Advanced Resistance Studies Identify Two Discrete Mechanisms in Staphylococcus aureus to Overcome Antibacterial Compounds that Target Biotin Protein Ligase

 ,
,  , , , ,
, , , ,
Abstract
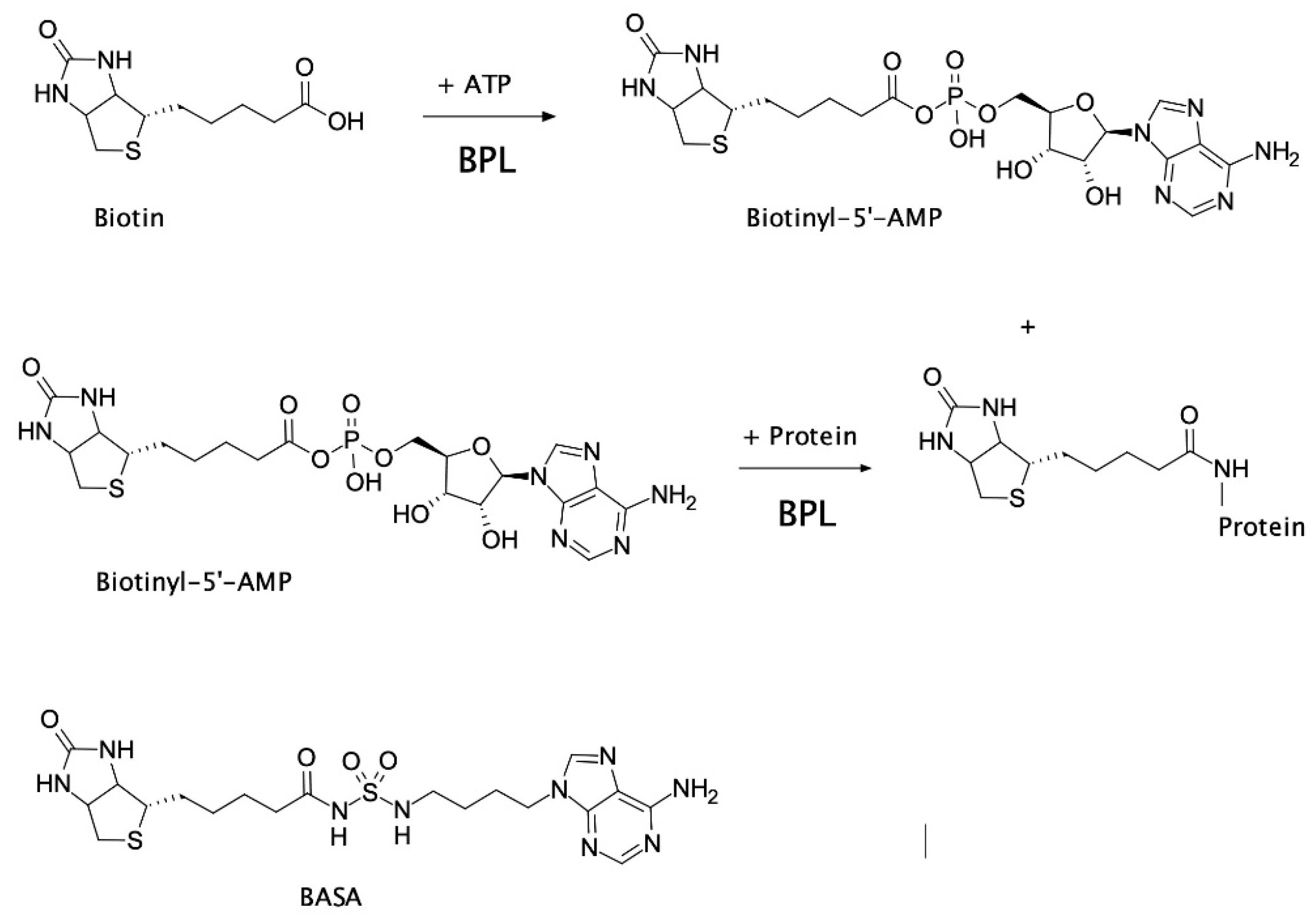
:1. Introduction
2. Results
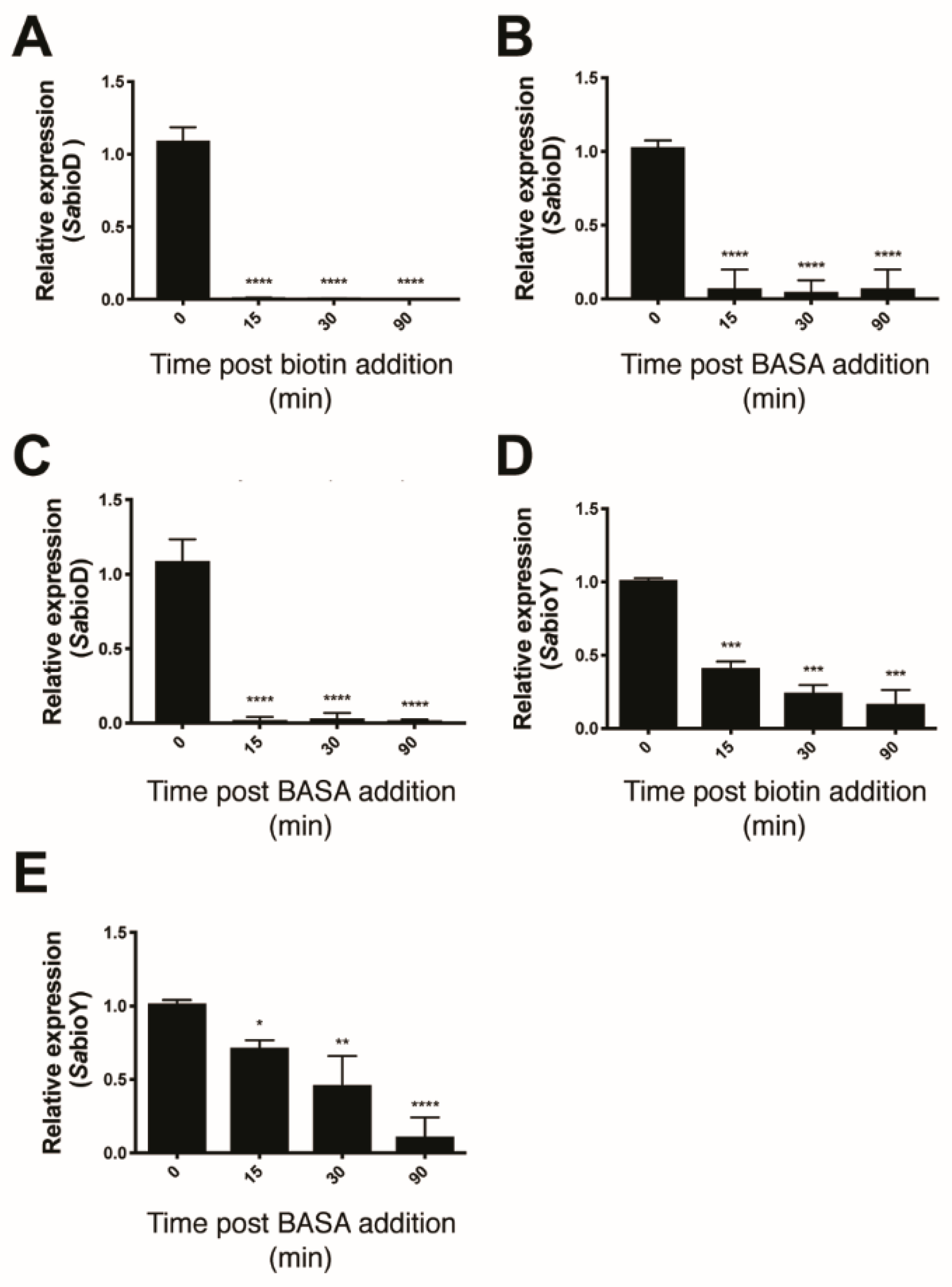
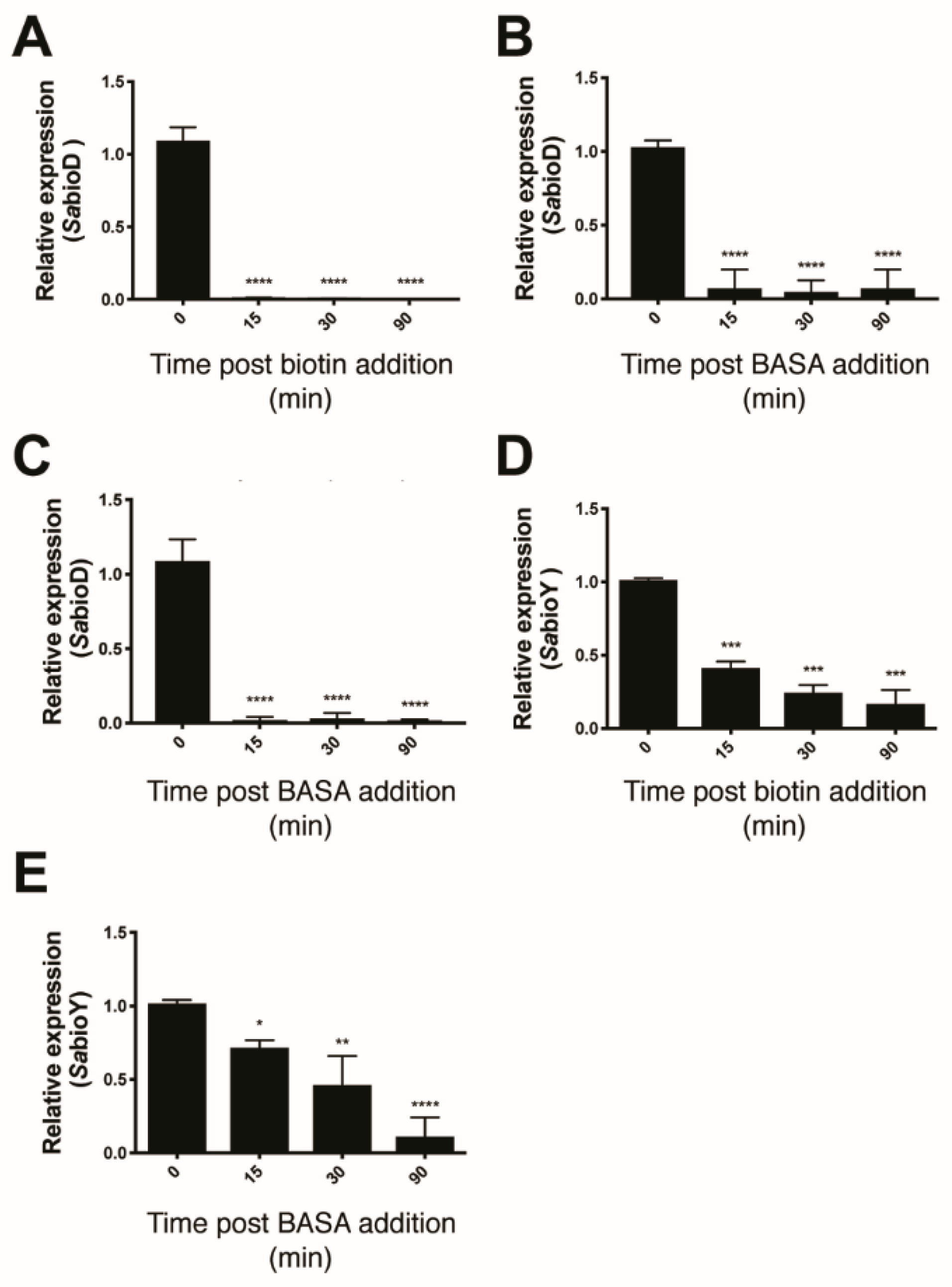
2.1. Biological Evaluation of BASA
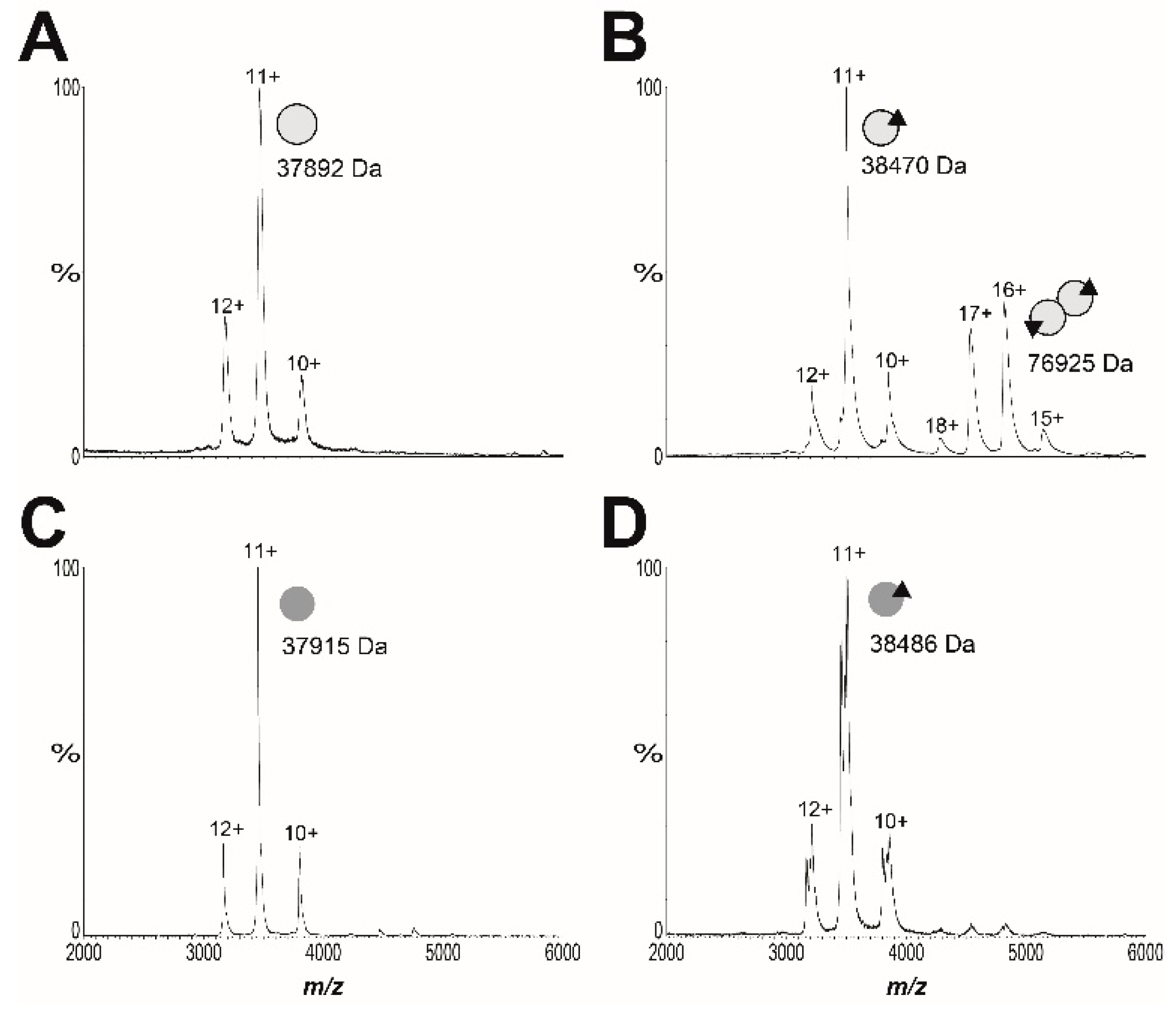
2.2. Mechanism of Action of BASA
2.3. Advanced Resistance Studies Identify Two Resistance Mechanisms
2.4. Resistance Mechanism 1: Deletion of pyc
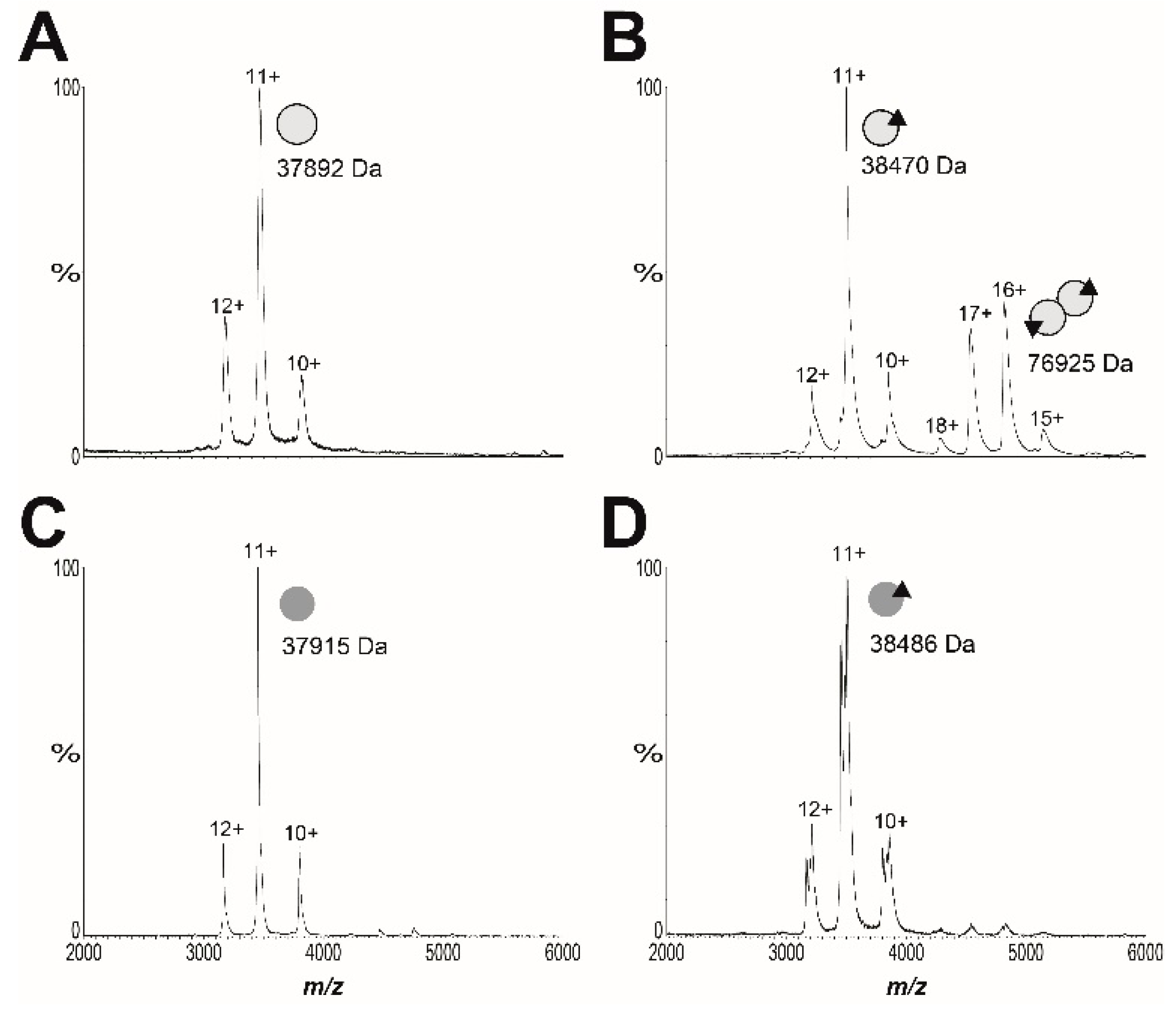
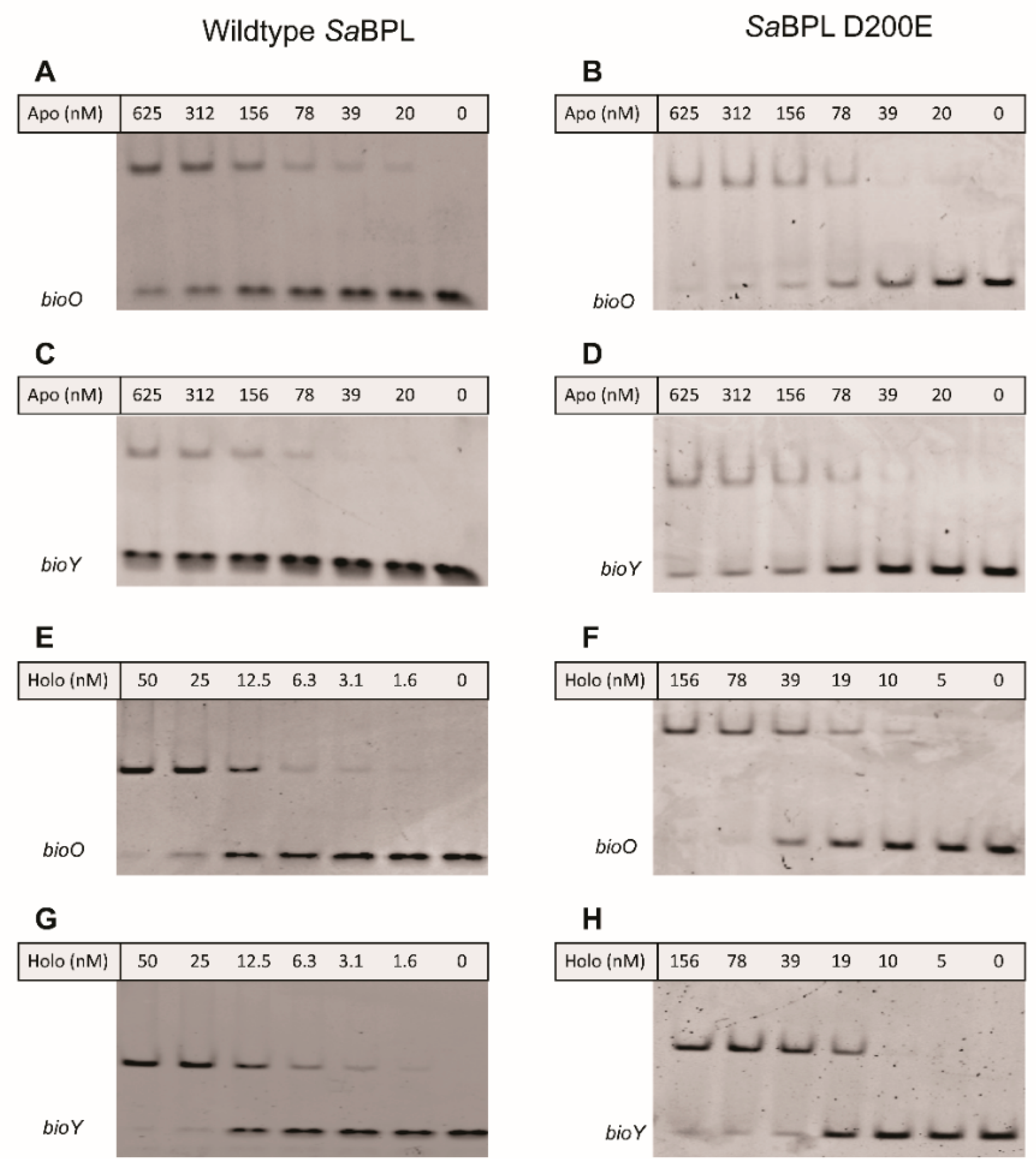
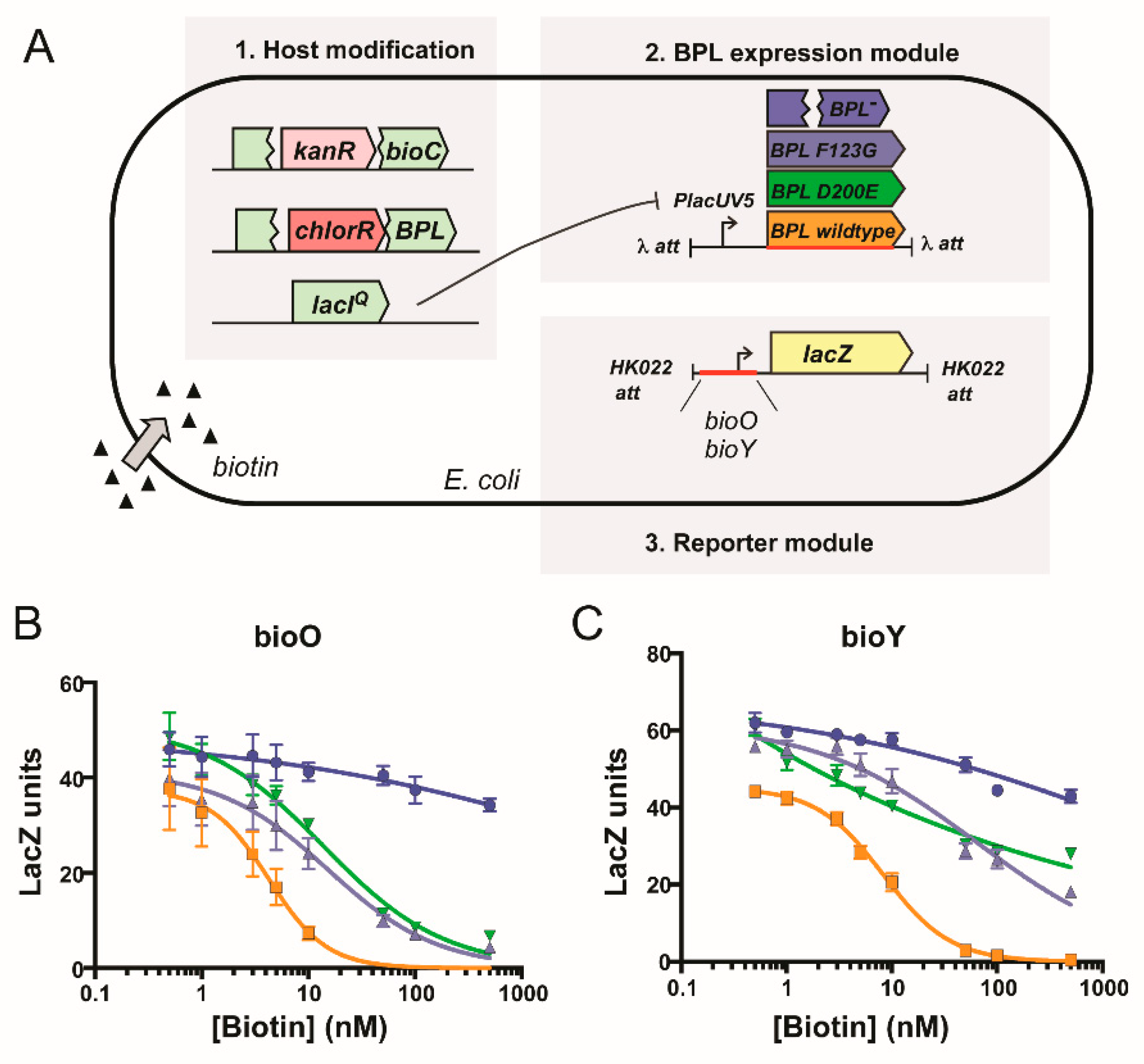
2.5. Resistance Mechanism 2: Missense Mutation in the BPL Target
3. Discussion
4. Materials and Methods
4.1. General Bacterial Culture and Molecular Biology Reagents
4.2. Quantification of Gene Expression Using qRT–PCR
4.3. Electrophoretic Mobility Shift Assay (EMSA)
4.4. Spontaneous Resistance Rate
4.5. Generation of Resistant Mutants by Serial Passaging
4.6. Genomic DNA Purification and Whole-Genome Sequencing
4.7. Bioinformatic Analysis
4.8. Antimicrobial Susceptibility Testing/Cross Resistance
4.9. Measurement of the Kinetics of Bacterial Growth
4.10. Protein Methods
4.11. Mass Spectrometry
4.12. Chromosomal Integration and β-Galactosidase Reporter Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cooper, M.A.; Shlaes, D. Fix the antibiotics pipeline. Nature. 2011, 472, 32. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.S.; Blaskovich, M.A.T.; Cooper, M.A. Antibiotics in the clinical pipeline at the end of 2015. J. Antibiot. 2016, 70, 3–24. [Google Scholar] [CrossRef] [PubMed]
- Rushton, J.; Stärk, K.D.C.; Ferreira, J.P. Antimicrobial Resistance; Organisation for Economic Co-Operation and Development (OECD): Paris, France, 2014. [Google Scholar]
- World Health Organisation. Global Action Plan on Antimicrobial Resistance; WHO: Geneva, Switzerland, 2015. [Google Scholar]
- Feng, J.; Paparella, A.; Booker, G.W.; Polyak, S.W.; Abell, A.D. Biotin Protein Ligase Is a Target for New Antibacterials. Antibiotics. 2016, 5, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paparella, A.; Da Costa, T.P.S.; Yap, M.; Tieu, W.; Wilce, M.; Booker, G.; Abell, A.; Polyak, S.W. Structure Guided Design of Biotin Protein Ligase Inhibitors for Antibiotic Discovery. Curr. Top. Med. Chem. 2013, 14, 4–20. [Google Scholar] [CrossRef]
- Lee, K.J.; Tieu, W.; Blanco-Rodriguez, B.; Paparella, A.S.; Yu, J.; Hayes, A.J.; Feng, J.; Marshall, A.C.; Noll, B.; Milne, R.; et al. Sulfonamide-Based Inhibitors of Biotin Protein Ligase as New Antibiotic Leads. ACS Chem. Boil. 2019, 14, 1990–1997. [Google Scholar] [CrossRef]
- Sternicki, L.; Wegener, K.L.; Bruning, J.B.; Booker, G.W.; Polyak, S.W. Mechanisms Governing Precise Protein Biotinylation. Trends Biochem. Sci. 2017, 42, 383–394. [Google Scholar] [CrossRef]
- Chaudhuri, R.; Allen, A.; Owen, P.J.; Shalom, G.; Stone, K.; Harrison, M.; A Burgis, T.; Lockyer, M.; Garcia-Lara, J.; Foster, S.J.; et al. Comprehensive identification of essential Staphylococcus aureus genes using Transposon-Mediated Differential Hybridisation (TMDH). BMC Genom. 2009, 10, 291. [Google Scholar] [CrossRef] [Green Version]
- Forsyth, R.A.; Haselbeck, R.J.; Ohlsen, K.L.; Yamamoto, R.T.; Xu, H.; Trawick, J.D.; Wall, D.; Wang, L.; Brown-Driver, V.; Froelich, J.M.; et al. A genome-wide strategy for the identification of essential genes in Staphylococcus aureus. Mol. Microbiol. 2002, 43, 1387–1400. [Google Scholar] [CrossRef]
- Payne, D.J.; Gwynn, M.N.; Holmes, D.J.; Pompliano, D.L. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 2006, 6, 29–40. [Google Scholar] [CrossRef]
- Tommasi, R.; Brown, D.G.; Walkup, G.K.; Manchester, J.I.; Miller, A.A. ESKAPEing the labyrinth of antibacterial discovery. Nat. Rev. Drug Discov. 2015, 14, 529–542. [Google Scholar] [CrossRef]
- Polyak, S.W.; Abell, A.D.; Wilce, M.C.J.; Zhang, L.; Booker, G.W. Structure, function and selective inhibition of bacterial acetyl-coa carboxylase. Appl. Microbiol. Biotechnol. 2011, 93, 983–992. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Rock, C. Bacterial fatty acid metabolism in modern antibiotic discovery. Biochim. et Biophys. Acta (BBA) - Mol. Cell Boil. Lipids 2016, 1862, 1300–1309. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, T.P.S.; Tieu, W.; Yap, M.Y.; Pendini, N.R.; Polyak, S.W.; Pedersen, D.S.; Morona, R.; Turnidge, J.D.; Wallace, J.C.; Wilce, M.C.J.; et al. Selective inhibition of Biotin Protein Ligase from Staphylococcus aureus*. J. Boil. Chem. 2012, 287, 17823–17832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duckworth, B.P.; Geders, T.; Tiwari, D.; Boshoff, H.I.; Sibbald, P.A.; Barry, C.E.; Schnappinger, D.; Finzel, B.; Aldrich, C.C. Bisubstrate Adenylation Inhibitors of Biotin Protein Ligase from Mycobacterium tuberculosis. Chem. Boil. 2011, 18, 1432–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, C.; Tiwari, D.; Wilson, D.J.; Seiler, C.L.; Schnappinger, D.; Aldrich, C.C. Bisubstrate Inhibitors of Biotin Protein Ligase in Mycobacterium tuberculosis Resistant to Cyclonucleoside Formation. ACS Med. Chem. Lett. 2013, 4, 1213–1217. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, T.P.S.; Tieu, W.; Yap, M.Y.; Zvarec, O.; Bell, J.M.; Turnidge, J.D.; Wallace, J.C.; Booker, G.W.; Wilce, J.A.; Abell, A.D.; et al. Biotin Analogues with Antibacterial Activity Are Potent Inhibitors of Biotin Protein Ligase. ACS Med. Chem. Lett. 2012, 3, 509–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, J.; Paparella, A.; Tieu, W.; Heim, D.; Clark, S.; Hayes, A.J.; Booker, G.W.; Polyak, S.W.; Abell, A.D. New Series of BPL Inhibitors To Probe the Ribose-Binding Pocket of Staphylococcus aureus Biotin Protein Ligase. ACS Med. Chem. Lett. 2016, 7, 1068–1072. [Google Scholar] [CrossRef] [Green Version]
- Benton, B.M.; Zhang, J.P.; Bond, S.; Pope, C.; Christian, T.; Lee, L.; Winterberg, K.M.; Schmid, M.B.; Buysse, J.M. Large-Scale Identification of Genes Required for Full Virulence of Staphylococcus aureus. J. Bacteriol. 2004, 186, 8478–8489. [Google Scholar] [CrossRef] [Green Version]
- Bae, T.; Banger, A.K.; Wallace, A.; Glass, E.M.; Åslund, F.; Schneewind, O.; Missiakas, M.M. Staphylococcus aureus virulence genes identified by bursa aurealis mutagenesis and nematode killing. Proc. Natl. Acad. Sci. USA 2004, 101, 12312–12317. [Google Scholar] [CrossRef] [Green Version]
- Rodionov, D.A.; Mironov, A.A.; Gelfand, M. Conservation of the Biotin Regulon and the BirA Regulatory Signal in Eubacteria and Archaea. Genome Res. 2002, 12, 1507–1516. [Google Scholar] [CrossRef] [Green Version]
- Satiaputra, J.; Eijkelkamp, B.; McDevitt, C.A.; Shearwin, K.; Booker, G.W.; Polyak, S.W. Biotin-mediated growth and gene expression in Staphylococcus aureus is highly responsive to environmental biotin. Appl. Microbiol. Biotechnol. 2018, 102, 3793–3803. [Google Scholar] [CrossRef] [PubMed]
- Satiaputra, J.; Shearwin, K.E.; Booker, G.; Polyak, S.W. Mechanisms of biotin-regulated gene expression in microbes. Synth. Syst. Biotechnol. 2016, 1, 17–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satiaputra, J.; Sternicki, L.M.; Hayes, A.J.; Pukala, T.L.; Booker, G.W.; Shearwin, K.; Polyak, S.W. Native mass spectrometry identifies an alternative DNA-binding pathway for BirA from Staphylococcus aureus. Sci. Rep. 2019, 9, 2767. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, T.P.S.; Yap, M.Y.; Perugini, M.; Wallace, J.C.; Abell, A.D.; Wilce, M.C.J.; Polyak, S.W.; Booker, G.W. Dual roles of F123 in protein homodimerization and inhibitor binding to biotin protein ligase from Staphylococcus aureus. Mol. Microbiol. 2013, 91, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Pendini, N.R.; Yap, M.Y.; Polyak, S.W.; Cowieson, N.P.; Abell, A.; Booker, G.W.; Wallace, J.C.; Wilce, J.A.; Wilce, M.C.J. Structural characterization of Staphylococcus aureus biotin protein ligase and interaction partners: An antibiotic target. Protein Sci. 2013, 22, 762–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, P.H.; Cronan, J.E.; Grøtli, M.; Beckett, D. The Biotin Repressor: Modulation of Allostery by Corepressor Analogs. J. Mol. Boil. 2004, 337, 857–869. [Google Scholar] [CrossRef] [PubMed]
- Wood, Z.A.; Weaver, L.H.; Brown, P.H.; Beckett, D.; Matthews, B.W. Co-repressor Induced Order and Biotin Repressor Dimerization: A Case for Divergent Followed by Convergent Evolution. J. Mol. Boil. 2006, 357, 509–523. [Google Scholar] [CrossRef]
- Paparella, A.; Lee, K.J.; Hayes, A.J.; Feng, J.; Feng, Z.; Cini, D.; Deshmukh, S.; Booker, G.W.; Wilce, M.C.J.; Polyak, S.W.; et al. Halogenation of Biotin Protein Ligase Inhibitors Improves Whole Cell Activity against Staphylococcus aureus. ACS Infect. Dis. 2017, 4, 175–184. [Google Scholar] [CrossRef]
- Tieu, W.; Jarrad, A.; Paparella, A.; Keeling, K.A.; Da Costa, T.P.S.; Wallace, J.C.; Booker, G.W.; Polyak, S.W.; Abell, A.D. Heterocyclic acyl-phosphate bioisostere-based inhibitors of Staphylococcus aureus biotin protein ligase. Bioorganic Med. Chem. Lett. 2014, 24, 4689–4693. [Google Scholar] [CrossRef]
- Tieu, W.; Polyak, S.W.; Paparella, A.; Yap, M.Y.; Da Costa, T.P.S.; Ng, B.; Wang, G.; Lumb, R.; Bell, J.M.; Turnidge, J.D.; et al. Improved Synthesis of Biotinol-5′-AMP: Implications for Antibacterial Discovery. ACS Med. Chem. Lett. 2014, 6, 216–220. [Google Scholar] [CrossRef] [Green Version]
- Bockman, M.R.; Engelhart, C.A.; Dawadi, S.; Larson, P.; Tiwari, D.; Ferguson, D.M.; Schnappinger, D.; Aldrich, C.C. Avoiding Antibiotic Inactivation in Mycobacterium tuberculosis by Rv3406 through Strategic Nucleoside Modification. ACS Infect. Dis. 2018, 4, 1102–1113. [Google Scholar] [CrossRef] [PubMed]
- Bockman, M.R.; Kalinda, A.S.; Petrelli, R.; De La Mora-Rey, T.; Tiwari, D.; Liu, F.; Dawadi, S.; Nandakumar, M.; Rhee, K.Y.; Schnappinger, D.; et al. Targeting Mycobacterium tuberculosis Biotin Protein Ligase (MtBPL) with Nucleoside-Based Bisubstrate Adenylation Inhibitors. J. Med. Chem. 2015, 58, 7349–7369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, L. Synergism Testing: Broth Microdilution Checkerboard and Broth Macrodilution Methods. In Clinical Microbiology Procedures Handbook, 3rd ed.; ASM Press: Washington, DC, USA, 2010; pp. 140–162. [Google Scholar]
- Odds, F.C. Synergy, antagonism, and what the chequerboard puts between them. J. Antimicrob. Chemother. 2003, 52, 1. [Google Scholar] [CrossRef] [PubMed]
- Blake, K.L.; Randall, C.; O’Neill, A. In Vitro Studies Indicate a High Resistance Potential for the Lantibiotic Nisin in Staphylococcus aureus and Define a Genetic Basis for Nisin Resistance. Antimicrob. Agents Chemother. 2011, 55, 2362–2368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sims, D.; Sudbery, I.; Ilott, N.; Heger, A.; Ponting, C.P. Sequencing depth and coverage: key considerations in genomic analyses. Nat. Rev. Genet. 2014, 15, 121–132. [Google Scholar] [CrossRef]
- Broeke-Smits, N.J.P.T.; Pronk, T.; Jongerius, I.; Bruning, O.; Wittink, F.R.; Breit, T.M.; Van Strijp, J.A.G.; Fluit, A.C.; Boel, C.H.E. Operon structure of Staphylococcus aureus. Nucleic Acids Res. 2010, 38, 3263–3274. [Google Scholar] [CrossRef] [Green Version]
- Rahman, M.M.; Hunter, H.N.; Prova, S.; Verma, V.; Qamar, A.; Golemi-Kotra, D. The Staphylococcus aureus Methicillin Resistance Factor FmtA Is a d-Amino Esterase That Acts on Teichoic Acids. mBio 2016. [Google Scholar] [CrossRef] [Green Version]
- Corrigan, R.M.; Abbott, J.; Burhenne, H.; Kaever, V.; Gründling, A. c-di-AMP Is a New Second Messenger in Staphylococcus aureus with a Role in Controlling Cell Size and Envelope Stress. PLOS Pathog. 2011, 7, e1002217. [Google Scholar] [CrossRef] [Green Version]
- Engman, J.; Rogstam, A.; Frees, D.; Ingmer, H.; Von Wachenfeldt, C. The YjbH Adaptor Protein Enhances Proteolysis of the Transcriptional Regulator Spx in Staphylococcus aureus. J. Bacteriol. 2011, 194, 1186–1194. [Google Scholar] [CrossRef] [Green Version]
- Fey, P.D.; Endres, J.L.; Yajjala, V.K.; Widhelm, T.J.; Boissy, R.J.; Bose, J.L.; Bayles, K.W. A Genetic Resource for Rapid and Comprehensive Phenotype Screening of Nonessential Staphylococcus aureus Genes. mBio 2013, 4, e00537-12. [Google Scholar] [CrossRef] [Green Version]
- Pendini, N.R.; Polyak, S.W.; Booker, G.W.; Wallace, J.C.; Wilce, J.A. Purification, crystallization and preliminary crystallographic analysis of biotin protein ligase from Staphylococcus aureus. Acta Crystallogr. Sect. F Struct. Boil. Cryst. Commun. 2008, 64, 520–523. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, F.; Cui, L.; Priest, D.; Endy, D.; Dodd, I.; Shearwin, K. One-Step Cloning and Chromosomal Integration of DNA. ACS Synth. Boil. 2013, 2, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Blount, K.; Breaker, R.R. Riboswitches as antibacterial drug targets. Nat. Biotechnol. 2006, 24, 1558–1564. [Google Scholar] [CrossRef]
- Pedrolli, D.; Matern, A.; Wang, J.; Ester, M.; Siedler, K.; Breaker, R.R.; Mack, M. A highly specialized flavin mononucleotide riboswitch responds differently to similar ligands and confers roseoflavin resistance to Streptomyces davawensis. Nucleic Acids Res. 2012, 40, 8662–8673. [Google Scholar] [CrossRef] [PubMed]
- Wilson, K.P.; Shewchuk, L.M.; Brennan, R.G.; Otsuka, A.J.; Matthews, B.W. Escherichia coli biotin holoenzyme synthetase/bio repressor crystal structure delineates the biotin- and DNA-binding domains. Proc. Natl. Acad. Sci. USA 1992, 89, 9257–9261. [Google Scholar] [CrossRef] [Green Version]
- Buoncristiani, M.R.; Howard, P.K.; Otsuka, A.J. DNA-binding and enzymatic domains of the bifunctional biotin operon repressor (BirA) of Escherichia. Gene 1986, 44, 255–261. [Google Scholar] [CrossRef]
- Hellman, L.; Fried, M. Electrophoretic mobility shift assay (EMSA) for detecting protein–nucleic acid interactions. Nat. Protoc. 2007, 2, 1849–1861. [Google Scholar] [CrossRef]
- Pope, C.F.; O’Sullivan, D.M.; McHugh, T.D.; Gillespie, S.H. A Practical Guide to Measuring Mutation Rates in Antibiotic Resistance. Antimicrob. Agents Chemother. 2008, 52, 1209–1214. [Google Scholar] [CrossRef] [Green Version]
- Weiner, O.; Se, L.; M, D. Faculty of 1000 evaluation for Mutations of Bacteria from Virus Sensitivity to Virus Resistance. F1000 - Post-publication peer review of the biomedical literature 2010, 28, 491–511. [Google Scholar]
- Young, K. In Vitro Antibacterial Resistance Selection and Quantitation. Curr. Protoc. Pharmacol. 2006, 34, 13A.6.1–13A.6.22. [Google Scholar] [CrossRef]
- Friedman, L.; Alder, J.D.; Silverman, J.A. Genetic Changes That Correlate with Reduced Susceptibility to Daptomycin in Staphylococcus aureus. Antimicrob. Agents Chemother. 2006, 50, 2137–2145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deatherage, D.E.; Barrick, J.E. Identification of mutations in laboratory-evolved microbes from next-generation sequencing data using breseq. Breast Cancer 2014, 1151, 165–188. [Google Scholar]
- Giachino, P.; Engelmann, S.; Bischoff, M. ςB Activity Depends on RsbU in Staphylococcus aureus. J. Bacteriol. 2001, 183, 1843–1852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brune, I.; Götker, S.; Schneider, J.; Rodionov, D.A.; Tauch, A. Negative transcriptional control of biotin metabolism genes by the TetR-type regulator BioQ in biotin-auxotrophic Corynebacterium glutamicum ATCC 13032. J. Biotechnol. 2012, 159, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Chapman-Smith, A.; Turner, D.L.; E Cronan, J.; Morris, T.W.; Wallace, J.C. Expression, biotinylation and purification of a biotin-domain peptide from the biotin carboxy carrier protein of Escherichia coli acetyl-CoA carboxylase. Biochem. J. 1994, 302, 881–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | MIC (μg/mL) |
|---|---|
| Staphylococcus aureus | |
| Methicillin-sensitive (n = 8) | 0.25–0.5 |
| Methicillin-resistant (n = 9) | 0.25–0.5 |
| Coagulase negative Staphylococci (n = 7) | 0.125–0.5 |
| S. aureus ATCC 49775 | 0.5 |
| S. aureus ATCC 49975 MBH + 10% FCS | 0.5 |
| S. aureus ATCC 49975 MHB + 20% FCS | 4 |
| Mycobacterium tuberculosis (n = 1) | 55 |
| Streptococcus pneumoniae (n = 6) | >32 |
| Enterocococcus faecalis (n = 3) | >128 |
| Enterococcus faecium (n = 5) | >128 |
| Escherichia coli (n = 1) | >128 |
| Cell lines | EC50 (μg/mL) |
| HepG2 | >250 |
| HEK293 | >250 |
| Strain | MIC μg/mL BASA | Doubling Time (Min) | Genotype |
|---|---|---|---|
| NCTC 8325 | 0.065 | 51.8 ± 2.1 | |
| B1 | 1 | 69.2 ± 11.6 | Δpyc trkA pyc Δ194bp (2761-2954/3453) SAOUHSC_01981 E20K |
| B2 | 0.5 | 64.5 ± 1.42 | Δpyc Δ94bp intergenic (SAOUHSC_01476 to 01477) presumed silent mutation (no rsbU) |
| B3 | 0.125 | 193.6 ± 14.3 *** | gdpP, yjbH yjbH Q213- |
| B4 | 0.25 | 53.5 ± 5.5 | Δpyc Δ113bp intergenic (SAOUHSC_01476 to 01477) |
| B5 | 0.25 | 68.8 ± 12.1 | greA SAOUHSC _01504 P15L |
| B6 | 1 | 41.9 ± 0.73 | Δpyc, rpoβ, gtfA homolog Δ139bp intergenic (SAOUHSC_01476 to 01477) rpoβ P626L SAOUHSC_02984 (gtfA-like) R296H |
| B7 | 4 | 67.1 ± 0.90 | Δpyc, birA D200E, fmtA pyc Δ200bp (917-1116/3453) Δ139bp intergenic (SAOUHSC_01476 to 01477) fmtA K163 |
| JE2 | 0.5 | ND | Parent strain, USA300 JE2 |
| NR-47297 | 4 | ND | JE2 Δpyc |
| NR-47439 | 4 | ND | JE2 ΔyjbH |
| NR-47565 | 0.5 | ND | JE2 ΔfmtA |
| NR-47335 | 0.5 | ND | JE2 ΔgtfA |
| NR-47331 | 0.5 | ND | JE2 ΔtrkA |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hayes, A.J.; Satiaputra, J.; Sternicki, L.M.; Paparella, A.S.; Feng, Z.; Lee, K.J.; Blanco-Rodriguez, B.; Tieu, W.; Eijkelkamp, B.A.; Shearwin, K.E.; et al. Advanced Resistance Studies Identify Two Discrete Mechanisms in Staphylococcus aureus to Overcome Antibacterial Compounds that Target Biotin Protein Ligase. Antibiotics 2020, 9, 165. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics9040165
Hayes AJ, Satiaputra J, Sternicki LM, Paparella AS, Feng Z, Lee KJ, Blanco-Rodriguez B, Tieu W, Eijkelkamp BA, Shearwin KE, et al. Advanced Resistance Studies Identify Two Discrete Mechanisms in Staphylococcus aureus to Overcome Antibacterial Compounds that Target Biotin Protein Ligase. Antibiotics. 2020; 9(4):165. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics9040165
Chicago/Turabian StyleHayes, Andrew J., Jiulia Satiaputra, Louise M. Sternicki, Ashleigh S. Paparella, Zikai Feng, Kwang J. Lee, Beatriz Blanco-Rodriguez, William Tieu, Bart A. Eijkelkamp, Keith E. Shearwin, and et al. 2020. "Advanced Resistance Studies Identify Two Discrete Mechanisms in Staphylococcus aureus to Overcome Antibacterial Compounds that Target Biotin Protein Ligase" Antibiotics 9, no. 4: 165. https://0-doi-org.brum.beds.ac.uk/10.3390/antibiotics9040165