Ib-M6 Antimicrobial Peptide: Antibacterial Activity against Clinical Isolates of Escherichia coli and Molecular Docking

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Antimicrobial Activity
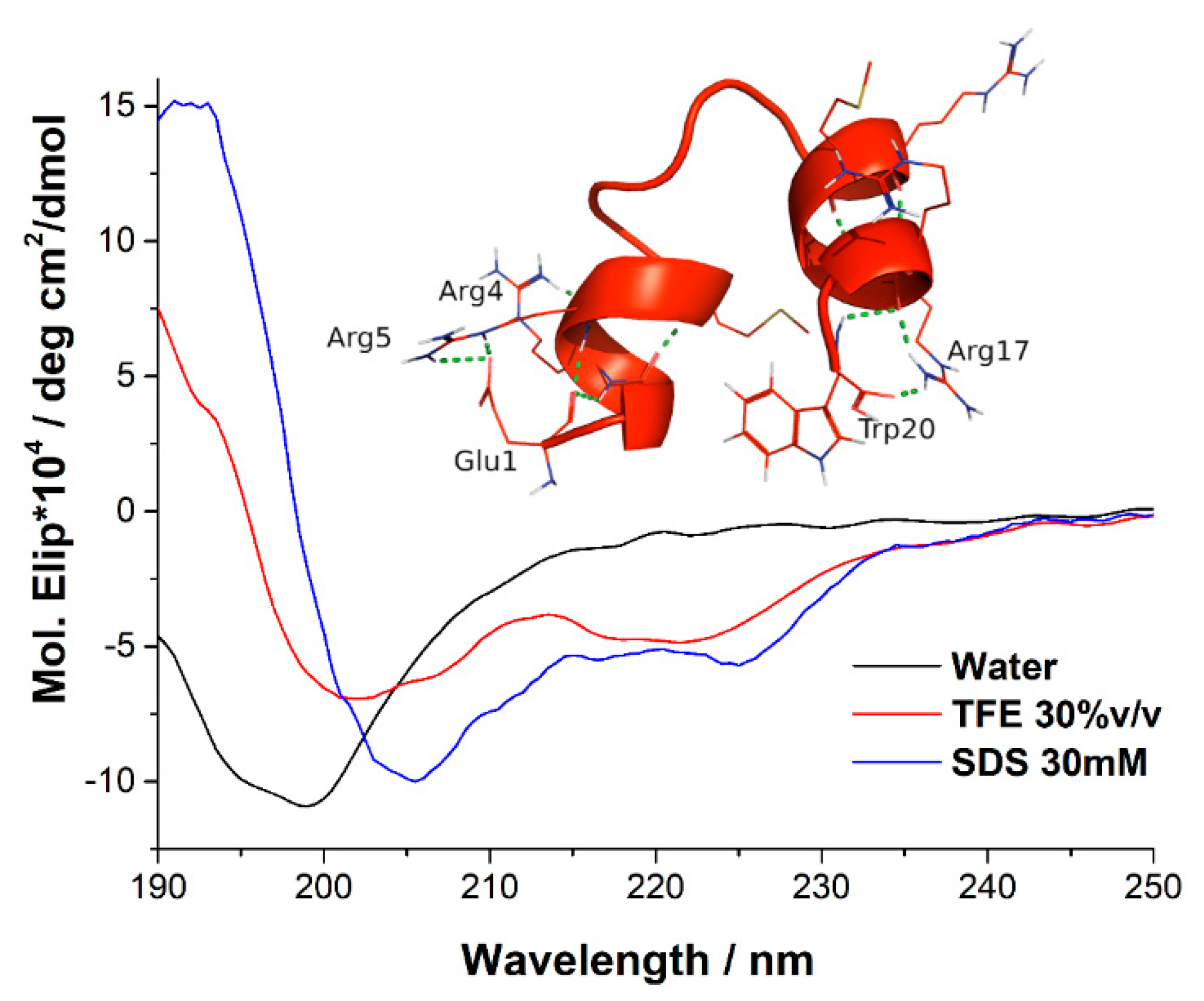
2.2. Circular Dicrhroism and 3D Structure
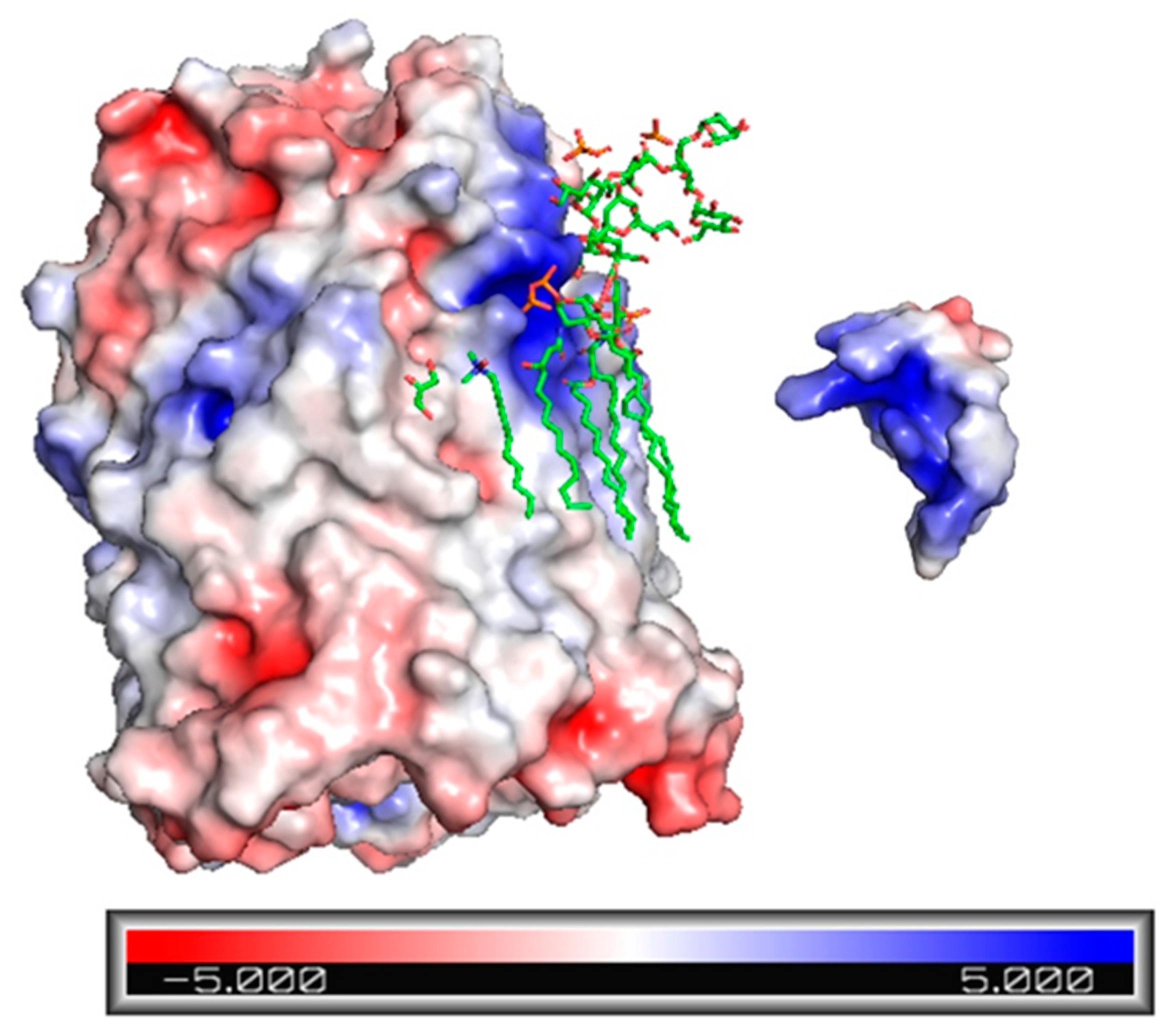
2.3. Electrostatic Potential Map of Ib-M6 Peptide and FhuA Protein
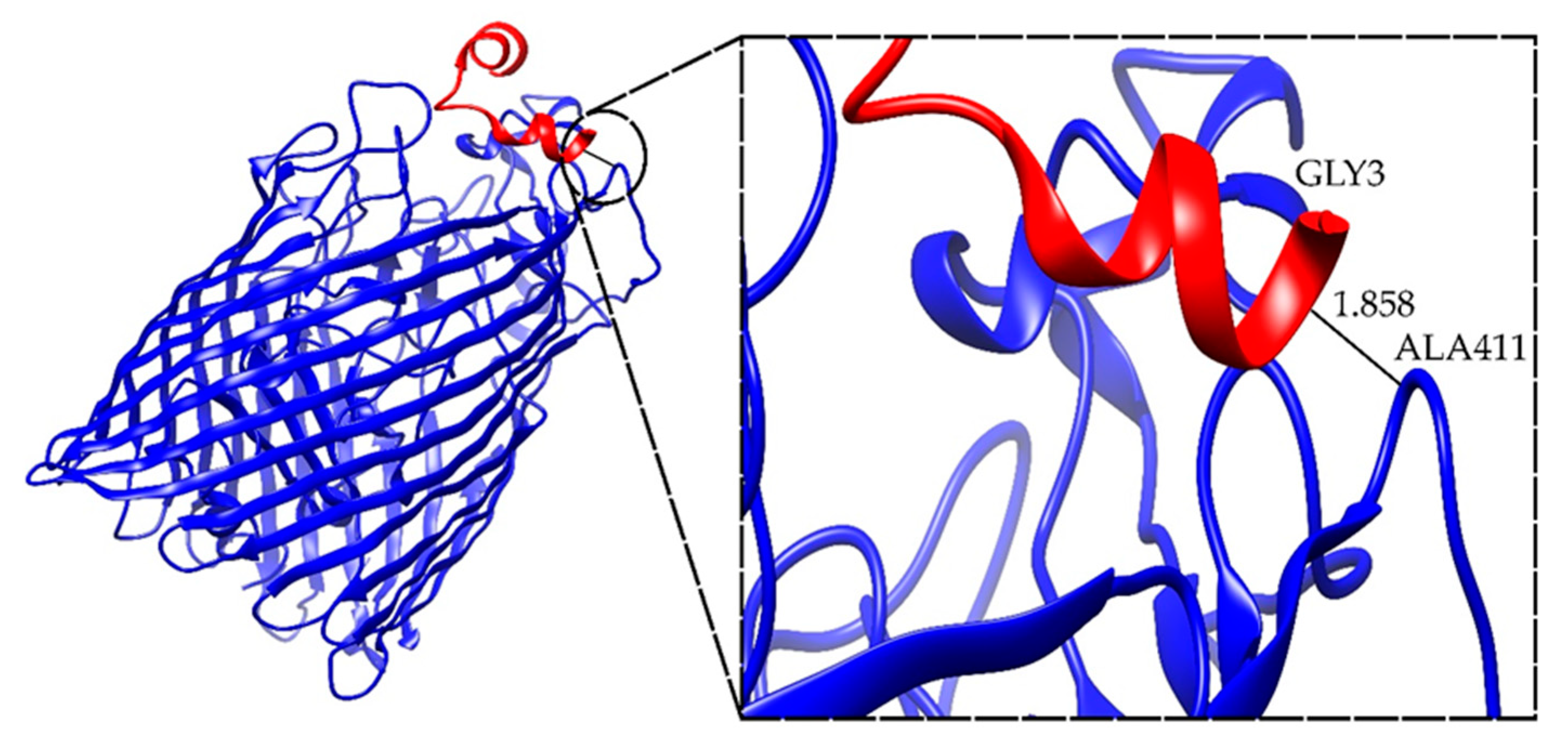
2.4. Docking Results for the Ib-M6 Peptide in Complex with FhuA Protein
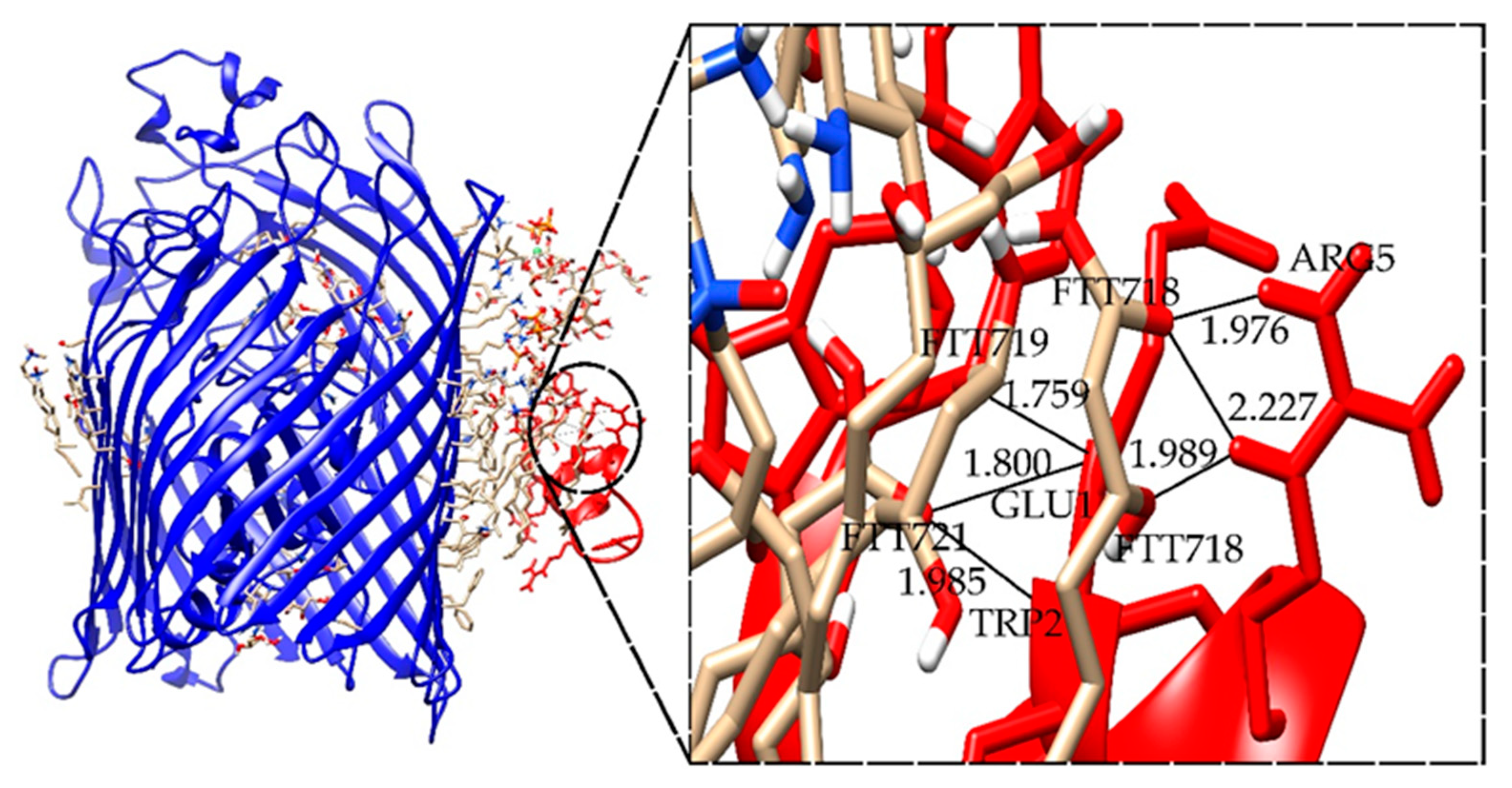
2.5. Docking Results for the Ib-M6 Peptide Interacting with Lipopolysaccharide–FhuA Complex
3. Discussion
4. Materials and Methods
4.1. Antimicrobial Agents and Bacterial Strains
4.2. Antimicrobial Activity of Ib-M6 Peptide
4.3. Circular Dichroism and Structure 3D of Peptide
4.4. Preparation of Ligand and Targets Molecules
4.5. Docking Procedure for the Ib-M6 Peptide in Complex with FhuA Protein
4.6. Docking Procedure for the Ib-M6 Peptide in Complex with Lipopolysaccharide–FhuA
4.7. Analysis of the Resulting Potential Complexes
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- O’Neill, J. Antimicrobial Resistance: Tackling a crisis for the health and wealth of nations. Rev. Antimicrob. Resist. 2014, 20, 1–16. [Google Scholar]
- UK Parliament. Antimicrobial Resistance; Health and Social Care Committee: House of Commons; UK Parliament: London, UK, 2018. [Google Scholar]
- La Organización Mundial de la Salud (OMS). Estrategia Mundial DE La OMS Para Contener La Resistencia a Los Antimicrobianos; La Organización Mundial de la Salud OMS: Geneva, Switzerland, 2001. [Google Scholar]
- WHO. Global Antimicrobial Resistance Surveillance System (GLASS) Report; Early Implementation 2016–2017; WHO: Geneva, Switzerland, 2017. [Google Scholar]
- Kaper, J.B. Pathogenic Escherichia coli. Int. J. Med. Microbiol. 2005, 295, 355–356. [Google Scholar] [CrossRef]
- Zhang, L.J.; Gallo, R.L. Antimicrobial peptides. Curr. Biol. 2016, 26, R1–R21. [Google Scholar] [CrossRef]
- Arnusch, C.J.; Pieters, R.J.; Breukink, E. Enhanced membrane pore formation through high-affinity targeted antimicrobial peptides. PLoS ONE 2012, 7, e39768. [Google Scholar] [CrossRef]
- Almaaytah, A.; Qaoud, M.T.; Mohammed, G.K.; Abualhaijaa, A.; Knappe, D.; Hoffmann, R.; Al-Balas, Q. Antimicrobial and antibiofilm activity of UP-5, an ultrashort antimicrobial peptide designed using only arginine and biphenylalanine. Pharmaceuticals 2018, 11, 3. [Google Scholar] [CrossRef] [Green Version]
- Travkova, O.G.; Moehwald, H.; Brezesinski, G. The interaction of antimicrobial peptides with membranes. Adv. Colloid Interface Sci. 2017, 247, 521–532. [Google Scholar] [CrossRef]
- Téllez, G.A.; Castaño, J.C. Péptidos antimicrobianos. Infectio 2010, 14, 55–67. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.T.; Haney, E.F.; Vogel, H.J. The expanding scope of antimicrobial peptide structures and their modes of action. Trends Biotechnol. 2011, 29, 464–472. [Google Scholar] [CrossRef]
- Lai, P.K.; Kaznessis, Y.N. Free energy calculations of microcin J25 variants binding to the FhuA receptor. J. Chem. theory Comput. 2017, 13, 3413–3423. [Google Scholar] [CrossRef]
- Farahmandian, N.; Sifipour, M.; Sefid, F. Article Structure Engineering of Fhua as a Vaccine. IIOAB J. 2016, 7, 352–358. [Google Scholar]
- Flórez-Castillo, J.M.; Perullini, M.; Jobbágy, M.; Jesús Cano Calle, H. Enhancing Antibacterial Activity Against Escherichia coli K-12 of Peptide Ib-AMP4 with Synthetic Analogues. Int. J. Pept. Res. Ther. 2014, 20, 365–369. [Google Scholar] [CrossRef]
- Balhara, V.; Schmidt, R.; Gorr, S.U.; Dewolf, C. Membrane selectivity and biophysical studies of the antimicrobial peptide GL13K. Biochim. Biophys. Acta Biomembr. 2013, 1828, 2193–2203. [Google Scholar] [CrossRef] [Green Version]
- Ashby, M.; Petkova, A.; Hilpert, K. Cationic antimicrobial peptides as potential new therapeutic agents in neonates and children: A review. Curr. Opin. Infect. Dis. 2014, 27, 258–267. [Google Scholar] [CrossRef]
- Raetz, C.R.H.; Reynolds, C.M.; Trent, M.S.; Bishop, R.E. Lipid A Modification Systems in Gram-Negative Bacteria. NIH Public Access 2007, 76, 295–329. [Google Scholar] [CrossRef] [Green Version]
- Chan, D.I.; Prenner, E.J.; Vogel, H.J. Tryptophan- and arginine-rich antimicrobial peptides: Structures and mechanisms of action. Biochim. Biophys. Acta Biomembr. 2006, 1758, 1184–1202. [Google Scholar] [CrossRef] [Green Version]
- Farfán-García, A.E.; Zhang, C.; Imdad, A.; Arias-Guerrero, M.Y.; Sánchez-Alvarez, N.T.; Shah, R.; Iqbal, J.; Tamborski, M.E.; Gómez-Duarte, O.G. Case-Control Pilot Study on Acute Diarrheal Disease in a Geographically Defined Pediatric Population in a Middle Income Country. Int. J. Pediatr. 2017. [Google Scholar] [CrossRef] [Green Version]
- CLSI Performance Standards for Antimicrobial Susceptibility Testing. Wayne PA Clin. Lab. Stand. Inst. 2019, 39, 1–285.
- Shen, Y.; Maupetit, J.; Derreumaux, P.; Tufféry, P. Improved PEP-FOLD approach for peptide and miniprotein structure prediction. J. Chem. Theory Comput. 2014, 10, 4745–4758. [Google Scholar] [CrossRef]
- Thévenet, P.; Shen, Y.; Maupetit, J.; Guyon, F.; Derreumaux, P.; Tufféry, P. PEP-FOLD: An updated de novo structure prediction server for both linear and disulfide bonded cyclic peptides. Nucleic Acids Res. 2012, 40, 288–293. [Google Scholar] [CrossRef] [Green Version]
- London, N.; Raveh, B.; Cohen, E.; Fathi, G.; Schueler-Furman, O. Rosetta FlexPepDock web server—High resolution modeling of peptide-protein interactions. Nucleic Acids Res. 2011, 39, W249–W253. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger, L. The PyMOL Molecular Graphics System, Version 1.8; Schrodinger LLC: New York, NY, USA, 2015. [Google Scholar]
- Alford, R.F.; Leaver-Fay, A.; Jeliazkov, J.R.; O’Meara, M.J.; DiMaio, F.P.; Park, H.; Shapovalov, M.V.; Renfrew, P.D.; Mulligan, V.K.; Kappel, K.; et al. The Rosetta All-Atom Energy Function for Macromolecular Modeling and Design. J. Chem. Theory Comput. 2017, 13, 3031–3048. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name Strain | MIC * (µM) | MBC * (µM) | |
|---|---|---|---|
| Ib-M6 | Streptomycin | ||
| EAEC_1 | 1.5 | 6.25 | 25 |
| EAEC_2 | 0.7 | 6.25 | 1.5 |
| ETEC_1 | 3.1 | 6.25 | 12.5 |
| ETEC_2 | 12.5 | 6.25 | > 100 |
| EPEC_1 | 3.1 | 6.25 | 6.25 |
| EPEC_2 | 1.5 | 100 | 3.1 |
| DAEC_1 | 6.25 | 200 | 12.5 |
| DAEC_2 | 3.1 | 50 | 6.25 |
| EIEC | 25 | 200 | 50 |
| STEC_1 | 12.5 | 50 | 25 |
| STEC_2 | 6.25 | 50 | 6.25 |
| Non-pathogenic_1 | 1.5 | 200 | 3.1 |
| Non-pathogenic_2 | 1.5 | 3.1 | 25 |
| Non-pathogenic_3 | 3.1 | 400 | 3.1 |
| Complex | I_sc (REU) |
|---|---|
| FhuA+M6_A | −8.928 |
| FhuA+M6_B | −7.407 |
| FhuA+M6_C | −8.334 |
| FhuA+M6_D | −7.099 |
| FhuA+M6_E | −10.305 |
| Complex | I_sc (REU) | Hydrogen Bonds | Length (Å) |
|---|---|---|---|
| FhuA+M6_A_1 | −42.545 | ALA411-GLY3 | 1.858 |
| FhuA+M6_A_2 | −41.622 | GLY548-ARG14 | 2.216 |
| FhuA+M6_A_3 | −41.466 | GLY548-ARG14 | 2.277 |
| FhuA+M6_B_1 | −33.700 | TYR307-GLY 8 | 1.849 |
| FhuA+M6_B_2 | −33.421 | TYR 307-GLY 8 | 2.616 |
| FhuA+M6_B_3 | −33.071 | TYR 307-GLY 8 | 2.363 |
| FhuA+M6_D_1 | −39.595 | PRO415-GLU1 | 1.843 |
| Complex | I_sc (REU) |
|---|---|
| LPS–FhuA+M6_A | −6.038 |
| LPS–FhuA+M6_B | −3.975 |
| LPS–FhuA+M6_C | −3.772 |
| LPS–FhuA+M6_D | −4.021 |
| LPS–FhuA+M6_E | −4.833 |
| Complex * | I_sc (REU) | Hydrogen Bonds ** | Length (Å) |
|---|---|---|---|
| LPS+M6_B_1 | −40.625 | FTT718-ARG5 | 1.976 |
| FTT718-ARG5 | 2.227 | ||
| FTT718-ARG5 | 1.989 | ||
| FTT719-GLU1 | 1.759 | ||
| FTT721-GLU1 | 1.800 | ||
| FTT721-TRP2 | 1.985 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flórez-Castillo, J.M.; Rondón-Villareal, P.; Ropero-Vega, J.L.; Mendoza-Espinel, S.Y.; Moreno-Amézquita, J.A.; Méndez-Jaimes, K.D.; Farfán-García, A.E.; Gómez-Rangel, S.Y.; Gómez-Duarte, O.G. Ib-M6 Antimicrobial Peptide: Antibacterial Activity against Clinical Isolates of Escherichia coli and Molecular Docking. Antibiotics 2020, 9, 79. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics9020079
Flórez-Castillo JM, Rondón-Villareal P, Ropero-Vega JL, Mendoza-Espinel SY, Moreno-Amézquita JA, Méndez-Jaimes KD, Farfán-García AE, Gómez-Rangel SY, Gómez-Duarte OG. Ib-M6 Antimicrobial Peptide: Antibacterial Activity against Clinical Isolates of Escherichia coli and Molecular Docking. Antibiotics. 2020; 9(2):79. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics9020079
Chicago/Turabian StyleFlórez-Castillo, J. M., P. Rondón-Villareal, J. L. Ropero-Vega, S. Y. Mendoza-Espinel, J. A. Moreno-Amézquita, K. D. Méndez-Jaimes, A. E. Farfán-García, S. Y. Gómez-Rangel, and Oscar Gilberto Gómez-Duarte. 2020. "Ib-M6 Antimicrobial Peptide: Antibacterial Activity against Clinical Isolates of Escherichia coli and Molecular Docking" Antibiotics 9, no. 2: 79. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics9020079