Silica gel matrix, with fluorescent indicator 254 nm, was used in analytical thin-layer chromatography (TLC on aluminium foils), and silica gel (particle size 40-63 µm, Merck) was used in flash chromatography on Biotage IsoleraTM One or Sepachrom Puriflash XS 420; visualizations were accomplished with UV light (λ 254 nm).
Synthesis
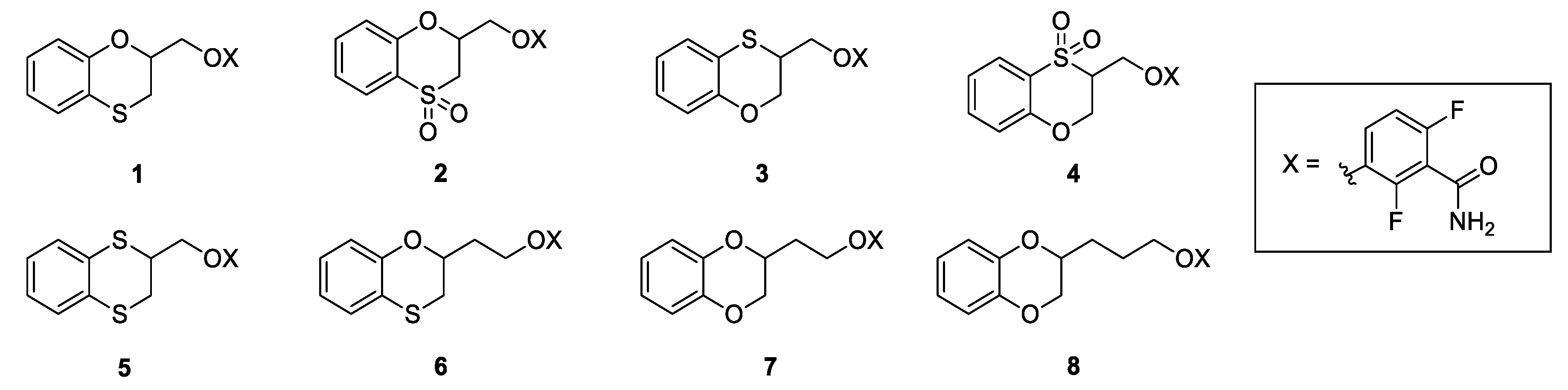
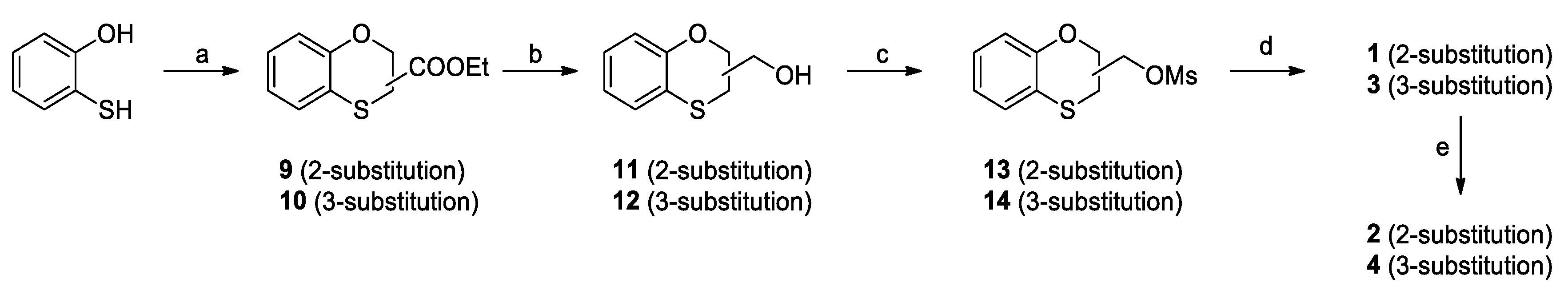
Ethyl 1,4-benzoxathian-2-carboxylate (9) and ethyl 1,4-benzoxathian-3-carboxylate (10): TEA (0.50 mL, 3.52 mmol) was added dropwise to a solution of ethyl-2,3-dibromopropionate (0.44 g, 1.76 mmol) into a mixture 1/1 of ACN and water (10 mL). After stirring at room temperature for 30 min, 1,2-mercaptophenol (0.22 g, 1.76 mmol) was slowly added. The reaction mixture was stirred at RT for 72 h, and then, it was diluted with diethyl ether and water. The organic phase was dried over Na2SO4, filtered and concentrated to give a yellow oil as the residue. The crude was purified by flash chromatography on silica gel, using cyclohexane/ethyl acetate 95/5 as the elution solvent; the yielded ratio between 9 and 10 was quantified by 1H NMR and resulted in 60% for 9 and of 40% for 10. Yield: 65%
Ethyl 1,4-benzoxathian-2-carboxylate (9). 1H NMR (300 MHz, CDCl3, δ): 7.04 (m, 2H), 6.96 (d, J = 7.7 Hz, 1H), 6.87 (t, J = 7.4 Hz, 1H), 4.99 (dd, J = 5.3, 3.8 Hz, 1H), 4.27 (q, J = 7.1 Hz, 2H), 3.29 (d, J = 3.8 Hz, 1H), 3.28 (d, J = 5.3 Hz, 1H), 1.28 ppm (t, J = 7.1 Hz, 3H).
Ethyl 1,4-benzoxathian-3-carboxylate (10). 1H NMR (300 MHz, CDCl3, δ): 7.06 (m, 1H), 7.00 (dd, J = 7.6, 2.0 Hz, 1H), 6.88 (t, J = 7.4 Hz, 2H), 4.56 (dd, J = 11.5, 3.0 Hz, 1H), 4.46 (dd, J = 11.5, 6.5 Hz, 1H), 4.25 (q, J = 7.1 Hz, 2H), 4.12 (dd, J = 6.5, 3.0 Hz, 1H), 1.30 ppm (t, J = 7.1 Hz, 3H).
2-hydroxymethyl-1,4-benzoxathiane (11) and 3-hydroxymethyl-1,4-benzoxathiane (12): LiAlH4 (85 mg, 2.23 mmol) was suspended in dry THF (5 mL) at 0 °C under nitrogen atmosphere. The mixture of 9 and 10 (0.5 g, 2.23 mmol) was dissolved in THF (5 mL) and was added to the reaction. The mixture was warmed to RT and stirred for 30 minutes; at completion, it was cooled to 0 °C and slowly quenched with ethyl Acetate (5 mL). Further ethyl acetate (10 mL) was added, the organic layer was washed with brine (3 × 10 mL), dried over Na2SO4 and concentrated under vacuum to give 0.38 g (81%) as a mixture of 60% 11 and 40% 12 as a colorless oil.
2-hydroxymethyl-1,4-benzoxathiane (11). 1H NMR (300 MHz, CDCl3, δ): 7.03 (m, 2H), 6.86 (m, 2H), 4.32 (m, 1H), 3.90 (dd, J = 11.7, 4.2 Hz, 1H), 3.84 (dd, J = 11.7, 5.6 Hz, 1H), 3.14 (dd, J = 13.0, 9.0 Hz, 1H), 2.97 ppm (dd, J = 13.0, 2.1 Hz, 1H).
3-hydroxymethyl-1,4-benzoxathiane (12). 1H NMR (300 MHz, CDCl3, δ): 7.02 (m, 2H), 6.86 (m, 2H), 4.50 (dd, J = 11.7, 4.2 Hz, 1H), 4.34 (dd, J = 11.7, 2.1 Hz, 1H), 3.88 (m, 2H), 3.42 ppm (ddd, J = 7.2, 4.2, 2.1 Hz, 1H).
2-mesyloxymethyl-1,4-benzoxathiane (13) and 3-mesyloxymethyl-1,4-benzoxathiane (14): Mesyl chloride (0.55 mL, 7.13 mmol) was added dropwise to a solution of 11 and 12 (1.0 g, 5.49 mmol) and TEA (1.0 mL, 7.13 mmol) in DCM (5 mL) at 0 °C. The mixture was stirred at room temperature for 3 h, diluted with DCM (20 mL), washed firstly with 10% aqueous NaHCO3 (10 mL), secondly with 10% aqueous HCl (10 mL) and finally with brine (10 mL), dried over Na2SO4, filtered and concentrated under vacuum to yield 1.58 g (80%) of 13 and 14 as an oil residue.
2-mesyloxymethyl-1,4-benzoxathiane (13). 1H NMR (300 MHz, CDCl3, δ): 7.04 (m, 2H), 6.88 (ddd, J = 14.2, 7.7, 1.2 Hz, 2H), 4.57 (m, 1H), 4.45 (m, 2H), 3.10 (s, 3H), 3.20–2.99 ppm (m, 2H).
3-mesyloxymethyl-1,4-benzoxathiane (14). 1H NMR (300 MHz, CDCl3, δ): 7.03 (m, 2H), 6.89 (m, 2H), 4.63 (ddd, J = 11.8, 3.2, 1.5 Hz, 1H), 4.42 (m, 2H), 4.24 (m, 1H), 3.59 (dddd, J = 9.5, 6.4, 3.2, 1.7 Hz, 1H), 3.07 ppm (s, 3H).
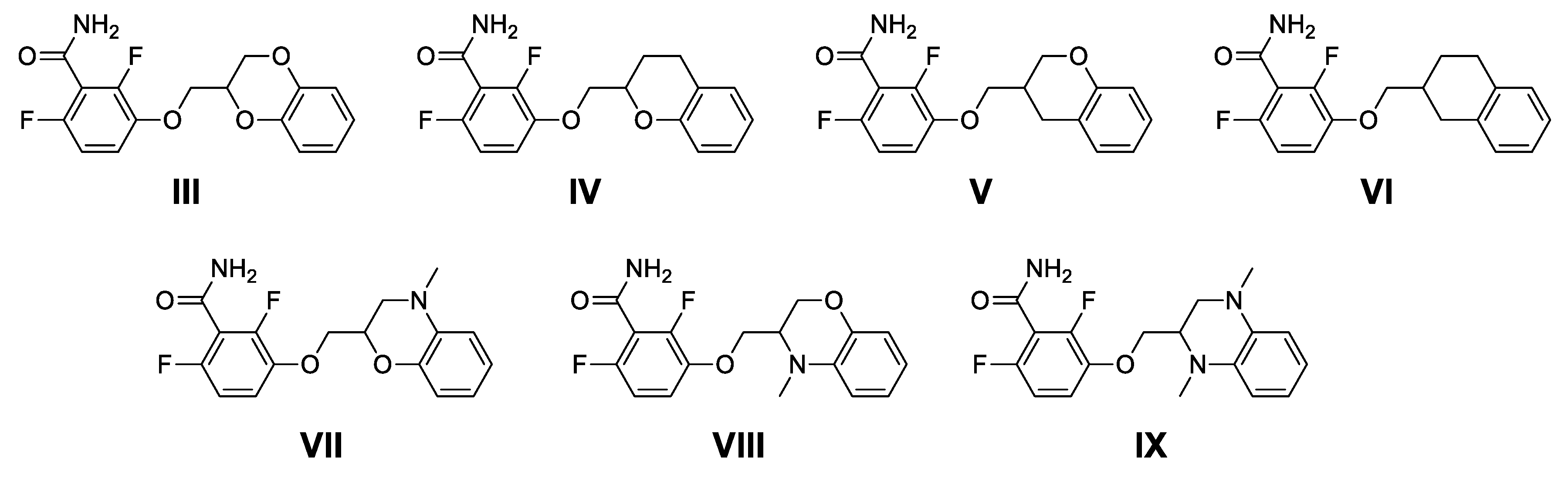
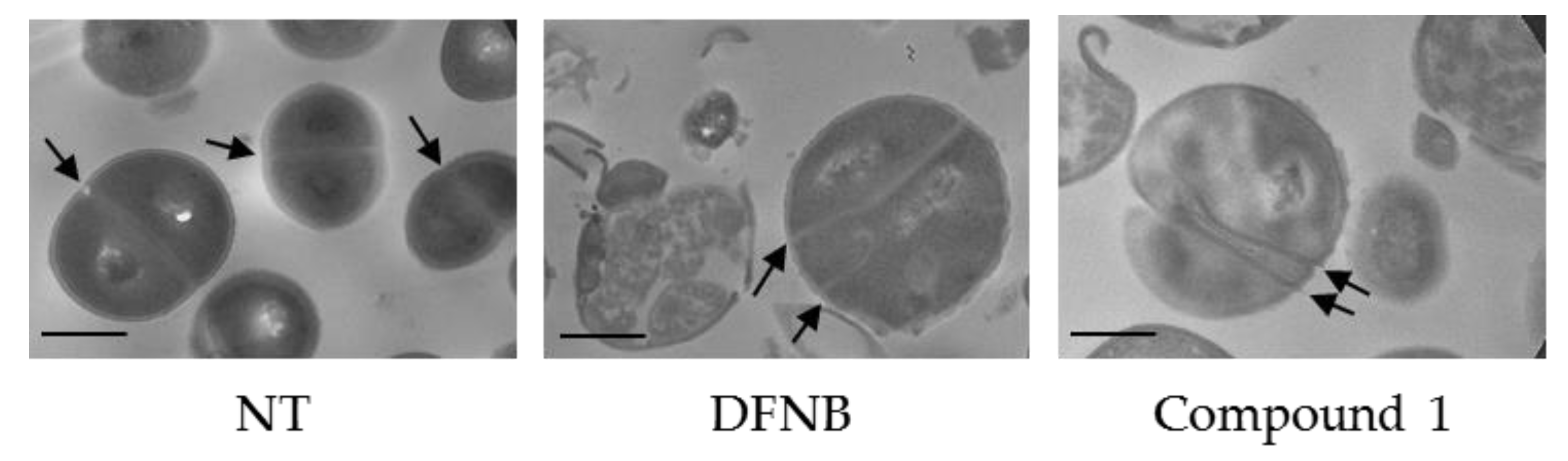
3-(1,4-benzoxathiane-2-yl)-2,6-difluorobenzamide (1) and 3-(1,4-benzoxathiane-3-yl)-2,6-difluorobenzamide (3): Potassium carbonate (0.42 g, 3.01 mmol) was added to a solution of 2,6-difluoro-3-hydroxybenzamide (0.50 g, 2.88 mmol) in dry DMF (2 mL). After stirring at room temperature for 30 min, a solution of 13 and 14 (0.71 g, 2.74 mmol) in DMF (2 mL) was added. The reaction mixture was stirred at 60 °C for 16 h, concentrated under vacuum, diluted with Ethyl Acetate (20 mL), washed with brine (3 × 10 mL), dried over Na2SO4, filtered and concentrated, to give a residue, which was purified by flash chromatography on silica gel. Elution with 9/1 Cyclohexane/Ethyl Acetate gave 1 (35%) and 3 (20%) as white solids. mp (1): 117 °C and mp (3): 109–112 °C.
3-(1,4-benzoxathiane-2-yl)-2,6-difluorobenzamide (1). Tr (HPLC): 13.3 min (A% = 98.0% at λ = 288 nm). 1H NMR (300 MHz, d6-DMSO, δ): 8.14 (bs, 1H), 7.85 (bs, 1H), 7.29 (m, 1H), 7.04 (m, 3H), 6.85 (m, 2H), 4.56 (m, 1H), 4.31 (m, 2H), 3.29 (dd, J = 14.1, 13.1 Hz, 1H), 3.15 ppm (dd, J = 13.1, 8.4 Hz, 1H). 13C NMR (75 MHz, d6-DMSO, δ): 161.7, 152.6 (dd, J = 240.5, 6.9 Hz), 151.2, 148.4 (dd, J = 247.4, 9.1 Hz), 143.2 (dd, J = 10.9, 2.8 Hz), 127.7, 126.2, 122.1, 118.8, 117.5, 117.1 (dd, J = 24.0, 20.6 Hz), 116.5 (d, J = 9.1 Hz), 111.5 (dd, J = 22.9, 3.4 Hz), 73.1, 71.2, 26.1 ppm.
3-(1,4-benzoxathiane-3-yl)-2,6-difluorobenzamide (3): Tr (HPLC): 13.5 min (A% = 95.1% at λ = 288 nm). 1H NMR (300 MHz, d6-DMSO, δ): 8.13 (bs, 1H), 7.85 (bs, 1H), 7.27 (m, 1H), 7.03 (m, 3H), 6.86 (m, 2H), 4.48 (dd, J = 11.8, 4.3 Hz, 1H), 4.24 (m, 3H), 3.88 ppm (m, 1H). 13C NMR (75 MHz, d6-DMSO, δ): 161.7, 152.6 (dd, J = 240.5, 6.8 Hz), 151.5, 148.3 (dd, J = 247.3, 8.0 Hz), 143.0 (dd, J = 10.9, 2.8 Hz), 127.7, 126.0, 122.5, 118.7, 117.3, 117.1 (dd, J = 24.6, 20.0 Hz), 116.4 (d, J = 9.2 Hz), 111.5 (dd, J = 22.9, 4.6 Hz), 69.3, 65.3, 37.4 ppm.
3-(1,4-benzoxathiane-4,4-dioxide-2-yl)-2,6-difluorobenzamide (2): m-CPBA (0.10 g, 0.59 mmol) was added to a cooled solution of 1 (0.10 g, 0.30 mmol) in acetone (5 mL) at 0 °C. The reaction mixture was warmed to RT and stirred for 2 h, then 10 mL of 10% aqueous NaHCO3 was added, followed by 15 mL of ethyl acetate. The organic phase was dried over Na2SO4, filtered, and concentrated to give an oily residue. The further treatment with DCM lets the precipitation of 0.03 g (28%) of 2 as a white solid. mp: 192 °C. Tr (HPLC): 10.1 min (A% = 95.9% at λ = 280 nm). 1H NMR (300 MHz, d6-DMSO, δ): 8.13 (bs, 1H), 7.85 (bs, 1H), 7.76 (dd, J = 8.0, 1.6 Hz, 1H), 7.57 (m, 1H), 7.31 (td, J = 9.3, 5.3 Hz, 1H), 7.20 (m, 1H), 7.14-7.06 (m, 2H), 5.10 (m, 1H), 4.49 (dd, J = 11.3, 3.2 Hz, 1H), 4.42 (dd, J = 11.3, 5.1 Hz, 1H), 4.03 (dd, J = 14.2, 1.6 Hz, 1H), 3.82 ppm (dd, J = 14.2, 12.0 Hz, 1H). 13C-NMR (75 MHz, d6-DMSO, δ): 162.0, 153.1, 152.7 (dd, J = 240.8, 6.7 Hz), 148.5 (dd, J = 247.5, 9.0 Hz), 142.9 (dd, J = 10.9, 3.4 Hz), 135.4, 125.7, 123.9, 122.9, 119.2, 117.1 (dd, J = 7.9, 1.9 Hz), 116.8 (dd, J = 24.7, 20.2 Hz), 111.7 (dd, J = 23.3, 3.7 Hz), 75.1, 71.0, 50.2 ppm.
3-(1,4-benzoxathiane-4,4-dioxide-3-yl)-2,6-difluorobenzamide (4): m-CPBA (0.06 g, 0.36 mmol) was added to a cooled solution of 2 (60 mg, 0.18 mmol) in acetone (5 mL) at 0 °C. The reaction mixture was warmed to RT and stirred for 2 h, then 10 mL of 10% aqueous NaHCO3 was added, followed by 15 mL of ethyl acetate. The organic phase was dried over Na2SO4, filtered, and concentrated to give an oily residue. The further treatment with DCM lets the precipitation of 0.03 g (28%) of 4 as a white solid. mp: 184 °C. Tr (HPLC): 10.3 min (A% = 97.9% at λ = 281 nm). 1H NMR (300 MHz, d6-DMSO, δ): 8.11 (bs, 1H), 7.82 (bs, 1H), 7.76 (dd, J = 8.0, 1.6 Hz, 1H), 7.56 (ddd, J = 8.9, 7.3, 1.6 Hz, 1H 1H), 7.31 (td, J = 9.3, 5.3 Hz, 1H), 7.20 (m, 1H), 7.12-6.99 (m, 2H), 4.87 (dd, J = 13.1, 2.0 Hz, 1H), 4.81 (dd, J = 13.1, 5.6 Hz, 1H), 4.53 (m, 1H), 4.44-4.27 ppm (m, 2H). 13C-NMR (75 MHz, d6-DMSO, δ): 161.5, 154.0, 152.8 (dd, J = 242.2, 6.7 Hz), 148.4 (dd, J = 249.8, 8.6 Hz), 142.7 (dd, J = 11.4, 3.4 Hz), 135.0, 126.0, 124.3, 122.8, 119.1, 117.1 (dd, J = 24.9, 19.9 Hz), 116.7 (d, J = 10.4 Hz), 111.5 (dd, J = 23.0, 4.0 Hz), 67.1, 64.3, 57.0 ppm.
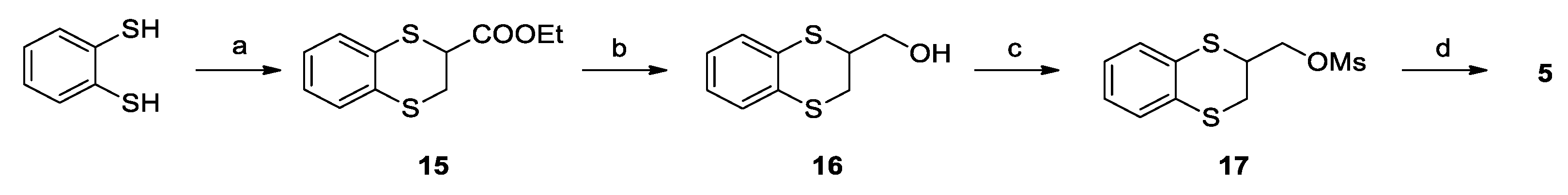
Ethyl 1,4-benzodithian-2-carboxylate (15): TEA (0.56 mL, 4.03 mmol) was added dropwise to a solution of ethyl-2,3-dibromopropionate (0.52 g, 2.01 mmol) in DMF (5 mL). After stirring at room temperature for 20 min; therefore, 1,2-benzenedithiol (0.26 g, 1.83 mmol) in 2 mL of DMF was slowly added. The reaction mixture was stirred at RT for 2 h, and then it was concentrated under vacuum and then diluted with 15 mL of ethyl acetate. The organic phase was washed with brine (3 × 10 mL), dried over Na2SO4, filtered, and concentrated to give 0.38 g (86%) of 15 as a yellow oil. 1H NMR (300 MHz, CDCl3, δ): 7.26 (m, 2H), 7.06 (m, 2H), 4.30 (t, J = 6.5 Hz, 1H), 4.23 (q, J = 7.2 Hz, 2H), 3.32 (d, J = 6.5 Hz, 2H), 1.28 ppm (t, J = 7.2 Hz, 3H).
2-hydroxymethyl-1,4-benzodithiane (16): LiAlH4 (60 mg, 1.58 mmol) was suspended in dry THF (2 mL) at 0 °C under nitrogen atmosphere. The solution of 15 (0.38 g, 1.58 mmol) in THF (5 mL) was slowly added to the reaction. The mixture was then warmed to RT and stirred for 1 h; at completion, it was cooled to 0 °C and slowly quenched with ethyl acetate (5 mL). Further ethyl acetate (10 mL) was added, the organic layer was washed with brine (3 × 10 mL), dried over Na2SO4 and concentrated under vacuum to give 0.26 g (84%) of 16 as a brown oil. 1H NMR (300 MHz, CDCl3, δ): 7.20 (m, 2H), 7.02 (m, 2H), 3.90 (dd, J = 11.1, 7.3 Hz, 1H), 3.81 (dd, J = 11.1, 6.0 Hz, 1H), 3.70 (m, 1H), 3.27 (dd, J = 13.4, 3.7 Hz, 1H), 3.10 ppm (dd, J = 13.4, 6.4 Hz, 1H).
2-mesyloxymethyl-1,4-benzodithiane (17): Mesyl chloride (0.14 mL, 1.76 mmol) was added dropwise to a solution of 16 (0.25 g, 1.26 mmol) and TEA (0.25 mL, 1.76 mmol) in DCM (4 mL) at 0 °C. The mixture was stirred at that temperature for 3 h, diluted with DCM (20 mL), washed firstly with 10% aqueous NaHCO3 (10 mL), secondly with 10% aqueous HCl (10 mL) and finally with brine (10 mL), dried over Na2SO4, filtered and concentrated under vacuum to yield 0.34 g (quantitative) of 17 as a pink oil. 1H NMR (300 MHz, CDCl3, δ): 7.18 (ddd, J = 9.4, 4.8, 2.3 Hz, 2H), 7.04 (m, 2H), 4.57 (t, J = 10.0 Hz, 1H), 4.40 (dd, J = 10.3, 5.1 Hz, 1H), 3.91 (m, 1H), 3.29 (dd, J = 14.0, 3.5 Hz, 1H), 3.19 (dd, J = 14.0, 5.1 Hz, 1H), 3.07 ppm (s, 3H).
3-(1,4-benzodithiane-2-yl)-2,6-difluorobenzamide (5): Potassium carbonate (0.19 g, 1.35 mmol) was added to a solution of 2,6-difluoro-3-hydroxybenzamide (0.22 g, 1.29 mmol) in dry DMF (2 mL). After stirring at room temperature for 30 min, a solution of 17 (0.34 g, 1.23 mmol) in DMF (2 mL) was added. The reaction mixture was stirred at 60 °C for 16 h, concentrated under vacuum, diluted with Ethyl Acetate (15 mL), washed with brine (3 × 10 mL), dried over Na2SO4, filtered and concentrated, to give a residue, which was purified by flash chromatography on silica gel. Elution with 6/4 Cyclohexane/Ethyl Acetate and further crystallization from chloroform gave 0.22 g (50%) of 5 as a white solid. mp: 134 °C. Tr (HPLC): 14.3 min (A% = 98.8% at λ = 268 nm). 1H NMR (300 MHz, d6-DMSO, δ): 8.14 (bs, 1H), 7.85 (bs, 1H), 7.26 (m, 1H), 7.21 (m, 2H), 7.05 (m, 3H), 4.35 (m, 1H), 4.21 (m, 1H), 4.09 (m, 1H), 3.30 (dd, J = 13.6, 3.0 Hz, 1H), 3.18 ppm (dd, J = 13.6, 6.1, 2.5 Hz, 1H). 13C NMR (75 MHz, d6-DMSO, δ): 161.6, 152.6 (dd, J = 240.4, 6.8 Hz), 148.4 (dd, J = 247.2, 8.3 Hz), 143.0 (dd, J = 10.8, 3.2 Hz), 131.5, 131.0, 129.5, 128.8, 126.4, 125.6, 117.1 (dd, J = 24.9, 20.4 Hz), 116.6 (dd, J = 9.3, 2.1 Hz), 111.5 (dd, J = 22.8, 3.9 Hz), 71.6, 41.6, 29.9 ppm.
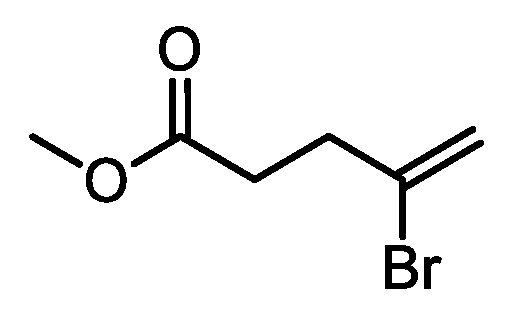
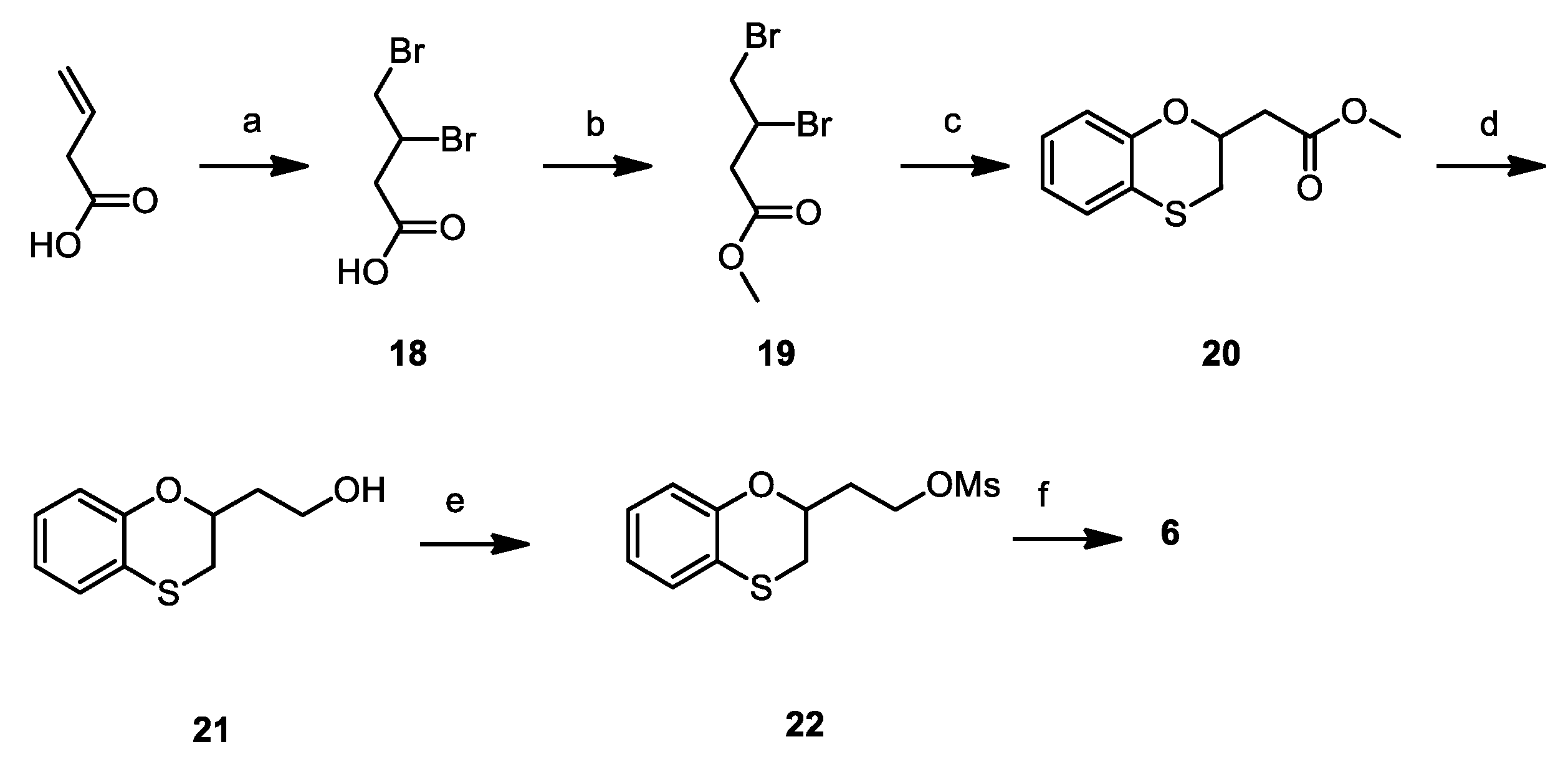
3,4-dibromobutyric acid (18): Bromine (2.98 mL, 58.1 mmol) was added dropwise to a solution of 3-butenoic acid (5.0 g, 58.1 mmol) in DCM (50 mL) at 0 °C. Once added, the reaction mixture was warmed to RT and kept stirring for 16 h. The reaction mixture was then quenched with aqueous 10% sodium thiosulfate solution, dried over Na2SO4, filtered and concentrated under reduced pressure, affording 13.25 g (93%) of 18 as a yellowish oil. 1H NMR (300 MHz, CDCl3, δ): 9.58 (bs, 1H); 4.48 (m, 1H), 3.92 (dd, J = 10.4, 4.3 Hz, 1H), 3.72 (t, J = 10.4 Hz, 1H), 3.44 (dd, J = 17.1, 3.8 Hz, 1H), 2.93 ppm (dd, J = 17.1, 9.2 Hz, 1H).
Methyl 3,4-dibromobutyrate (19): 0.5 mL of H2SO4 conc. was added dropwise to a solution of 18 (13.25 g, 53.8 mmol) and trimethyl orthoformate (11.35 g, 107.0 mmol) in methanol (150 mL) at 0 °C. Once added, the reaction mixture was warmed to RT and then refluxed, keeping stirring, for 16 h. The reaction mixture was then concentrated under reduced pressure, resumed with ethyl acetate (150 mL), washed with aqueous 10% NaHCO3 (2 × 100 mL) and brine (100 mL), dried over Na2SO4, filtered and concentrated under vacuum, affording 10.9 g (78%) of 19 as a yellowish oil. 1H NMR (300 MHz, CDCl3, δ): 4.50 (m, 1H), 3.91 (dd, J = 10.4, 4.3 Hz, 1H), 3.76 (s, 3H), 3.72 (dd, J = 10.2, 2.7 Hz, 1H), 3.34 (dd, J = 17.1, 3.8 Hz, 1H), 2.87 ppm (dd, J = 16.7, 9.2 Hz, 1H).
Methyl (1,4-benzoxathian-2-yl)-acetate (20): Methyl 3,4-dibromobutirrate (1.50 g, 5.77 mmol) was added dropwise to a solution of 1,2-mercaptophenol (0.73 g, 5.77 mmol) and TEA (1.16 mL, 11.54 mmol) in acetonitrile/water 1:1 (15 mL) and stirred at room temperature for 18 h. The reaction mixture is then added with ethyl acetate (15 mL), washed firstly with 10% aqueous NaOH (15 mL) and lastly with brine (15 mL). The organic phase is then dried over NaSO4, filtered and concentrated under vacuum, affording 1.01 g (78%) of 20 as a yellowish oil. 1H NMR (300 MHz, CDCl3, δ): δ 7.15–6.93 (m, 2H), 6.94–6.74 (m, 2H), 4.70 (m, 1H), 3.74 (s, 3H), 3.14 (dd, J = 13.0, 2.3 Hz, 1H), 3.04 (dd, J = 13.0, 7.6 Hz, 1H), 2.87 (dd, J = 15.8, 6.7 Hz, 1H), 2.74 ppm (dd, J = 15.8, 6.5 Hz, 1H).
2-hydroxyethyl-1,4-benzoxathiane (21): LiAlH4 (30 mg, 1.78 mmol) was suspended in dry THF (2 mL) at 0 °C under nitrogen atmosphere. The solution of 20 (0.18 g, 0.71 mmol) in THF (5 mL) was slowly added to the reaction. The mixture was then warmed to RT and stirred for 1 h; at completion, it was cooled to 0 °C and slowly quenched with ethyl acetate (5 mL). Further ethyl acetate (10 mL) was added, the organic layer was washed with brine (3 × 10 mL), dried over Na2SO4 and concentrated under vacuum to give 0.13 g (92%) of 21 as an orange oil. 1H NMR (300 MHz, CDCl3, δ): δ 7.10–6.91 (m, 2H), 6.91–6.75 (m, 2H), 4.44 (m, 1H), 4.02–3.79 (m, 2H), 3.12–2.95 (m, 2H), 2.15–1.87 (m, 2H), 1.78 ppm (bs, 1H).
2-mesyloxyethyl-1,4-benzoxathiane (22): Mesyl chloride (0.24 mL, 3.12 mmol) was added dropwise to a solution of 21 (0.51 g, 2.59 mmol) and TEA (0.44 mL, 3.12 mmol) in DCM (5 mL) at 0 °C. The mixture was stirred at that temperature for 3 h, diluted with DCM (20 mL), washed firstly with 10% aqueous NaHCO3 (10 mL), secondly with 10% aqueous HCl (10 mL) and finally with brine (10 mL), dried over Na2SO4, filtered and concentrated under vacuum to yield 0.71 g (quantitative) of 22 as a yellowish oil. 1H NMR (300 MHz, CDCl3, δ): δ 7.11–6.93 (m, 2H), 6.85 (m, 2H), 4.61–4.32 (m, 3H), 3.04–3.01 (m, 2H), 3.03 (s, 3H), 2.18 ppm (m, 2H).
3-(1,4-benzoxathian-2-yl)ethoxy)-2,6-difluorobenzamide (6): Potassium carbonate (0.40 g, 2.86 mmol) was added to a solution of 2,6-difluoro-3-hydroxybenzamide (0.49 g, 2.73 mmol) in dry DMF (5 mL). After stirring at room temperature for 30 min, a solution of 22 (0.70 g, 2.55 mmol) in DMF (5 mL) was added. The reaction mixture was stirred at 60 °C for 16 h, concentrated under vacuum, diluted with ethyl acetate (30 mL), washed with brine (3 × 20 mL), dried over Na2SO4, filtered and concentrated, to give a residue, which was purified by flash chromatography on silica gel. Elution with 1/1 cyclohexane/ethyl acetate and further crystallization from IPA gave 0.36 g (39%) of 6 as a white solid. mp: 109.75 °C. Tr (HPLC): 14.1 min (A% = 99.4% at λ = 282 nm). 1H NMR (300 MHz, d6-DMSO, δ): 8.09 (bs, 1H), 7.82 (bs, 1H), 7.26 (dt, J = 9.3, 5.3 Hz, 1H), 7.10–6.94 (m, 3H), 6.89–6.74 (m, 2H), 4.38 (tdd, J = 8.1, 5.2, 2.1 Hz, 1H), 4.29–4.22 (m, 2H), 3.27 (dd, J = 13.2, 2.1 Hz, 1H), 3.07 (dd, J = 13.2, 8.1 Hz, 1H), 2.27–2.05 ppm (m, 2H). 13C NMR (75 MHz, d6-DMSO, δ): 161.8, 152.3 (dd, J = 241.2, 6.8 Hz), 151.3, 148.3 (dd, J = 248.4, 8.4 Hz), 143.3 (dd, J = 10.9, 3.3 Hz), 127.5, 126.0, 121.9, 118.8, 117.7, 117.0 (dd, J = 24.9, 20.5 Hz), 115.9 (dd, J = 9.4, 2.5 Hz), 111.4 (dd, J = 22.9, 4.0 Hz), 71.5, 66.0, 34.1, 29.1 ppm.
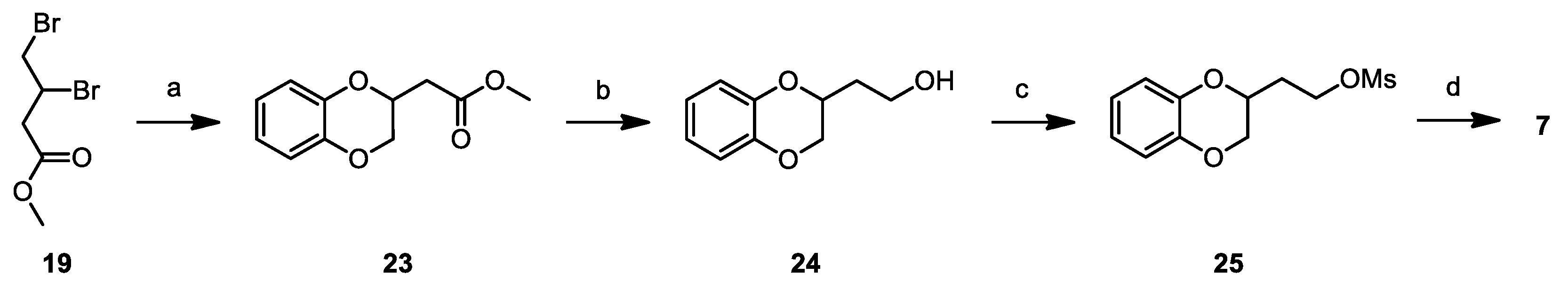
Methyl 1,4-benzodioxane-2-yl-acetate (23): Potassium carbonate (4.78 g, 34.00 mmol) was added to a solution of catechol (1.2 g, 11.00 mmol) in acetone (20 mL). After stirring at room temperature for 30 min, a solution of 19 (3.00 g, 11.00 mmol) in acetone (20 mL) was added. The reaction mixture was stirred at 60 °C for 16 h, concentrated under vacuum, diluted with Ethyl Acetate (50 mL), washed with brine (3 × 40 mL), dried over Na2SO4, filtered and concentrated, to give a residue, which was purified by flash chromatography on silica gel. Elution with 8/2 Cyclohexane/Ethyl Acetate gave 1.98 g (87%) of 23 as a colourless oil. 1H NMR (300 MHz, CDCl3, δ): 6.86 (m, 4H); 4.63 (tt, J = 6.7, 3.3 Hz, 1H), 4.32 (dd, J = 11.3, 2.2 Hz, 1H), 4.00 (dd, J = 11.3, 6.8 Hz, 1H), 3.75 (s, 3H), 2.80 (dd, J = 16.1, 6.8 Hz, 1H), 2.65 ppm (dd, J = 16.1, 6.7 Hz, 1H).
2-hydroxyethyl-1,4-benzodioxane (24): LiAlH4 (0.37 g, 10.00 mmol) was suspended in dry THF (2 mL) at 0 °C under nitrogen atmosphere. A solution of 23 (1.98 g, 9.5 mmol) in THF (5 mL) was added dropwise to the mixture; once added, the reaction was warmed to RT and stirred for 1 h; at completion, it was cooled to 0 °C and slowly quenched with ethyl acetate (10 mL). Further ethyl acetate (10 mL) was added, the organic layer was washed with brine (3 × 20 mL), dried over Na2SO4 and concentrated under vacuum to give 1.60 g (93%) of 24 as a colorless oil. 1H NMR (300 MHz, CDCl3, δ): 6.87 (m, 4H), 4.38 (m, 1H), 4.28 (dd, J = 11.3, 2.2 Hz, 1H), 3.94 (m, 3H), 1.91 ppm (m, 2H).
2-mesiloxyethyl-1,4-benzodioxane (25): Mesyl chloride (1.00 mL, 13.32 mmol) was added dropwise to a solution of 24 (1.60 g, 8.88 mmol) and TEA (1.80 mL, 13.32 mmol) in DCM (20 mL) at 0 °C. The mixture was warmed to RT and stirred at that temperature for 2 h, diluted with further DCM (20 mL), washed firstly with 10% aqueous NaHCO3 (30 mL), secondly with 10% aqueous HCl (30 mL) and finally with brine (30 mL), dried over Na2SO4, filtered and concentrated under vacuum to yield 2.10 g (91%) of 25 as yellowish oil. 1H NMR (300 MHz, CDCl3, δ): 6.87 (m, 4H), 4.48 (ddd, J = 10.2, 7.0, 5.1 Hz, 2H), 4.34 (m, 1H), 4.27 (dd, J = 11.4, 2.3 Hz, 1H), 3.96 (dd, J = 11.4, 7.0 Hz, 1H), 3.05 (s, 3H), 2.09 ppm (m, 2H).
3-(1,4-benzodioxane-2-yl)ethoxy)-2,6-difluorobenzamide (7): Potassium carbonate (0.42 g, 3.01 mmol) was added to a solution of 2,6-difluoro-3-hydroxybenzamide (0.50 g, 2.88 mmol) in dry DMF (5 mL). After stirring at room temperature for 30 min, a solution of 25 (0.71 g, 2.75 mmol) in DMF (5 mL) was added. The reaction mixture was stirred at 60 °C for 16 h, concentrated under vacuum, diluted with ethyl acetate (30 mL), washed with brine (3 × 20 mL), dried over Na2SO4, filtered and concentrated, to give a residue which was crystallized from 3/2 cyclohexane/ethyl Acetate, accomplishing 0.50 g (54%) of 7 as a white solid. mp: 96 °C. Tr (HPLC): 13.2 min (A% = 98.0% at λ = 278 nm). 1H NMR (300 MHz, d6-DMSO, δ): 8.12 (bs, 1H), 7.85 (bs, 1H), 7.27 (td, J = 9.4, 5.3 Hz, 1H), 7.07 (t, J = 8.9 Hz, 1H), 6.84 (m, 4H), 4.37 (dd, J = 16.8, 5.7 Hz, 2H), 4.24 (t, J = 6.1 Hz, 2H), 3.97 (dd, J = 11.2, 7.2 Hz, 1H), 2.08 ppm (dd, J = 11.0, 6.2 Hz, 2H). 13C NMR (75 MHz, d6-DMSO, δ): 161.8, 152.3 (dd, J = 239.9, 6.8 Hz), 148.3 (dd, J = 246.9, 8.4 Hz), 143.4, 143.3 (d, J = 10.8 Hz), 143.2, 121.8, 121.7, 117.6, 117.3, 117.1 (dd, J = 25.6, 21.0 Hz), 115.9 (d, J = 7.1 Hz), 111.4 (dd, J = 22.8, 3.9 Hz), 70.5, 67.6, 66.8, 30.4 ppm.
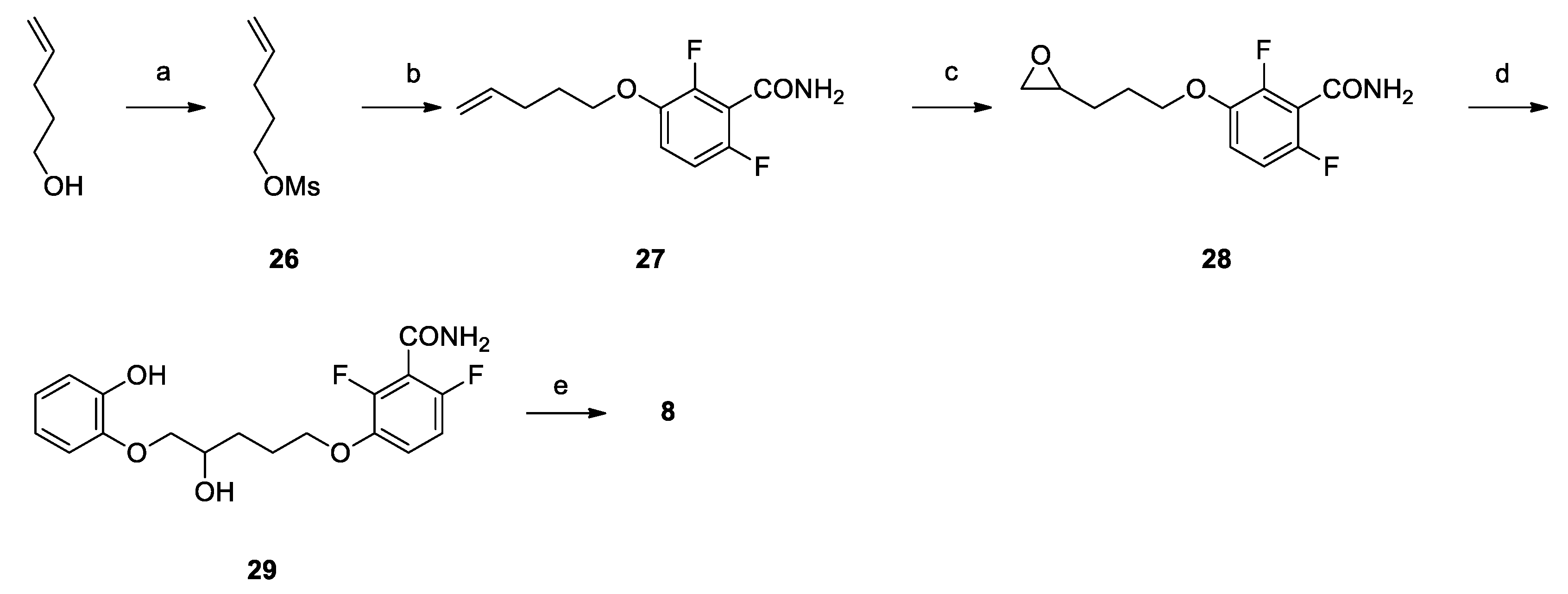
Pent-4-en-1-yl methanesulfonate (26): Mesyl chloride (1.08 mL, 13.93 mmol) was added dropwise to a solution of 4-penten-1-ol (1.00 g, 11.61 mmol) and TEA (1.90 mL, 13.93 mmol) in DCM (10 mL) at −15 °C. The mixture was stirred at that temperature for 3 h, diluted with DCM (20 mL), washed firstly with 10% aqueous NaHCO3 (10 mL), secondly with 10% aqueous HCl (10 mL) and finally with brine (10 mL), dried over Na2SO4, filtered and concentrated under vacuum to yield 1.52 g (80%) of 26 as a colorless oil. 1H NMR (300 MHz, CDCl3, δ): 5.78 (ddt, J = 16.9, 10.2, 6.7 Hz, 1H), 5.12–4.97 (m, 2H), 4.23 (t, J = 6.5 Hz, 2H), 3.00 (s, 3H), 2.25–2.12 (m, 2H), 1.94–1.79 ppm (m, 2H).
2,6-difluoro-3-(pent-4-en-1-yloxy)benzamide (27): Potassium carbonate (0.48 g, 3.48 mmol) was added to a solution of 2,6-difluoro-3-hydroxybenzamide (0.57 g, 3.32 mmol) in dry DMF (5 mL). After stirring at room temperature for 30 min, a solution of 26 (0.52 g, 3.17 mmol) in DMF (5 mL) was added. The reaction mixture was stirred at 80 °C for 16 h, concentrated under vacuum, diluted with ethyl acetate (30 mL), washed firstly with 10% aqueous NaHCO3 (10 mL), secondly with 10% aqueous HCl (10 mL) and finally with brine (10 mL), dried over Na2SO4, filtered and concentrated, to give 0.71 g (93%) of 27 as a brown oil. 1H NMR (300 MHz, CDCl3, δ): 6.99 (td, J = 9.1, 5.2 Hz, 1H), 6.86 (td, J = 9.1, 1.9 Hz, 1H), 5.98 (bs, 2H), 5.83 (ddt, J = 16.9, 10.2, 7.0 Hz, 1H), 5.12–4.92 (m, 2H), 4.01 (t, J = 6.4 Hz, 2H), 2.24 (dd, J = 14.1, 7.0 Hz, 2H), 1.98–1.81 ppm (m, 2H).
2,6-difluoro-3-(3-(oxiran-2-yl)propoxy)benzamide (28):m-CPBA (0.31 g, 1.78 mmol) was added to a solution of 27 (0.43 g, 1.78 mmol) in DCM (5 mL) at 0 °C. The reaction mixture was stirred at room temperature for 18 h, diluted with DCM, washed with 10% aqueous NaHCO3 (2 × 15 mL) dried over NaSO4, filtered and concentrated to give 0.42 g (91%) of 28 as a colorless oil 1H NMR (300 MHz, CDCl3, δ): 7.00 (td, J = 9.1, 5.2 Hz, 1H), 6.86 (td, J = 9.1, 1.9 Hz, 1H), 6.16 (bs, 1H), 6.03 (bs, 1H), 4.19–3.94 (m, 2H), 3.07–2.89 (m, 1H), 2.78 (dd, J = 4.8, 4.1 Hz, 1H), 2.51 (dd, J = 4.8, 2.7 Hz, 1H), 2.03–1.74 (m, 2H), 1.74–1.43 ppm (m, 2H).
2,6-difluoro-3-((4-hydroxy-5-(2-hydroxyphenoxy)pentyl)oxy)benzamide (29): NaH (0.04 g, 1.63 mmol) was suspended in dry DMF (5 mL) at 0 °C under nitrogen atmosphere. A solution of catechol (0.18 g, 1.63 mmol) in DMF (5 mL) was added dropwise and, after stirring at room temperature for 30 minutes, a solution of 28 (0.42 g, 1.63 mmol) in DMF (5 mL) was then added dropwise. The reaction mixture is heated at 100 °C and stirred for 72 h. The mixture was then cooled to RT and slowly quenched with 1/1 ethyl acetate and 10% aqueous HCl (40 mL). The phases were separated and the organic one was washed with brine (10 mL), dried over NaSO4, filtered and concentrated to yield 0.46 g (77%) of 29 as a reddish oil. 1H NMR (300 MHz, d6-DMSO, δ): 8.62 (bs, 1H), 8.08 (bs, 1H), 7.80 (s, 1H), 7.20 (td, J = 9.3, 5.3 Hz, 1H), 7.03 (td, J = 9.3, 1.9 Hz, 1H), 6.89 (d, J = 6.9 Hz, 1H), 6.79–6.60 (m, 3H), 5.00 (s, 1H), 4.11–3.94 (m, 2H), 3.94–3.78 (m, 2H), 3.72 (dd, J = 8.8, 6.3 Hz, 1H), 1.95–1.45 ppm (m, 4H).
3-(1,4-benzodioxane-2-yl)propoxy)-2,6-difluorobenzamide (8): Under nitrogen atmosphere, to a solution of triphenylphosphine (0.48 g, 1.84 mmol) in THF (2 mL), DIAD (0.36 mL, 1.84 mmol) and a solution of 28 (0.45 g, 1.23 mmol) in THF (1 mL) were added dropwise at 0 °C. The reaction mixture was stirred at reflux for 24 h, concentrated under vacuum, diluted with ethyl acetate (10 mL), washed firstly with brine (10 mL), secondly with 10% aqueous NaOH (10 mL) and lastly with brine (10 mL), dried over NaSO4, filtered and concentrated, to give a residue which was purified by flash chromatography on silica gel. Elution with 1/1 cyclohexane/ethyl acetate gave 0.05 g (11%) of 8 as a white wax. Tr (HPLC): 13.7 min (A% = 99.2% at λ = 279 nm). 1H NMR (300 MHz, d6-DMSO, δ): 8.11 (bs, 1H), 7.83 (bs, 1H), 7.20 (td, J = 9.3, 5.3 Hz, 1H), 7.04 (td, J = 9.0, 1.8 Hz, 1H), 6.87–6.70 (m, 4H), 4.31 (dd, J = 11.4, 2.2 Hz, 1H), 4.25–4.14 (m, 1H), 4.08 (t, J = 6.3 Hz, 2H), 3.87 (dd, J = 11.4, 7.5 Hz, 1H), 2.00–1.79 (m, 2H), 1.79–1.59 ppm (m, 2H). 13C NMR (75 MHz, d6-DMSO, δ): 161.81, 152.13 (dd, J = 240.8, 6.8 Hz), 148.29 (dd, J = 248.2, 8.5 Hz), 143.46, 143.50 (dd, J =10.6, 3.2 Hz), 143.45, 121.72, 121.51, 117.51, 117.23, 117.0 (dd, J = 23.0, 18.6 Hz), 115.83 (dd, J = 9.3, 2.5 Hz), 111.33 (dd, J = 22.8, 4.0 Hz), 72.80, 69.59, 67.67, 27.22, 24.68 ppm.

 ,
, 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




