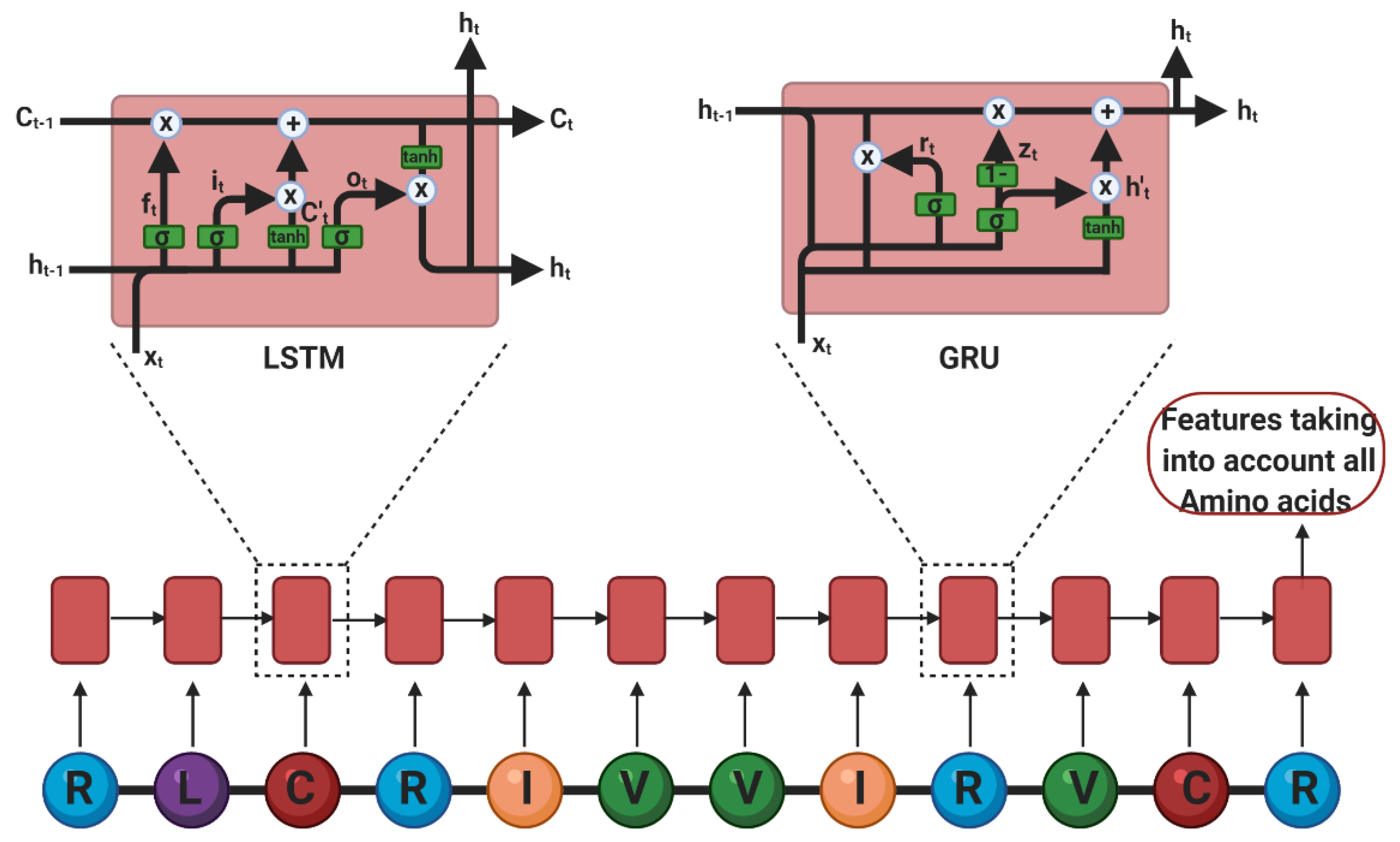
Deep Learning
3.2. Non-Rational
3.2.1. Phage Display
3.2.2. Bacterial Display
3.2.3. Yeast Surface Display
3.2.4. Library Screening
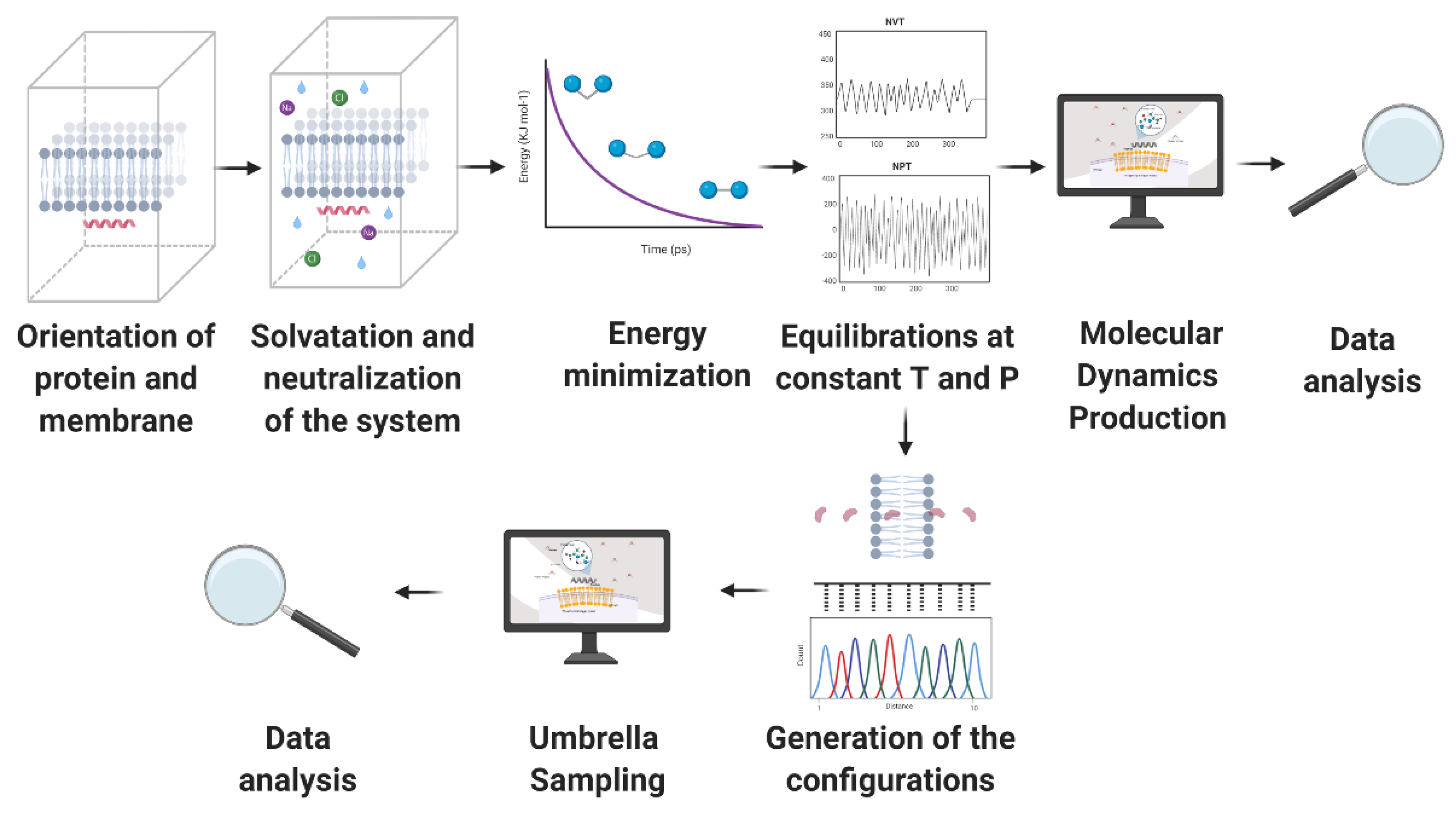
4. Molecular Dynamics (MD)
4.1. Configuration of the System
4.2. Molecular Dynamics (MD) Simulations Method
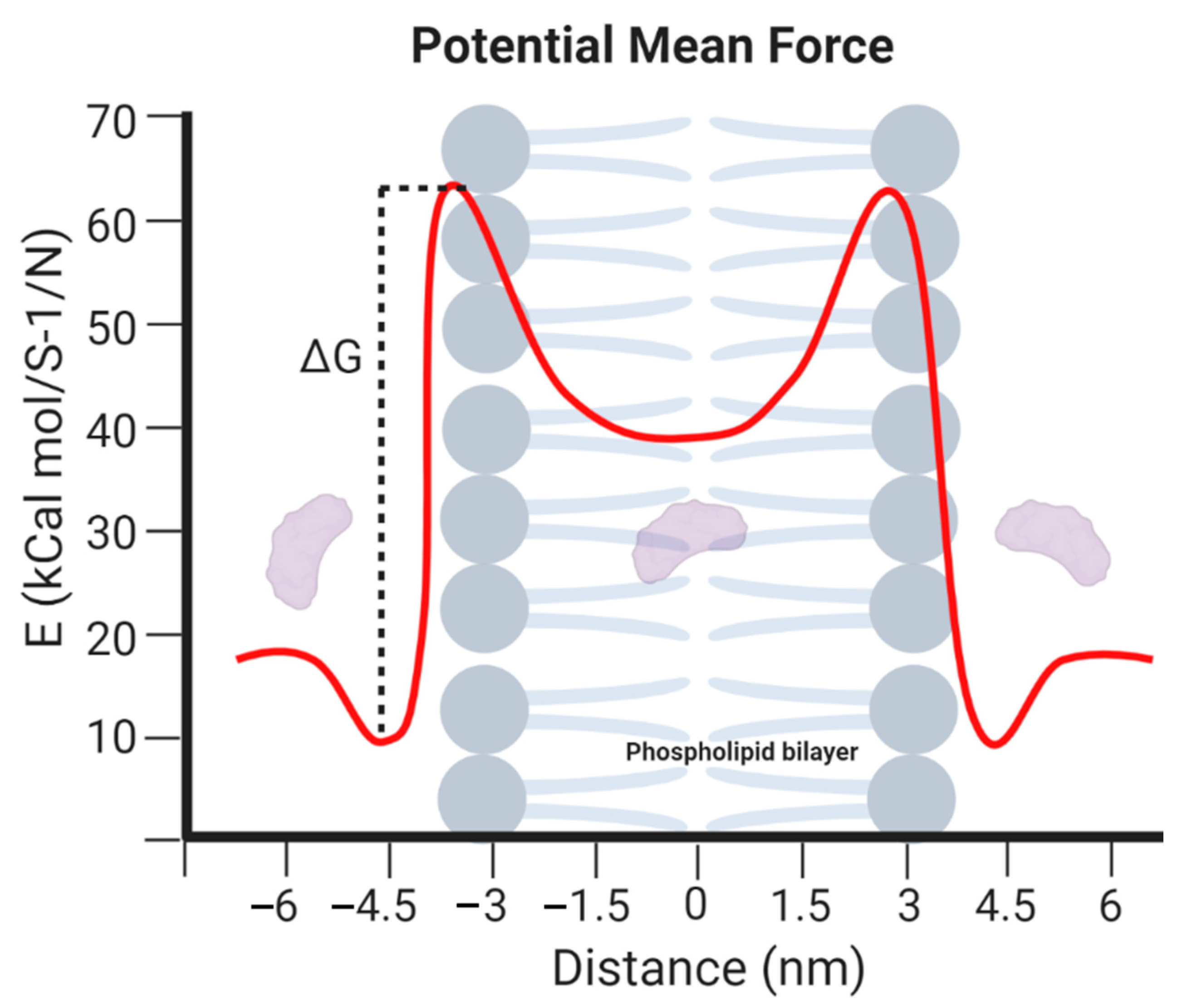
4.3. Information Provided by the MD Simulations
5. Microfluidic Approaches
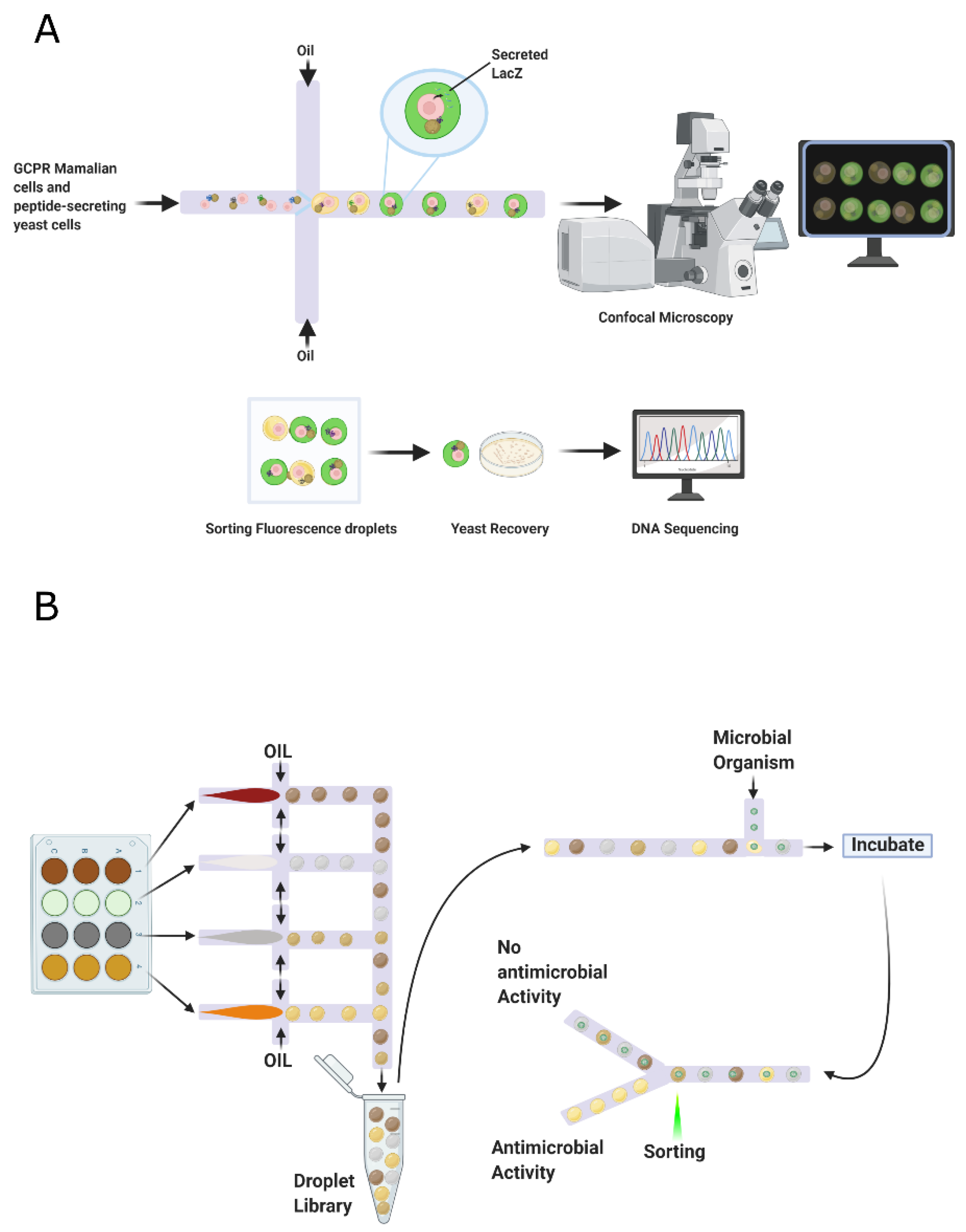
5.1. Droplet-Based Screening
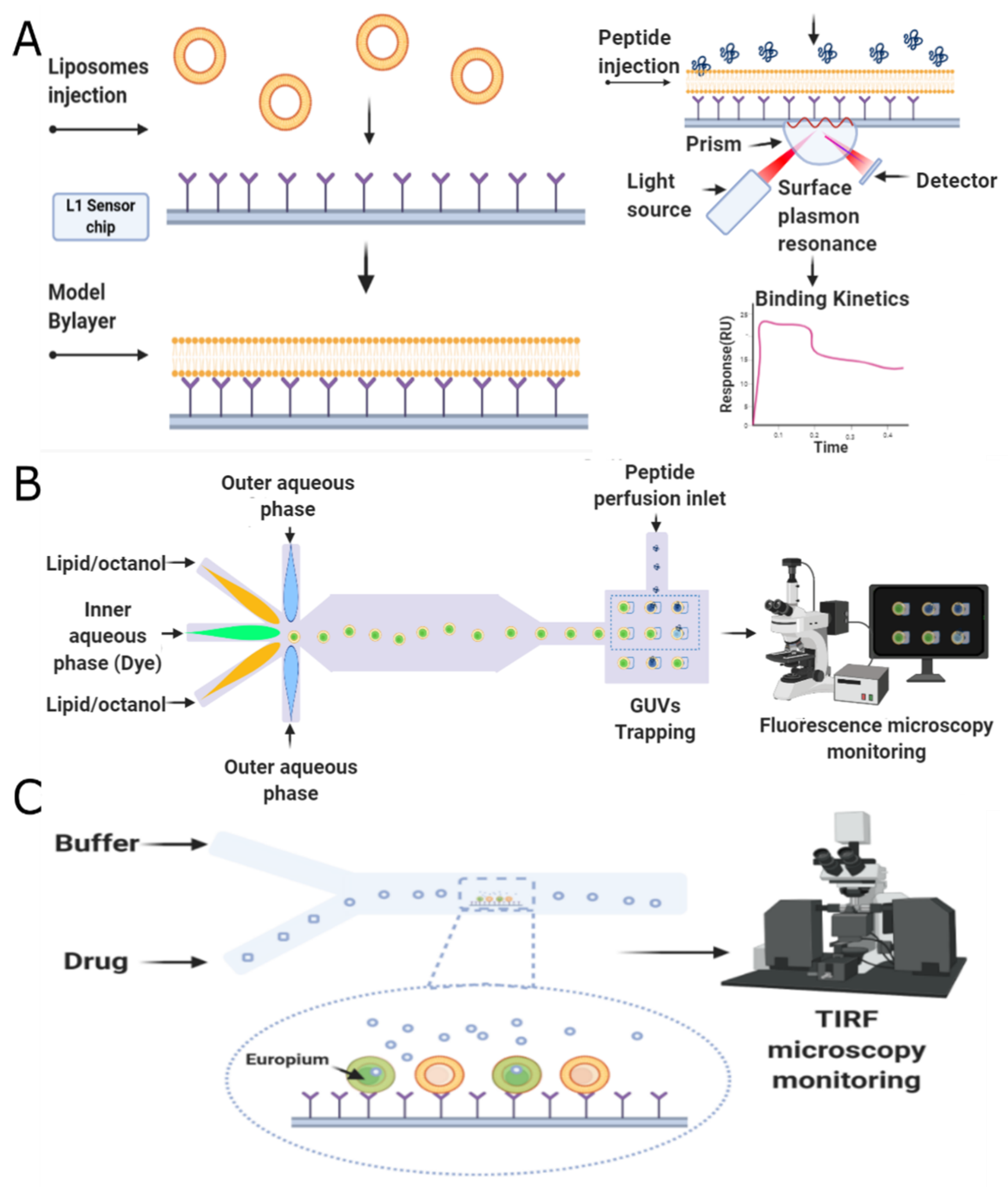
5.2. Membrane-Based Screening
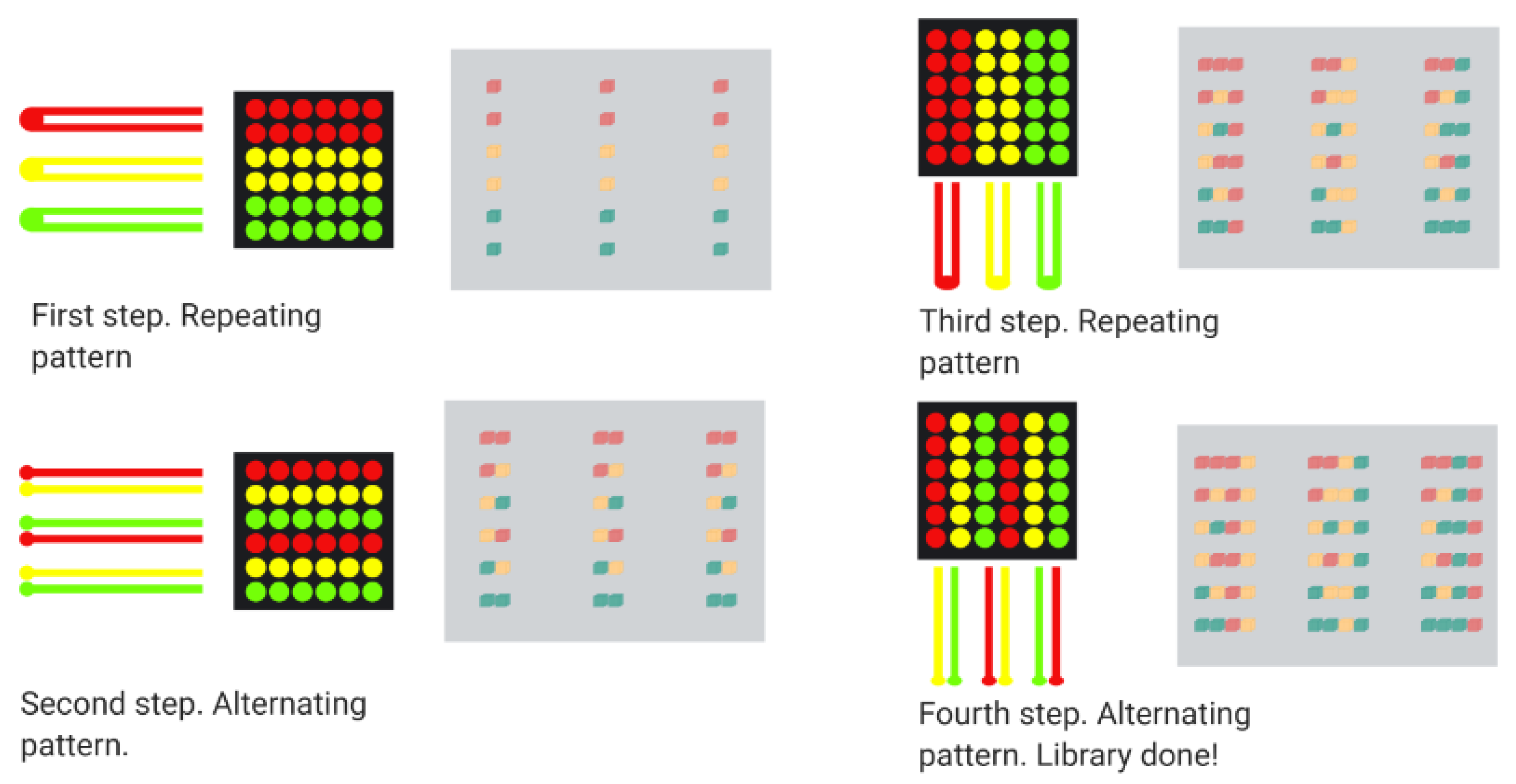
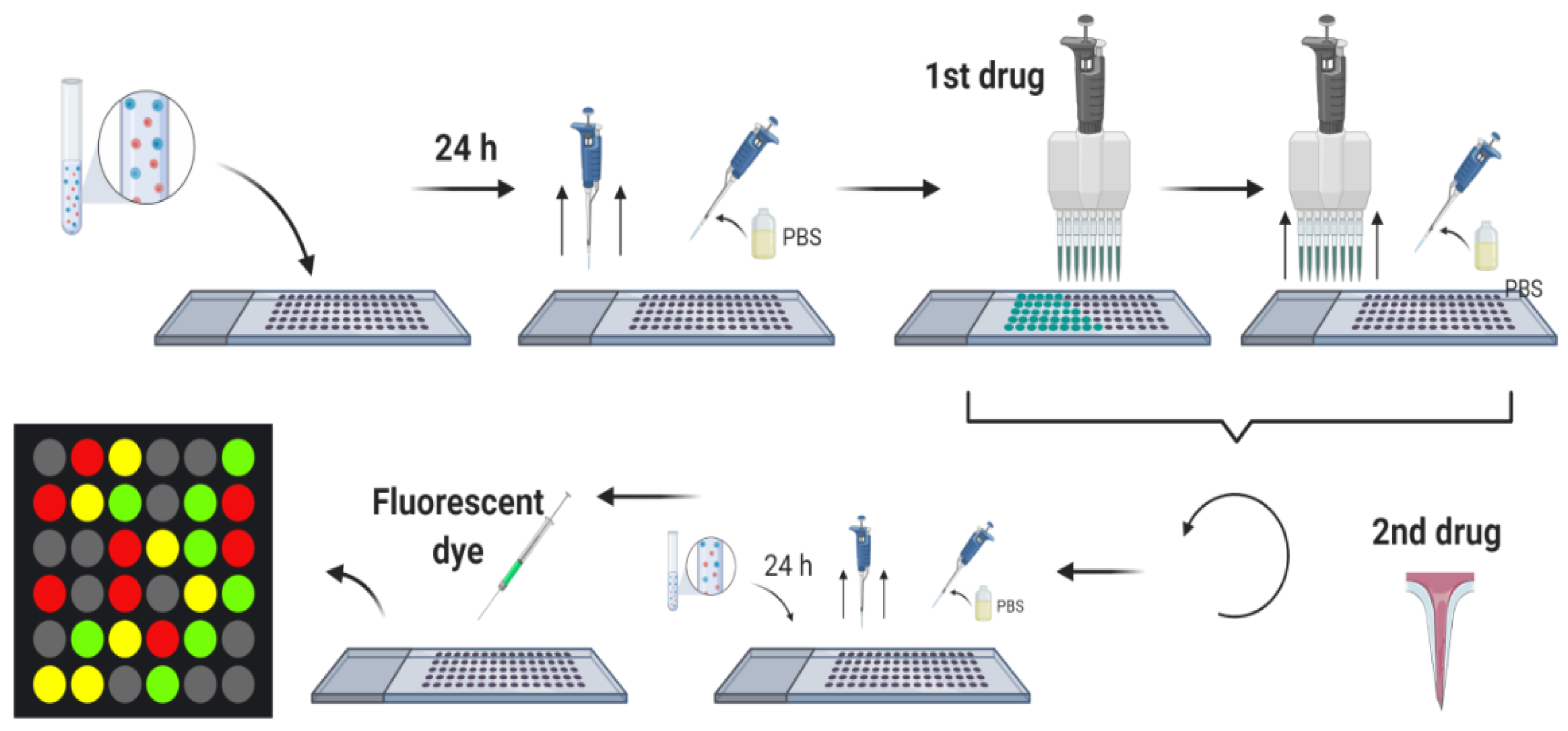
5.3. Combinatorial Microarray Screening
6. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Naylor, N.R.; Atun, R.; Zhu, N.; Kulasabanathan, K.; Silva, S.; Chatterjee, A.; Knight, G.M.; Robotham, J.V. Estimating the burden of antimicrobial resistance: A systematic literature review. Antimicrob. Resist. Infect. Control 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Stokowski, L.A. Antimicrobial Resistance: A Primer. Available online: https://www.medscape.com/viewarticle/729196 (accessed on 3 November 2020).
- Mulani, M.S.; Kamble, E.E.; Kumkar, S.N.; Tawre, M.S.; Pardesi, K.R. Emerging Strategies to Combat ESKAPE Pathogens in the Era of Antimicrobial Resistance: A Review. Front. Microbiol. 2019, 10, 539. [Google Scholar] [CrossRef] [PubMed]
- Ventola, C.L. The Antibiotic Resistance Crisis: Part 1: Causes and Threats. Pharm. Ther. 2015, 40, 277–283. [Google Scholar]
- World Health Organization (WHO). Antimicrobial Resistance. Available online: https://www.who.int/health-topics/antimicrobial-resistance (accessed on 3 November 2020).
- Sakeena, M.H.F.; Bennett, A.A.; McLachlan, A.J. Enhancing pharmacists’ role in developing countries to overcome the challenge of antimicrobial resistance: A narrative review. Antimicrob. Resist. Infect. Control 2018, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Center for Disease Control and Prevention (CDC). Antibiotic-Resistant Germs: New Threats. Available online: https://www.cdc.gov/drugresistance/index.html (accessed on 3 November 2020).
- El-Mahallawy, H.A.; Hassan, S.S.; El-Wakil, M.; Moneer, M.M. Bacteremia due to ESKAPE pathogens: An emerging problem in cancer patients. J. Egypt. Natl. Cancer Inst. 2016, 28, 157–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marturano, J.E.; Lowery, T.J. ESKAPE Pathogens in Bloodstream Infections Are Associated with Higher Cost and Mortality but Can Be Predicted Using Diagnoses Upon Admission. Open Forum Infect. Dis. 2019, 6. [Google Scholar] [CrossRef]
- Pendleton, J.N.; Gorman, S.P.; Gilmore, B.F. Clinical relevance of the ESKAPE pathogens. Expert Rev. Anti Infect. Ther. 2013, 11, 297–308. [Google Scholar] [CrossRef]
- Tang, M.W.; Shafer, R.W. HIV-1 Antiretroviral Resistance. Drugs 2012, 72, e1–e25. [Google Scholar] [CrossRef] [Green Version]
- Goldhill, D.H.; Te Velthuis, A.J.; Fletcher, R.A.; Langat, P.; Zambon, M.; Lackenby, A.; Barclay, W.S. The mechanism of resistance to favipiravir in influenza. Proc. Natl. Acad. Sci. USA 2018, 115, 11613–11618. [Google Scholar] [CrossRef] [Green Version]
- Lázár, V.; Martins, A.; Spohn, R.; Daruka, L.; Grézal, G.; Fekete, G.; Számel, M.; Jangir, P.K.; Kintses, B.; Csörgő, B.; et al. Antibiotic-resistant bacteria show widespread collateral sensitivity to antimicrobial peptides. Nat. Microbiol. 2018, 3, 718–731. [Google Scholar] [CrossRef] [Green Version]
- Bechinger, B.; Gorr, S.-U. Antimicrobial Peptides: Mechanisms of Action and Resistance. J. Dent. Res. 2016, 96, 254–260. [Google Scholar] [CrossRef] [Green Version]
- Anunthawan, T.; De La Fuente-Núñez, C.; Hancock, R.E.; Klaynongsruang, S. Cationic amphipathic peptides KT2 and RT2 are taken up into bacterial cells and kill planktonic and biofilm bacteria. Biochim. Biophys. Acta Biomembr. 2015, 1848, 1352–1358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malanovic, N.; Lohner, K. Gram-positive bacterial cell envelopes: The impact on the activity of antimicrobial peptides. Biochim. Biophys. Acta Biomembr. 2016, 1858, 936–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bechinger, B. The SMART model: Soft Membranes Adapt and Respond also Transiently, in the presence of antimicrobial peptides. J. Pept. Sci. 2014, 21, 346–355. [Google Scholar] [CrossRef] [Green Version]
- Boas, L.C.P.V.; Campos, M.L.; Berlanda, R.L.A.; de Carvalho Neves, N.; Franco, O.L. Antiviral peptides as promising therapeutic drugs. Cell. Mol. Life Sci. 2019, 76, 3525–3542. [Google Scholar] [CrossRef] [PubMed]
- Waghu, F.H.; Joseph, S.; Ghawali, S.; Martis, E.A.; Madan, T.; Venkatesh, K.V.; Idicula-Thomas, S. Designing antibacterial peptides with enhanced killing kinetics. Front. Microbiol. 2018, 9, 325. [Google Scholar] [CrossRef]
- Torres, M.D.; Sothiselvam, S.; Lu, T.K.; de la Fuente-Nunez, C. Peptide design principles for antimicrobial applications. J. Mol. Biol. 2019, 431, 3547–3567. [Google Scholar] [CrossRef]
- Dean, S.N.; Walper, S.A. Variational Autoencoder for Generation of Antimicrobial Peptides. ACS Omega 2020, 5, 20746–20754. [Google Scholar] [CrossRef]
- Lin, E.; Lin, C.H.; Lane, H.Y. Relevant Applications of Generative Adversarial Networks in Drug Design and Discovery: Molecular De Novo Design Dimensionality Reduction, and De Novo Peptide and Protein Design. Molecules 2020, 25, 3250. [Google Scholar] [CrossRef]
- Kalafatovic, D.; Mauša, G.; Todorovski, T.; Giralt, E. Algorithm-supported, mass and sequence diversity-oriented random peptide library design. J. Cheminform. 2019, 11, 25. [Google Scholar] [CrossRef] [Green Version]
- Henninot, A.; Collins, J.C.; Nuss, J.M. The current state of peptide drug discovery: Back to the future? J. Med. Chem. 2018, 61, 1382–1414. [Google Scholar] [CrossRef]
- Liu, R.; Li, X.; Lam, K.S. Combinatorial chemistry in drug discovery. Curr. Opin. Chem. Biol. 2017, 38, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Hamzeh-Mivehroud, M.; Alizadeh, A.A.; Morris, M.B.; Church, W.B.; Dastmalchi, S. Phage display as a technology delivering on the promise of peptide drug discovery. Drug Discov. Today 2013, 18, 1144–1157. [Google Scholar] [CrossRef] [PubMed]
- Guralp, S.A.; Murgha, Y.E.; Rouillard, J.M.; Gulari, E. From design to screening: A new antimicrobial peptide discovery pipeline. PLoS ONE 2013, 8, e59305. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.; Choi, J.H.; Xu, Z. Microbial cell-surface display. Trends Biotechnol. 2003, 21, 45–52. [Google Scholar] [CrossRef]
- Lane, N.; Kahanda, I. DeepACPpred: A Novel Hybrid CNN-RNN Architecture for Predicting Anti-Cancer Peptides. In Advances in Intelligent Systems and Computing; Springer International Publishing: Cham, Switzerland, 2020; pp. 60–69. [Google Scholar]
- Müller, A.T.; Hiss, J.A.; Schneider, G. Recurrent Neural Network Model for Constructive Peptide Design. J. Chem. Inf. Model. 2018, 58, 472–479. [Google Scholar] [CrossRef]
- Goh, G.B.; Siegel, C.; Vishnu, A.; Hodas, N. Using Rule-Based Labels for Weak Supervised Learning. In Proceedings of the 24th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, London, UK, 19–23 August 2018; Association for Computing Machinery: New York, NY, USA, 2018. [Google Scholar]
- Tiwary, S.; Levy, R.; Gutenbrunner, P.; Soto, F.S.; Palaniappan, K.K.; Deming, L.; Berndl, M.; Brant, A.; Cimermancic, P.; Cox, J. High-quality MS/MS spectrum prediction for data-dependent and data-independent acquisition data analysis. Nat. Methods 2019, 16, 519–525. [Google Scholar] [CrossRef]
- Goodfellow, I.; Bengio, Y.; Courville, A.; Bengio, Y. Deep Learning; MIT Press Cambridge: Cambridge, MA, USA, 2016; Volume 1. [Google Scholar]
- Wahnström, G. Molecular Dynamics Lecture Notes; Chalmers University of Technology: Gothenburg, Sweden, 2018. [Google Scholar]
- Feig, M.; Nawrocki, G.; Yu, I.; Wang, P.-H.; Sugita, Y. Challenges and opportunities in connecting simulations with experiments via molecular dynamics of cellular environments. J. Phys. Conf. Ser. 2018, 1036. [Google Scholar] [CrossRef]
- Allen, M.P. Introduction to Molecular Dynamics Simulation. In Computational Soft Matter: From Synthetic Polymers to Proteins; John von Neumann Institute for Computing (NIC): Jülich, Germany, 2004. [Google Scholar]
- Rathore, N.; Pablo, J.J. de Monte Carlo simulation of proteins through a random walk in energy space. J. Chem. Phys. 2002, 116, 7225–7230. [Google Scholar] [CrossRef] [Green Version]
- Gofman, Y.; Haliloglu, T.; Ben-Tal, N. Monte-Carlo Simulations of Peptide-Membrane Interactions: Web-Server. Biophys. J. 2010, 98, 487a. [Google Scholar] [CrossRef] [Green Version]
- Karplus, M.; McCammon, J.A. Molecular dynamics simulations of biomolecules. Nat. Struct. Biol. 2002, 9, 646–652. [Google Scholar] [CrossRef] [PubMed]
- Reif, M.; Zacharias, M. Computer Modelling and Molecular Dynamics Simulation of Biomolecules. In Biomolecular and Bioanalytical Techniques; John Wiley & Sons Ltd.: New York, NY, USA, 2019; pp. 501–535. [Google Scholar]
- Alder, B.J.; Wainwright, T.E. Phase Transition for a Hard Sphere System. J. Chem. Phys. 1957, 27, 1208–1209. [Google Scholar] [CrossRef] [Green Version]
- Hollingsworth, S.A.; Dror, R.O. Molecular Dynamics Simulation for All. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández, E.R.; Zetina, L.M.M.; Vega, G.T.; Rocha, M.G.; Ochoa, L.F.R.; Fernandez, R.L. Molecular Dynamics: From basic techniques to applications (A Molecular Dynamics Primer). In AIP Conference Proceedings; AIP: College Park, MD, USA, 2008. [Google Scholar]
- Aliaga, L.C.R.; Lima, L.V.P.C.; Domingues, G.M.B.; Bastos, I.N.; Evangelakis, G.A. Experimental and molecular dynamics simulation study on the glass formation of Cu-Zr-Al alloys. Mater. Res. Express 2019, 6. [Google Scholar] [CrossRef]
- Chen, J. The Development and Comparison of Molecular Dynamics Simulation and Monte Carlo Simulation. IOP Conf. Ser. Earth Environ. Sci. 2018, 128. [Google Scholar] [CrossRef]
- Neyts, E.C.; Bogaerts, A. Combining molecular dynamics with Monte Carlo simulations: Implementations and applications. Theor. Chem. Acc. Belg. 2012, 132. [Google Scholar] [CrossRef]
- Kikuchi, K.; Yoshida, M.; Maekawa, T.; Watanabe, H. Metropolis Monte Carlo method as a numerical technique to solve the FokkerPlanck equation. Chem. Phys. Lett. 1991, 185, 335–338. [Google Scholar] [CrossRef]
- Cuendet, M.A.; van Gunsteren, W.F. On the calculation of velocity-dependent properties in molecular dynamics simulations using the leapfrog integration algorithm. J. Chem. Phys. 2007, 127. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [Green Version]
- Spoel, D.V.D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Brooks, B.R.; Brooks, C.L.; Mackerell, A.D.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [Green Version]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Smith, W.; Yong, C.W.; Rodger, P.M. DL_POLY: Application to molecular simulation. Mol. Simul. 2002, 28, 385–471. [Google Scholar] [CrossRef]
- FrantzDale, B.; Plimpton, S.J.; Shephard, M.S. Software components for parallel multiscale simulation: An example with LAMMPS. Eng. Comput. 2009, 26, 205–211. [Google Scholar] [CrossRef]
- Hernández-Rodríguez, M.; Rosales-Hernández, M.C.; Mendieta-Wejebe, J.E.; Martínez-Archundia, M.; Basurto, J.C. Current Tools and Methods in Molecular Dynamics (MD) Simulations for Drug Design. Curr. Med. Chem. 2016, 23, 3909–3924. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Seara, H.; Róg, T. Molecular Dynamics Simulations of Lipid Bilayers: Simple Recipe of How to Do It. In Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2012; pp. 407–429. [Google Scholar]
- Langham, A.; Kaznessis, Y.N. Molecular Simulations of Antimicrobial Peptides. In Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2009; pp. 267–285. [Google Scholar]
- Shahane, G.; Ding, W.; Palaiokostas, M.; Orsi, M. Physical properties of model biological lipid bilayers: Insights from all-atom molecular dynamics simulations. J. Mol. Model. 2019, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bharadwaj, P.; Solomon, T.; Malajczuk, C.J.; Mancera, R.L.; Howard, M.; Arrigan, D.W.M.; Newsholme, P.; Martins, R.N. Role of the cell membrane interface in modulating production and uptake of Alzheimers beta amyloid protein. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1639–1651. [Google Scholar] [CrossRef]
- Szlasa, W.; Zendran, I.; Zalesińska, A.; Tarek, M.; Kulbacka, J. Lipid composition of the cancer cell membrane. J. Bioenerg. Biomembr. 2020, 52, 321–342. [Google Scholar] [CrossRef]
- Revin, V.V.; Gromova, N.V.; Revina, E.S.; Martynova, M.I.; Seikina, A.I.; Revina, N.V.; Imarova, O.G.; Solomadin, I.N.; Tychkov, A.Y.; Zhelev, N. Role of Membrane Lipids in the Regulation of Erythrocytic Oxygen-Transport Function in Cardiovascular Diseases. BioMed Res. Int. 2016, 2016, 3429604. [Google Scholar] [CrossRef] [Green Version]
- Fu, Y.; Luo, J.; Qin, J.; Yang, M. Screening techniques for the identification of bioactive compounds in natural products. J. Pharm. Biomed. Anal. 2019, 168, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Hsu, S.-C.; Han, S.-I.; Thapa, H.R.; Guzman, A.R.; Browne, D.R.; Tatli, M.; Devarenne, T.P.; Stern, D.B.; Han, A. High-throughput droplet microfluidics screening platform for selecting fast-growing and high lipid-producing microalgae from a mutant library. Plant Direct 2017, 1, e00011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barata, D.; van Blitterswijk, C.; Habibovic, P. High-throughput screening approaches and combinatorial development of biomaterials using microfluidics. Acta Biomater. 2016, 34, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, A.M.; Hsieh, K.; Wang, T.-H. Droplet microfluidics for high-sensitivity and high-throughput detection and screening of disease biomarkers. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2018, 10. [Google Scholar] [CrossRef]
- Zhu, X.-D.; Shi, X.; Wang, S.-W.; Chu, J.; Zhu, W.-H.; Ye, B.-C.; Zuo, P.; Wang, Y.-H. High-throughput screening of high lactic acid-producing Bacillus coagulans by droplet microfluidic based flow cytometry with fluorescence activated cell sorting. RSC Adv. 2019, 9, 4507–4513. [Google Scholar] [CrossRef] [Green Version]
- Longwell, C.K.; Labanieh, L.; Cochran, J.R. High-throughput screening technologies for enzyme engineering. Curr. Opin. Biotechnol. 2017, 48, 196–202. [Google Scholar] [CrossRef]
- Prodanović, R.; Ung, W.L.; Đurđić, K.I.; Fischer, R.; Weitz, D.A.; Ostafe, R. A high-throughput screening system based on droplet microfluidics for glucose oxidase gene libraries. Molecules 2020, 25, 2418. [Google Scholar] [CrossRef]
- Mashaghi, S.; Abbaspourrad, A.; Weitz, D.A.; van Oijen, A.M. Droplet microfluidics: A tool for biology chemistry and nanotechnology. TrAC Trends Anal. Chem. 2016, 82, 118–125. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Yang, X.; Liu, L.; Zhou, P.; Zhou, J.; Shi, X.; Wang, Y. A microarray platform designed for high-throughput screening the reaction conditions for the synthesis of micro/nanosized biomedical materials. Bioact. Mater. 2020, 5, 286–296. [Google Scholar] [CrossRef]
- Holland-Moritz, D.A.; Wismer, M.K.; Mann, B.F.; Farasat, I.; Devine, P.; Guetschow, E.D.; Mangion, I.; Welch, C.J.; Moore, J.C.; Sun, S.; et al. Mass Activated Droplet Sorting (MADS) Enables High-Throughput Screening of Enzymatic Reactions at Nanoliter Scale. Angew. Chem. Int. Ed. 2020, 59, 4470–4477. [Google Scholar] [CrossRef]
- Lim, J.W.; Shin, K.S.; Moon, J.; Lee, S.K.; Kim, T. A Microfluidic Platform for High-Throughput Screening of Small Mutant Libraries. Anal. Chem. 2016, 88, 5234–5242. [Google Scholar] [CrossRef] [PubMed]
- Che, Y.-J.; Wu, H.-W.; Hung, L.-Y.; Liu, C.-A.; Chang, H.-Y.; Wang, K.; Lee, G.-B. An integrated microfluidic system for screening of phage-displayed peptides specific to colon cancer cells and colon cancer stem cells. Biomicrofluidics 2015, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahlapuu, M.; Håkansson, J.; Ringstad, L.; Björn, C. Antimicrobial Peptides: An Emerging Category of Therapeutic Agents. Front. Cell. Infect. Microbiol. 2016, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, P.; Kizhakkedathu, J.; Straus, S. Antimicrobial Peptides: Diversity Mechanism of Action and Strategies to Improve the Activity and Biocompatibility In Vivo. Biomolecules 2018, 8, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mojsoska, B.; Jenssen, H. Peptides and Peptidomimetics for Antimicrobial Drug Design. Pharmaceuticals 2015, 8, 366–415. [Google Scholar] [CrossRef]
- Waghu, F.H.; Barai, R.S.; Gurung, P.; Idicula-Thomas, S. CAMPR3: A database on sequences, structures and signatures of antimicrobial peptides: Table 1. Nucleic Acids Res. 2015, 44, D1094–D1097. [Google Scholar] [CrossRef] [Green Version]
- Pirtskhalava, M.; Amstrong, A.A.; Grigolava, M.; Chubinidze, M.; Alimbarashvili, E.; Vishnepolsky, B.; Gabrielian, A.; Rosenthal, A.; Hurt, D.E.; Tartakovsky, M. DBAASP v3: Database of antimicrobial/cytotoxic activity and structure of peptides as a resource for development of new therapeutics. Nucleic Acids Res. 2020. [Google Scholar] [CrossRef]
- Kang, X.; Dong, F.; Shi, C.; Liu, S.; Sun, J.; Chen, J.; Li, H.; Xu, H.; Lao, X.; Zheng, H. DRAMP 2.0, an updated data repository of antimicrobial peptides. Sci. Data 2019, 6. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Z.; Tharmalingam, N.; Liu, Q.; Jayamani, E.; Kim, W.; Fuchs, B.B.; Zhang, R.; Vilcinskas, A.; Mylonakis, E. Synergistic efficacy of Aedes aegypti antimicrobial peptide cecropin A2 and tetracycline against Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [Green Version]
- Martin, G.E.; Boudreau, R.M.; Couch, C.; Becker, K.A.; Edwards, M.J.; Caldwell, C.C.; Gulbins, E.; Seitz, A. Sphingosine’s role in epithelial host defense: A natural antimicrobial and novel therapeutic. Biochimie 2017, 141, 91–96. [Google Scholar] [CrossRef]
- Sedaghati, M.; Ezzatpanah, H.; Boojar, M.M.A.; Ebrahimi, M.T.; Kobarfard, F. Isolation and identification of some antibacterial peptides in the plasmin-digest of ββββ-casein. LWT Food Sci. Technol. 2016, 68, 217–225. [Google Scholar] [CrossRef]
- Harmouche, N.; Aisenbrey, C.; Porcelli, F.; Xia, Y.; Nelson, S.E.D.; Chen, X.; Raya, J.; Vermeer, L.; Aparicio, C.; Veglia, G.; et al. Solution and solid-state nuclear magnetic resonance structural investigations of the antimicrobial designer peptide GL13K in membranes. Biochemistry 2017, 56, 4269–4278. [Google Scholar] [CrossRef] [PubMed]
- Speck-Planche, A.; Kleandrova, V.V.; Ruso, J.M.; DS Cordeiro, M.N. First multitarget chemo-Bioinformatic model to enable the discovery of antibacterial peptides against multiple gram-positive pathogens. J. Chem. Inf. Model. 2016, 56, 588–598. [Google Scholar] [CrossRef] [PubMed]
- Bayer, A.; Lammel, J.; Tohidnezhad, M.; Lippross, S.; Behrendt, P.; Klüter, T.; Pufe, T.; Cremer, J.; Jahr, H.; Rademacher, F.; et al. The antimicrobial peptide human beta-defensin-3 is induced by platelet-released growth factors in primary keratinocytes. Mediat. Inflamm. 2017, 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juretić, D.; Vukičević, D.; Tossi, A. Tools for designing amphipathic helical antimicrobial peptides. In Antimicrobial Peptides; Springer: Cham, Switzerland, 2017; pp. 23–34. [Google Scholar]
- Seyfi, R.; Kahaki, F.A.; Ebrahimi, T.; Montazersaheb, S.; Eyvazi, S.; Babaeipour, V.; Tarhriz, V. Antimicrobial peptides (AMPs): Roles, functions and mechanism of action. Int. J. Pept. Res. Ther. 2019, 1451–1463. [Google Scholar] [CrossRef]
- Wang, C.-K.; Shih, L.-Y.; Chang, K.Y. Large-scale analysis of antimicrobial activities in relation to amphipathicity and charge reveals novel characterization of antimicrobial peptides. Molecules 2017, 22, 2037. [Google Scholar] [CrossRef] [Green Version]
- Chew, M.-F.; Poh, K.-S.; Poh, C.-L. Peptides as therapeutic agents for dengue virus. Int. J. Med. Sci. 2017, 14, 1342–1359. [Google Scholar] [CrossRef] [Green Version]
- Sadredinamin, M.; Mehrnejad, F.; Hosseini, P.; Doustdar, F. Antimicrobial Peptides (AMPs). Nov. Biomed. 2016, 4, 70–76. [Google Scholar]
- Da Mata, É.C.G.; Mourão, C.B.F.; Rangel, M.; Schwartz, E.F. Antiviral activity of animal venom peptides and related compounds. J. Venom. Anim. Toxins Incl. Trop. Dis. 2017, 23, 3. [Google Scholar] [CrossRef] [Green Version]
- Rautenbach, M.; Troskie, A.M.; Vosloo, J.A. Antifungal peptides: To be or not to be membrane active. Biochimie 2016, 130, 132–145. [Google Scholar] [CrossRef]
- Faruck, M.O.; Yusof, F.; Chowdhury, S. An overview of antifungal peptides derived from insect. Peptides 2016, 80, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Muhialdin, B.J.; Hassan, Z.; Bakar, F.A.; Saari, N. Identification of antifungal peptides produced by Lactobacillus plantarum IS10 grown in the MRS broth. Food Control 2016, 59, 27–30. [Google Scholar] [CrossRef]
- Mor, A. Multifunctional host defense peptides: Antiparasitic activities. FEBS J. 2009, 276, 6474–6482. [Google Scholar] [CrossRef] [PubMed]
- Lacerda, A.F.; Pelegrini, P.B.; de Oliveira, D.M.; Vasconcelos, É.A.; Grossi-de-Sá, M.F. Anti-parasitic Peptides from Arthropods and their Application in Drug Therapy. Front. Microbiol. 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Pretzel, J.; Mohring, F.; Rahlfs, S.; Becker, K. Antiparasitic Peptides. In Advances in Biochemical Engineering/Biotechnology; Springer: Berlin/Heidelberg, Germany, 2013; pp. 157–192. [Google Scholar]
- Peptide Library Design Guide. Available online: https://www.genscript.com/peptide-library-design-guide.html (accessed on 3 November 2020).
- Peptide Libraries—ProteoGenix. Available online: https://www.proteogenix.science/custom-peptide-synthesis/peptide-libraries/ (accessed on 3 November 2020).
- Bozovičar, K.; Bratkovič, T. Evolving a Peptide: Library Platforms and Diversification Strategies. Int. J. Mol. Sci. 2020, 21, 215. [Google Scholar] [CrossRef] [Green Version]
- Russo, A.; Scognamiglio, P.L.; Enriquez, R.P.H.; Santambrogio, C.; Grandori, R.; Marasco, D.; Giordano, A.; Scoles, G.; Fortuna, S. In Silico Generation of Peptides by Replica Exchange Monte Carlo: Docking-Based Optimization of Maltose-Binding-Protein Ligands. PLoS ONE 2015, 10, e133571. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Chaudhary, K.; Chauhan, J.S.; Nagpal, G.; Kumar, R.; Sharma, M.; Raghava, G.P.S. An in-silico platform for predicting screening and designing of antihypertensive peptides. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [Green Version]
- Stokes, J.M.; Yang, K.; Swanson, K.; Jin, W.; Cubillos-Ruiz, A.; Donghia, N.M.; MacNair, C.R.; French, S.; Carfrae, L.A.; Bloom-Ackermann, Z.; et al. A Deep Learning Approach to Antibiotic Discovery. Cell 2020, 180, 688–702. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Quan, Z.; Wang, Z.-J.; Huang, H.; Zeng, X. A novel molecular representation with BiGRU neural networks for learning atom. Brief. Bioinform. 2019. [Google Scholar] [CrossRef]
- Zohora, F.T.; Rahman, M.Z.; Tran, N.H.; Xin, L.; Shan, B.; Li, M. DeepIso: A Deep Learning Model for Peptide Feature Detection from LC-MS map. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef]
- Wu, C.; Gao, R.; Zhang, Y.; Marinis, Y.D. PTPD: Predicting therapeutic peptides by deep learning and word2vec. BMC Bioinform. 2019, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veltri, D.; Kamath, U.; Shehu, A. Deep learning improves antimicrobial peptide recognition. Bioinformatics 2018, 34, 2740–2747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, H.-C.; You, Z.-H.; Zhou, X.; Cheng, L.; Li, X.; Jiang, T.-H.; Chen, Z.-H. ACP-DL: A Deep Learning Long Short-Term Memory Model to Predict Anticancer Peptides Using High-Efficiency Feature Representation. Mol. Ther. Nucleic Acids 2019, 17, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, S.; Moran, M.F.; Ma, B. Prediction of LC-MS/MS Properties of Peptides from Sequence by Deep Learning. Mol. Cell. Proteom. 2019, 18, 2099–2107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armenteros, J.J.A.; Salvatore, M.; Emanuelsson, O.; Winther, O.; von Heijne, G.; Elofsson, A.; Nielsen, H. Detecting sequence signals in targeting peptides using deep learning. Life Sci. Alliance 2019, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, J.; Bhadra, P.; Li, A.; Sethiya, P.; Qin, L.; Tai, H.K.; Wong, K.H.; Siu, S.W.I. Deep-AmPEP30: Improve Short Antimicrobial Peptides Prediction with Deep Learning. Mol. Ther. Nucleic Acids 2020, 20, 882–894. [Google Scholar] [CrossRef]
- Goswami, S. Impact of Data Quality on Deep Neural Network Training. Available online: https://arxiv.org/abs/2002.03732 (accessed on 3 November 2020).
- Boopathi, V.; Subramaniyam, S.; Malik, A.; Lee, G.; Manavalan, B.; Yang, D.-C. mACPpred: A Support Vector Machine-Based Meta-Predictor for Identification of Anticancer Peptides. Int. J. Mol. Sci. 2019, 20, 1964. [Google Scholar] [CrossRef] [Green Version]
- Giguère, S.; Laviolette, F.; Marchand, M.; Tremblay, D.; Moineau, S.; Liang, X.; Biron, É.; Corbeil, J. Machine Learning Assisted Design of Highly Active Peptides for Drug Discovery. PLoS Comput. Biol. 2015, 11, e1004074. [Google Scholar] [CrossRef]
- Li, C.; Sutherland, D.; Hammond, S.A.; Yang, C.; Taho, F.; Bergman, L.; Houston, S.; Warren, R.L.; Wong, T.; Hoang, L.M.N.; et al. AMPlify: Attentive deep learning model for discovery of novel antimicrobial peptides effective against WHO priority pathogens. bioRxiv 2020. [Google Scholar] [CrossRef]
- Zeng, W.-F.; Zhou, X.-X.; Zhou, W.-J.; Chi, H.; Zhan, J.; He, S.-M. MS/MS Spectrum Prediction for Modified Peptides Using pDeep2 Trained by Transfer Learning. Anal. Chem. 2019, 91, 9724–9731. [Google Scholar] [CrossRef]
- Mikolov, T.; Sutskever, I.; Chen, K.; Corrado, G.S.; Dean, J. Distributed Representations of Words and Phrases and their Compositionality. In Advances in Neural Information Processing Systems (NIPS 2013); Burges, C.J.C., Bottou, L., Welling, M., Eds.; Curran Associates Inc: Red Hook, NY, USA, 2013. [Google Scholar]
- Hamid, M.-N.; Friedberg, I. Identifying antimicrobial peptides using word embedding with deep recurrent neural networks. Bioinformatics 2018, 35, 2009–2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wardah, W.; Dehzangi, A.; Taherzadeh, G.; Rashid, M.A.; Khan, M.G.M.; Tsunoda, T.; Sharma, A. Predicting protein-peptide binding sites with a deep convolutional neural network. J. Theor. Biol. 2020, 496, 110278. [Google Scholar] [CrossRef] [PubMed]
- Schneider, P.; Müller, A.T.; Gabernet, G.; Button, A.L.; Posselt, G.; Wessler, S.; Hiss, J.A.; Schneider, G. Hybrid Network Model for Deep Learning of Chemical Data: Application to Antimicrobial Peptides. Mol. Inform. 2017, 36. [Google Scholar] [CrossRef] [PubMed]
- Sepp Hochreiter, J.S. Long short-term memory. Neural Comput. 1997, 9. [Google Scholar] [CrossRef]
- Nagarajan, D.; Nagarajan, T.; Roy, N.; Kulkarni, O.; Ravichandran, S.; Mishra, M.; Chakravortty, D.; Chandra, N. Computational antimicrobial peptide design and evaluation against multidrug-resistant clinical isolates of bacteria. J. Biol. Chem. 2017, 293, 3492–3509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grisoni, F.; Neuhaus, C.S.; Gabernet, G.; Müller, A.T.; Hiss, J.A.; Schneider, G. Designing Anticancer Peptides by Constructive Machine Learning. ChemMedChem 2018, 13, 1300–1302. [Google Scholar] [CrossRef]
- Cho, K.; Van Merriënboer, B.; Gulcehre, C.; Bahdanau, D.; Bougares, F.; Schwenk, H.; Bengio, Y. Learning Phrase Representations using RNN Encoder–Decoder for Statistical Machine Translation. In Proceedings of the Conference on Empirical Methods in Natural Language Processing (ENMLP); Moschitti, A., Pang, B., Daelemans, W., Eds.; Association for Computational Linguistics: Stroudsburg, PA, USA, 2014. [Google Scholar]
- Puentes, P.R.; Valderrama, N.; González, C.; Daza, L.; Muñoz-Camargo, C.; Cruz, J.C.; Arbeláez, P. PharmaNet: Pharmaceutical discovery with deep recurrent neural networks. bioRxiv 2020. [Google Scholar] [CrossRef]
- Chen, E.Y.; Xu, H.; Gordonov, S.; Lim, M.P.; Perkins, M.H.; Ma’ayan, A. Expression2Kinases: mRNA profiling linked to multiple upstream regulatory layers. Bioinformatics 2012, 28, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Chicco, D.; Jurman, G. The advantages of the Matthews correlation coefficient (MCC) over F1 score and accuracy in binary classification evaluation. BMC Genom. 2020, 21. [Google Scholar] [CrossRef] [Green Version]
- Burdukiewicz, M.; Sidorczuk, K.; Rafacz, D.; Pietluch, F.; Chilimoniuk, J.; Rödiger, S.; Gagat, P. Proteomic Screening for Prediction and Design of Antimicrobial Peptides with AmpGram. Int. J. Mol. Sci. 2020, 21, 4310. [Google Scholar] [CrossRef]
- Chung, C.R.; Kuo, T.R.; Wu, L.C.; Lee, T.Y.; Horng, J.T. Characterization and identification of antimicrobial peptides with different functional activities. Brief. Bioinform. 2019, 21, 1098–1114. [Google Scholar] [CrossRef] [PubMed]
- Barman, R.; Deshpande, S.; Agarwal, S.; Inamdar, U.; Devare, M.; Patil, A. Transfer Learning for Small Dataset. In Proceedings of the National Conference on Machine Learning, Mumbai, India, 26 March 2019; ResearchGate: Berlin, Germany, 2019. [Google Scholar]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef] [PubMed]
- Thwala, L.N.; Préat, V.; Csaba, N.S. Emerging delivery platforms for mucosal administration of biopharmaceuticals: A critical update on nasal, pulmonary and oral routes. Expert Opin. Drug Deliv. 2017, 14, 23–36. [Google Scholar] [CrossRef] [PubMed]
- GenScript. Random Library. Available online: https://www.genscript.com/random_library.html (accessed on 3 November 2020).
- Han, L.; Zhao, Y.; Cui, S.; Liang, B. Redesigning of microbial cell surface and its application to whole-cell biocatalysis and biosensors. Appl. Biochem. Biotechnol. 2018, 185, 396–418. [Google Scholar] [CrossRef] [PubMed]
- Bawazer, L.A. From DNA to genetically evolved technology. MRS Bull. 2013, 38, 509. [Google Scholar] [CrossRef]
- Sioud, M. Phage Display Libraries: From binders to targeted drug delivery and human therapeutics. Mol. Biotechnol. 2019, 61, 286–303. [Google Scholar] [CrossRef]
- Aghebati-Maleki, L.; Bakhshinejad, B.; Baradaran, B.; Motallebnezhad, M.; Aghebati-Maleki, A.; Nickho, H.; Yousefi, M.; Majidi, J. Phage display as a promising approach for vaccine development. J. Biomed. Sci. 2016, 23, 66. [Google Scholar] [CrossRef] [Green Version]
- Mimmi, S.; Maisano, D.; Quinto, I.; Iaccino, E. Phage display: An overview in context to drug discovery. Trends Pharmacol. Sci. 2019, 40, 87–91. [Google Scholar] [CrossRef]
- Principi, N.; Silvestri, E.; Esposito, S. Advantages and limitations of bacteriophages for the treatment of bacterial infections. Front. Pharmacol. 2019, 10, 513. [Google Scholar] [CrossRef] [Green Version]
- Tao, P.; Zhu, J.; Mahalingam, M.; Batra, H.; Rao, V.B. Bacteriophage T4 nanoparticles for vaccine delivery against infectious diseases. Adv. Drug Deliv. Rev. 2019, 145, 57–72. [Google Scholar] [CrossRef]
- Domingo-Calap, P.; Delgado-Martínez, J. Bacteriophages: Protagonists of a Post-Antibiotic Era. Antibiotics 2018, 7, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandenheuvel, D.; Lavigne, R.; Brüssow, H. Bacteriophage therapy: Advances in formulation strategies and human clinical trials. Annu. Rev. Virol. 2015, 2, 599–618. [Google Scholar] [CrossRef]
- Nemudraya, A.A.; Richter, V.A.; Kuligina, E.V. Phage peptide libraries as a source of targeted ligands. Acta Nat. 2016, 8, 48–57. [Google Scholar] [CrossRef]
- Nie, D.; Hu, Y.; Chen, Z.; Li, M.; Hou, Z.; Luo, X.; Mao, X.; Xue, X. Outer membrane protein A (OmpA) as a potential therapeutic target for Acinetobacter baumannii infection. J. Biomed. Sci. 2020, 27, 26. [Google Scholar] [CrossRef] [Green Version]
- Chaturvedi, D.; Mahalakshmi, R. Folding determinants of transmembrane ββββ-barrels using engineered OMP chimeras. Biochemistry 2018, 57, 1987–1996. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, D.; Mahalakshmi, R. Transmembrane β-barrels: Evolution, folding and energetics. Biochim. Biophys. Acta Biomembr. 2017, 1859, 2467–2482. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Wang, K.; Chi, X.; Zhou, L.; Li, J.; Liu, L.; Zheng, Q.; Wang, Y.; Yu, H.; Gu, Y.; et al. Construction of a bacterial surface display system based on outer membrane protein F. Microb. Cell Fact. 2019, 18, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Angelini, A.; Chen, T.F.; de Picciotto, S.; Yang, N.J.; Tzeng, A.; Santos, M.S.; Van Deventer, J.A.; Traxlmayr, M.W.; Wittrup, K.D. Protein engineering and selection using yeast surface display. In Yeast Surface Display; Springer: Cham, Switzerland, 2015; pp. 3–36. [Google Scholar]
- Cherf, G.M.; Cochran, J.R. Applications of yeast surface display for protein engineering. In Yeast Surface Display; Springer: Cham, Switzerland, 2015; pp. 155–175. [Google Scholar]
- Linciano, S.; Pluda, S.; Bacchin, A.; Angelini, A. Molecular evolution of peptides by yeast surface display technology. MedChemComm 2019, 10, 1569–1580. [Google Scholar] [CrossRef] [PubMed]
- Ueda, M. Establishment of cell surface engineering and its development. Biosci. Biotechnol. Biochem. 2016, 80, 1243–1253. [Google Scholar] [CrossRef] [Green Version]
- Boder, E.T.; Wittrup, K.D. Yeast surface display for screening combinatorial polypeptide libraries. Nat. Biotechnol. 1997, 15, 553–557. [Google Scholar] [CrossRef]
- Wu, C.-H.; Liu, I.-J.; Lu, R.-M.; Wu, H.-C. Advancement and applications of peptide phage display technology in biomedical science. J. Biomed. Sci. 2016, 23, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohrabi, C.; Foster, A.; Tavassoli, A. Methods for generating and screening libraries of genetically encoded cyclic peptides in drug discovery. Nat. Rev. Chem. 2020, 1–12. [Google Scholar] [CrossRef]
- Hussain, S.; Joo, J.; Kang, J.; Kim, B.; Braun, G.B.; She, Z.-G.; Kim, D.; Mann, A.P.; Mölder, T.; Teesalu, T.; et al. Antibiotic-loaded nanoparticles targeted to the site of infection enhance antibacterial efficacy. Nat. Biomed. Eng. 2018, 2, 95–103. [Google Scholar] [CrossRef]
- Tucker, A.T.; Leonard, S.P.; DuBois, C.D.; Knauf, G.A.; Cunningham, A.L.; Wilke, C.O.; Trent, M.S.; Davies, B.W. Discovery of next-generation antimicrobials through bacterial self-screening of surface-displayed peptide libraries. Cell 2018, 172, 618–628. [Google Scholar] [CrossRef]
- Rodrigues, C.J.C.; Sanches, J.M.; Carvalho, C.C.C.R. de Determining transaminase activity in bacterial libraries by time-lapse imaging. Chem. Commun. 2019, 55, 13538–13541. [Google Scholar] [CrossRef]
- Hosokawa, M.; Hoshino, Y.; Nishikawa, Y.; Hirose, T.; Yoon, D.H.; Mori, T.; Sekiguchi, T.; Shoji, S.; Takeyama, H. Droplet-based microfluidics for high-throughput screening of a metagenomic library for isolation of microbial enzymes. Biosens. Bioelectron. 2015, 67, 379–385. [Google Scholar] [CrossRef] [Green Version]
- Colin, P.-Y.; Kintses, B.; Gielen, F.; Miton, C.M.; Fischer, G.; Mohamed, M.F.; Hyvönen, M.; Morgavi, D.P.; Janssen, D.B.; Hollfelder, F. Ultrahigh-throughput discovery of promiscuous enzymes by picodroplet functional metagenomics. Nat. Commun. 2015, 6, 1–12. [Google Scholar] [CrossRef]
- Romero, P.A.; Tran, T.M.; Abate, A.R. Dissecting enzyme function with microfluidic-based deep mutational scanning. Proc. Natl. Acad. Sci. USA 2015, 112, 7159–7164. [Google Scholar] [CrossRef] [Green Version]
- Beneyton, T.; Thomas, S.; Griffiths, A.D.; Nicaud, J.-M.; Drevelle, A.; Rossignol, T. Droplet-based microfluidic high-throughput screening of heterologous enzymes secreted by the yeast Yarrowia lipolytica. Microb. Cell Fact. 2017, 16. [Google Scholar] [CrossRef] [Green Version]
- Elmezayen, A.D.; Al-Obaidi, A.; Şahin, A.T.; Yelekçi, K. Drug repurposing for coronavirus (COVID-19): In silico screening of known drugs against coronavirus 3CL hydrolase and protease enzymes. J. Biomol. Struct. Dyn. 2020, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Rognan, D. The impact of in silico screening in the discovery of novel and safer drug candidates. Pharmacol. Ther. 2017, 175, 47–66. [Google Scholar] [CrossRef] [PubMed]
- Saw, P.E.; Song, E.-W. Phage display screening of therapeutic peptide for cancer targeting and therapy. Protein Cell 2019, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dzwinel, W.; Kitowski, J.; Mościński, J. Checker Board Periodic Boundary Conditions in Molecular Dynamics Codes. Mol. Simul. 1991, 7, 171–179. [Google Scholar] [CrossRef]
- Tieleman, D.P.; Berendsen, H.J.C. Molecular dynamics simulations of a fully hydrated dipalmitoylphosphatidylcholine bilayer with different macroscopic boundary conditions and parameters. J. Chem. Phys. 1996, 105, 4871–4880. [Google Scholar] [CrossRef] [Green Version]
- Sajadi, F.; Rowley, C.N. Simulations of lipid bilayers using the CHARMM36 force field with the TIP3P-FB and TIP4P-FB water models. PeerJ 2018, 6, e5472. [Google Scholar] [CrossRef]
- Gajula, M.N.V.; Kumar, A.; Ijaq, J. Protocol for Molecular Dynamics Simulations of Proteins. Bio Protocol 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Cao, Z.; Bian, Y.; Hu, G.; Wang, J.; Zhou, Y. Molecular Dynamics Simulations of Human Antimicrobial Peptide LL-37 in Model POPC and POPG Lipid Bilayers. Int. J. Mol. Sci. 2018, 19, 1186. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Schlamadinger, D.E.; Kim, J.E.; McCammon, J.A. Comparative molecular dynamics simulations of the antimicrobial peptide CM15 in model lipid bilayers. Biochim. Biophys. Acta Biomembr. 2012, 1818, 1402–1409. [Google Scholar] [CrossRef] [Green Version]
- Catte, A.; Wilson, M.R.; Walker, M.; Oganesyan, V.S. Antimicrobial action of the cationic peptide, chrysophsin-3: A coarse-grained molecular dynamics study. Soft Matter 2018, 14, 2796–2807. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- UniProt Consortium. A worldwide hub of protein knowledge. Nucleic Acids Res. 2018, 47, D506–D515. [Google Scholar]
- Morshedian, A.; Razmara, J.; Lotfi, S. A novel approach for protein structure prediction based on an estimation of distribution algorithm. Soft Comput. 2018, 23, 4777–4788. [Google Scholar] [CrossRef]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protocols 2010, 5, 725–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling prediction and analysis. Nat. Protocols 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorn, M.; e Silva, M.B.; Buriol, L.S.; Lamb, L.C. Three-dimensional protein structure prediction: Methods and computational strategies. Comput. Biol. Chem. 2014, 53, 251–276. [Google Scholar] [CrossRef]
- Appelt, C.; Eisenmenger, F.; Kühne, R.; Schmieder, P.; Söderhäll, J.A. Interaction of the Antimicrobial Peptide Cyclo (RRWWRF) with Membranes by Molecular Dynamics Simulations. Biophys. J. 2005, 89, 2296–2306. [Google Scholar] [CrossRef] [Green Version]
- González, M.A. Force fields and molecular dynamics simulations. Ec. Thémat. Soc. Fr. Neutron. 2011, 12, 169–200. [Google Scholar] [CrossRef]
- Ewald, P.P. Die Berechnung optischer und elektrostatischer Gitterpotentiale. Ann. Phys. 1921, 369, 253–287. [Google Scholar] [CrossRef] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Kawata, M.; Mikami, M. Rapid calculation of two-dimensional Ewald summation. Chem. Phys. Lett. 2001, 340, 157–164. [Google Scholar] [CrossRef]
- Slattery, W.L.; Doolen, G.D.; DeWitt, H.E. Improved equation of state for the classical one-component plasma. Phys. Rev. A 1980, 21, 2087–2095. [Google Scholar] [CrossRef]
- Monticelli, L.; Tieleman, D.P. Force Fields for Classical Molecular Dynamics. In Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2012; pp. 197–213. [Google Scholar]
- Chen, Y.; Chen, S. Application Research of the Gears Predictor-Corrector Algorithms in a Molecular Dynamics Simulation to the EXP-6 Potential Function of Liquid Helium. In 2014 International Conference on Mechatronics Electronic, Industrial and Control Engineering; Atlantis Press: Paris, France, 2014. [Google Scholar]
- Jefferies, D.; Khalid, S. Molecular Simulations of Complex Membrane Models. In Modeling of Microscale Transport in Biological Processes; Elsevier: Amsterdam, The Netherlands, 2017; pp. 1–18. [Google Scholar]
- Yesylevskyy, S.O.; Schäfer, L.V.; Sengupta, D.; Marrink, S.J. Polarizable Water Model for the Coarse-Grained MARTINI Force Field. PLoS Comput. Biol. 2010, 6. [Google Scholar] [CrossRef] [Green Version]
- Izadi, S.; Anandakrishnan, R.; Onufriev, A.V. Building Water Models: A Different Approach. J. Phys. Chem. Lett. 2014, 5, 3863–3871. [Google Scholar] [CrossRef] [Green Version]
- Lippert, R.A.; Predescu, C.; Ierardi, D.J.; Mackenzie, K.M.; Eastwood, M.P.; Dror, R.O.; Shaw, D.E. Accurate and efficient integration for molecular dynamics simulations at constant temperature and pressure. J. Chem. Phys. 2013, 139. [Google Scholar] [CrossRef]
- Tobias, D.J.; Martyna, G.J.; Klein, M.L. Molecular dynamics simulations of a protein in the canonical ensemble. J. Phys. Chem. 1993, 97, 12959–12966. [Google Scholar] [CrossRef]
- Andersen, H.C. Molecular dynamics simulations at constant pressure and/or temperature. J. Chem. Phys. 1980, 72, 2384–2393. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef] [Green Version]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [Green Version]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Grest, G.S.; Kremer, K. Molecular dynamics simulation for polymers in the presence of a heat bath. Phys. Rev. A 1986, 33, 3628–3631. [Google Scholar] [CrossRef]
- Moradi, S.; Nowroozi, A.; Shahlaei, M. Shedding light on the structural properties of lipid bilayers using molecular dynamics simulation: A review study. RSC Adv. 2019, 9, 4644–4658. [Google Scholar] [CrossRef] [Green Version]
- Shahane, G.; Ding, W.; Palaiokostas, M.; Azevedo, H.S.; Orsi, M. Interaction of Antimicrobial Lipopeptides with Bacterial Lipid Bilayers. J. Membr. Biol. 2019, 252, 317–329. [Google Scholar] [CrossRef] [Green Version]
- Neale, C.; Hsu, J.C.Y.; Yip, C.M.; Pomès, R. Indolicidin Binding Induces Thinning of a Lipid Bilayer. Biophys. J. 2014, 106, L29–L31. [Google Scholar] [CrossRef] [Green Version]
- Wagle, S.; Georgiev, V.N.; Robinson, T.; Dimova, R.; Lipowsky, R.; Grafmüller, A. Interaction of SNARE Mimetic Peptides with Lipid bilayers: Effects of Secondary Structure Bilayer Composition and Lipid Anchoring. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef]
- Woo, S.Y.; Lee, H. All-atom simulations and free-energy calculations of coiled-coil peptides with lipid bilayers: Binding strength structural transition, and effect on lipid dynamics. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- Xing, C.; Faller, R. Density imbalances and free energy of lipid transfer in supported lipid bilayers. J. Chem. Phys. 2009, 131. [Google Scholar] [CrossRef]
- Payne, E.M.; Holland-Moritz, D.A.; Sun, S.; Kennedy, R.T. High-throughput screening by droplet microfluidics: Perspective into key challenges and future prospects. Lab Chip 2020, 20, 2247–2262. [Google Scholar] [CrossRef]
- Du, G.; Fang, Q.; Toonder, J.M.J. den Microfluidics for cell-based high throughput screening platforms—A review. Anal. Chim. Acta 2016, 903, 36–50. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Shen, J. Review of membranes in microfluidics. J. Chem. Technol. Biotechnol. 2016, 92, 271–282. [Google Scholar] [CrossRef]
- Guo, M.T.; Rotem, A.; Heyman, J.A.; Weitz, D.A. Droplet microfluidics for high-throughput biological assays. Lab Chip 2012, 12, 2146–2155. [Google Scholar] [CrossRef]
- Griffiths, A.D.; Tawfik, D.S. Miniaturising the laboratory in emulsion droplets. Trends Biotechnol. 2006, 24, 395–402. [Google Scholar] [CrossRef]
- Thorsen, T.; Roberts, R.W.; Arnold, F.H.; Quake, S.R. Dynamic Pattern Formation in a Vesicle-Generating Microfluidic Device. Phys. Rev. Lett. 2001, 86, 4163–4166. [Google Scholar] [CrossRef] [Green Version]
- Wong, I.; Ho, C.-M. Surface molecular property modifications for poly(dimethylsiloxane) (PDMS) based microfluidic devices. Microfluid. Nanofluid. 2009, 7. [Google Scholar] [CrossRef] [Green Version]
- Pellegrino, M.; Sciambi, A.; Treusch, S.; Durruthy-Durruthy, R.; Gokhale, K.; Jacob, J.; Chen, T.X.; Geis, J.A.; Oldham, W.; Matthews, J.; et al. High-throughput single-cell DNA sequencing of acute myeloid leukemia tumors with droplet microfluidics. Genome Res. 2018, 28, 1345–1352. [Google Scholar] [CrossRef] [Green Version]
- Köster, S.; Angilè, F.E.; Duan, H.; Agresti, J.J.; Wintner, A.; Schmitz, C.; Rowat, A.C.; Merten, C.A.; Pisignano, D.; Griffiths, A.D.; et al. Drop-based microfluidic devices for encapsulation of single cells. Lab Chip 2008, 8, 1110–1115. [Google Scholar] [CrossRef]
- Chaipan, C.; Pryszlak, A.; Dean, H.; Poignard, P.; Benes, V.; Griffiths, A.D.; Merten, C.A. Single-Virus Droplet Microfluidics for High-Throughput Screening of Neutralizing Epitopes on HIV Particles. Cell Chem. Biol. 2017, 24, 751–757. [Google Scholar] [CrossRef] [Green Version]
- Azizi, M.; Zaferani, M.; Cheong, S.H.; Abbaspourrad, A. Pathogenic Bacteria Detection Using RNA-Based Loop-Mediated Isothermal-Amplification-Assisted Nucleic Acid Amplification via Droplet Microfluidics. ACS Sens. 2019, 4, 841–848. [Google Scholar] [CrossRef]
- Kaushik, A.M.; Hsieh, K.; Chen, L.; Shin, D.J.; Liao, J.C.; Wang, T.-H. Accelerating bacterial growth detection and antimicrobial susceptibility assessment in integrated picoliter droplet platform. Biosens. Bioelectron. 2017, 97, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Abate, A.R.; Hung, T.; Sperling, R.A.; Mary, P.; Rotem, A.; Agresti, J.J.; Weiner, M.A.; Weitz, D.A. DNA sequence analysis with droplet-based microfluidics. Lab Chip 2013, 13, 4864–4869. [Google Scholar] [CrossRef] [Green Version]
- Mesbah, K.; Thai, R.; Bregant, S.; Malloggi, F. DMF-MALDI: Droplet based microfluidic combined to MALDI-TOF for focused peptide detection. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Safa, N.; Vaithiyanathan, M.; Sombolestani, S.; Charles, S.; Melvin, A.T. Population-based analysis of cell-penetrating peptide uptake using a microfluidic droplet trapping array. Anal. Bioanal. Chem. 2019, 411, 2729–2741. [Google Scholar] [CrossRef]
- Yaginuma, K.; Aoki, W.; Miura, N.; Ohtani, Y.; Aburaya, S.; Kogawa, M.; Nishikawa, Y.; Hosokawa, M.; Takeyama, H.; Ueda, M. High-throughput identification of peptide agonists against GPCRs by co-culture of mammalian reporter cells and peptide-secreting yeast cells using droplet microfluidics. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [Green Version]
- Sjostrom, S.L.; Bai, Y.; Huang, M.; Liu, Z.; Nielsen, J.; Joensson, H.N.; Svahn, H.A. High-throughput screening for industrial enzyme production hosts by droplet microfluidics. Lab Chip 2014, 14, 806–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheen, J. Signal Transduction in Maize and Arabidopsis Mesophyll Protoplasts. Plant Physiol. 2001, 127, 1466–1475. [Google Scholar] [CrossRef] [PubMed]
- Best, R.J.; Lyczakowski, J.J.; Abalde-Cela, S.; Yu, Z.; Abell, C.; Smith, A.G. Label-Free Analysis and Sorting of Microalgae and Cyanobacteria in Microdroplets by Intrinsic Chlorophyll Fluorescence for the Identification of Fast Growing Strains. Anal. Chem. 2016, 88, 10445–10451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murchie, E.H.; Lawson, T. Chlorophyll fluorescence analysis: A guide to good practice and understanding some new applications. J. Exp. Bot. 2013, 64, 3983–3998. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.; Boehm, C.R.; Hibberd, J.M.; Abell, C.; Haseloff, J.; Burgess, S.J.; Reyna-Llorens, I. Droplet-based microfluidic analysis and screening of single plant cells. PLoS ONE 2018, 13, e196810. [Google Scholar] [CrossRef] [Green Version]
- Schaich, M.; Cama, J.; Nahas, K.A.; Sobota, D.; Sleath, H.; Jahnke, K.; Deshpande, S.; Dekker, C.; Keyser, U.F. An Integrated Microfluidic Platform for Quantifying Drug Permeation across Biomimetic Vesicle Membranes. Mol. Pharm. 2019, 16, 2494–2501. [Google Scholar] [CrossRef]
- Joshi, S.; Hussain, M.T.; Roces, C.B.; Anderluzzi, G.; Kastner, E.; Salmaso, S.; Kirby, D.J.; Perrie, Y. Microfluidics based manufacture of liposomes simultaneously entrapping hydrophilic and lipophilic drugs. Int. J. Pharm. 2016, 514, 160–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nahas, K.A.; Cama, J.; Schaich, M.; Hammond, K.; Deshpande, S.; Dekker, C.; Ryadnov, M.G.; Keyser, U.F. A microfluidic platform for the characterisation of membrane active antimicrobials. Lab Chip 2019, 19, 837–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funakoshi, K.; Suzuki, H.; Takeuchi, S. Lipid Bilayer Formation by Contacting Monolayers in a Microfluidic Device for Membrane Protein Analysis. Anal. Chem. 2006, 78, 8169–8174. [Google Scholar] [CrossRef]
- Zagnoni, M.; Sandison, M.E.; Morgan, H. Microfluidic array platform for simultaneous lipid bilayer membrane formation. Biosens. Bioelectron. 2009, 24, 1235–1240. [Google Scholar] [CrossRef] [PubMed]
- Hall, K.; Aguilar, M.I. Surface Plasmon Resonance Spectroscopy for Studying the Membrane Binding of Antimicrobial Peptides. In Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2010; pp. 213–223. [Google Scholar]
- Šakanovič, A.; Hodnik, V.; Anderluh, G. Surface Plasmon Resonance for Measuring Interactions of Proteins with Lipids and Lipid Membranes. In Methods in Molecular Biology; Springer: New York, NY, USA, 2019; pp. 53–70. [Google Scholar]
- Lam, K.S.; Renil, M. From combinatorial chemistry to chemical microarray. Curr. Opin. Chem. Biol. 2002, 6, 353–358. [Google Scholar] [CrossRef]
- Lam, K.S.; Salmon, S.E.; Hersh, E.M.; Hruby, V.J.; Kazmierski, W.M.; Knapp, R.J. A new type of synthetic peptide library for identifying ligand-binding activity. Nature 1991, 354, 82–84. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wei, Z.; Zhang, D.; Ma, H.; Wang, Z.; Bu, X.; Li, M.; Geng, L.; Lausted, C.; Hood, L.; et al. Rapid Screening of Peptide Probes through In Situ Single-Bead Sequencing Microarray. Anal. Chem. 2014, 86, 11854–11859. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Wang, X.; Song, A.; Bao, T.; Lam, K.S. Development and Applications of Topologically Segregated Bilayer Beads in One-bead One-compound Combinatorial Libraries. QSAR Comb. Sci. 2005, 24, 1127–1140. [Google Scholar] [CrossRef]
- Li, J.; Carney, R.P.; Liu, R.; Fan, J.; Zhao, S.; Chen, Y.; Lam, K.S.; Pan, T. Microfluidic Print-to-Synthesis Platform for Efficient Preparation and Screening of Combinatorial Peptide Microarrays. Anal. Chem. 2018, 90, 5833–5840. [Google Scholar] [CrossRef]
- Li, J.; Zhao, S.; Yang, G.; Liu, R.; Xiao, W.; Disano, P.; Lam, K.S.; Pan, T. Combinatorial Peptide Microarray Synthesis Based on Microfluidic Impact Printing. ACS Comb. Sci. 2018, 21, 6–10. [Google Scholar] [CrossRef] [Green Version]
- Churski, K.; Kaminski, T.S.; Jakiela, S.; Kamysz, W.; Baranska-Rybak, W.; Weibel, D.B.; Garstecki, P. Rapid screening of antibiotic toxicity in an automated microdroplet system. Lab Chip 2012, 12, 1629–1637. [Google Scholar] [CrossRef]
- Cao, J.; Kürsten, D.; Schneider, S.; Knauer, A.; Günther, P.M.; Köhler, J.M. Uncovering toxicological complexity by multi-dimensional screenings in microsegmented flow: Modulation of antibiotic interference by nanoparticles. Lab Chip 2012, 12, 474–484. [Google Scholar] [CrossRef]
- MacConnell, A.B.; Price, A.K.; Paegel, B.M. An Integrated Microfluidic Processor for DNA-Encoded Combinatorial Library Functional Screening. ACS Comb. Sci. 2017, 19, 181–192. [Google Scholar] [CrossRef]
- Du, G.-S.; Pan, J.-Z.; Zhao, S.-P.; Zhu, Y.; den Toonder, J.M.J.; Fang, Q. Cell-Based Drug Combination Screening with a Microfluidic Droplet Array System. Anal. Chem. 2013, 85, 6740–6747. [Google Scholar] [CrossRef]
- Mee, R.P.; Auton, T.R.; Morgan, P.J. Design of active analogues of a 15-residue peptide using D-optimal design, QSAR and a combinatorial search algorithm. J. Pept. Res. 2009, 49, 89–102. [Google Scholar] [CrossRef]
- Ma, R.; Wong, S.W.; Ge, L.; Shaw, C.; Siu, S.W.; Kwok, H.F. In Vitro and MD Simulation Study to Explore Physicochemical Parameters for Antibacterial Peptide to Become Potent Anticancer Peptide. Mol. Ther. Oncol. 2020, 16, 7–19. [Google Scholar] [CrossRef] [Green Version]
- Velasco-Bolom, J.L.; Corzo, G.; Garduño-Juárez, R. Molecular dynamics simulation of the membrane binding and disruption mechanisms by antimicrobial scorpion venom-derived peptides. J. Biomol. Struct. Dyn. 2017, 36, 2070–2084. [Google Scholar] [CrossRef]
- Lyu, Y.; Xiang, N.; Zhu, X.; Narsimhan, G. Potential of mean force for insertion of antimicrobial peptide melittin into a pore in mixed DOPC/DOPG lipid bilayer by molecular dynamics simulation. J. Chem. Phys. 2017, 146. [Google Scholar] [CrossRef]
- Fields, F.R.; Freed, S.D.; Carothers, K.E.; Hamid, M.N.; Hammers, D.E.; Ross, J.N.; Kalwajtys, V.R.; Gonzalez, A.J.; Hildreth, A.D.; Friedberg, I.; et al. Novel antimicrobial peptide discovery using machine learning and biophysical selection of minimal bacteriocin domains. Drug Dev. Res. 2019, 81, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Luo, Q.; Wu, J. Label-free discrimination of membrane-translocating peptides on porous silicon microfluidic biosensors. Biomicrofluidics 2016, 10. [Google Scholar] [CrossRef] [Green Version]
- Bao, P.; Paterson, D.A.; Harrison, P.L.; Miller, K.; Peyman, S.; Jones, J.C.; Gleeson, H.F. Lipid coated liquid crystal droplets for the on-chip detection of antimicrobial peptides. Lab Chip 2019, 19, 1082–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gotanda, M.; Kamiya, K.; Osaki, T.; Fujii, S.; Misawa, N.; Miki, N.; Takeuchi, S. Sequential generation of asymmetric lipid vesicles using a pulsed-jetting method in rotational wells. Sens. Actuators B Chem. 2018, 261, 392–397. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Amino Acid Sequence | Biological Activity | Method |
|---|---|---|---|
| CAMEL0 [243] | KWKLFKKIGAVLKVL | Antimicrobial | MD |
| CAMEL17 [243] | KWNLNGNINAVLKVL | Antimicrobial | MD |
| LL-37 [175] | LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES | Antimicrobial | MD |
| AcrAP1 [244] | FLFSLIPHAISGLISAFK | Antimicrobial | MD |
| Chrys-3 [177] | FIGLLISAGKAIHDLIRRRH | Antimicrobial | MD |
| Pin2 [245] | FWGALAKGALKLIPSLFSSFSKKD | Antimicrobial | MD |
| Melittin [246] | GIGAVLKVLTTGLPALISWIKRKRQQ | Antimicrobial | MD |
| Putative Bacteriocin 3 [247] | IKKIGKKAAKKVIVKAIQAI | Antimicrobial | DL |
| Putative Bacteriocin 4 [247] | KKIGKKAAKKVIVKAIQAIV | Antimicrobial | DL |
| RaCa-1 [116] | GLLDIIKTTGKDFAVKILDNLKCKLAGGCPP | Antimicrobial | DL |
| RaCa-2 [116] | FFPIIARLAAKVIPSLVCAVTKKC | Antimicrobial | DL |
| RaCa-3 [116] | GLWETIKTTGKSIALNLLDKIKCKIAGGCPP | Antimicrobial | DL |
| RaCa-7 [116] | FFPRVLPLANKFLPTIYCALPKSVGN | Antimicrobial | DL |
| Cecropin A [248] | KWKLFKKIEKVGQNIRDGIIKAGPAVAVVGQATQIAK-NH2 | Translocating/Antimicrobial | MF |
| Cecropin B [228] | KWKVFKKIEKMGRNIRNGIVKAGPAIAVLGEAKAL-NH2 | Antimicrobial | MF |
| Smp43 [249] | GVWDWIKKTAGKIWNSEPVKALKSQALNAAKNFVAEKIGATPS | Antimicrobial | MF |
| Cinnamycin [250] | CRQSCSFGPFTFVCDGNTK | Antimicrobial | MF |
| RWRWR [218] | Ac-RWVRVpGO(FAM)WIRQ-NH2 | Traslocating | MF |
| OWRWR [218] | Ac-OWVRVpGO(FAM)WIRQ-NH2 | Traslocating | MF |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Puentes, P.R.; Henao, M.C.; Torres, C.E.; Gómez, S.C.; Gómez, L.A.; Burgos, J.C.; Arbeláez, P.; Osma, J.F.; Muñoz-Camargo, C.; Reyes, L.H.; et al. Design, Screening, and Testing of Non-Rational Peptide Libraries with Antimicrobial Activity: In Silico and Experimental Approaches. Antibiotics 2020, 9, 854. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics9120854
Puentes PR, Henao MC, Torres CE, Gómez SC, Gómez LA, Burgos JC, Arbeláez P, Osma JF, Muñoz-Camargo C, Reyes LH, et al. Design, Screening, and Testing of Non-Rational Peptide Libraries with Antimicrobial Activity: In Silico and Experimental Approaches. Antibiotics. 2020; 9(12):854. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics9120854
Chicago/Turabian StylePuentes, Paola Ruiz, María C. Henao, Carlos E. Torres, Saúl C. Gómez, Laura A. Gómez, Juan C. Burgos, Pablo Arbeláez, Johann F. Osma, Carolina Muñoz-Camargo, Luis H. Reyes, and et al. 2020. "Design, Screening, and Testing of Non-Rational Peptide Libraries with Antimicrobial Activity: In Silico and Experimental Approaches" Antibiotics 9, no. 12: 854. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics9120854